

Abstract
Background
Arsenic is both a human carcinogen and a chemotherapeutic agent, but the mechanism of neither arsenic-induced carcinogenesis nor tumor selective cytotoxicity is clear. Using a model cell line in which p53 expression is regulated exogenously in a tetracycline-off system (TR9-7 cells), our laboratory has shown that arsenite disrupts mitosis and that p53-deficient cells [p53(−)], in contrast to p53-expressing cells [p53(+)], display greater sensitivity to arsenite-induced mitotic arrest and apoptosis.
Objective
Our goal was to examine the role p53 plays in protecting cells from arsenite-induced mitotic arrest.
Methods
p53(+) and p53(−) cells were synchronized in G2 phase using Hoechst 33342 and released from synchrony in the presence or absence of 5 μM sodium arsenite.
Results
Mitotic index analysis demonstrated that arsenite treatment delayed exit from G2 in p53(+) and p53(−) cells. Arsenite-treated p53(+) cells exited mitosis normally, whereas p53(−) cells exited mitosis with delayed kinetics. Microarray analysis performed on mRNAs of cells exposed to arsenite for 0 and 3 hr after release from G2 phase synchrony showed that arsenite induced inhibitor of DNA binding-1 (ID1) differentially in p53(+)and p53(−) cells. Immunoblotting con-firmed that ID1 induction was more extensive and sustained in p53(+) cells.
Conclusions
p53 promotes mitotic exit and leads to more extensive ID1 induction by arsenite. ID1 is a dominant negative inhibitor of transcription that represses cell cycle regulatory genes and is elevated in many tumors. ID1 may play a role in the survival of arsenite-treated p53(+) cells and contribute to arsenic carcinogenicity.
Keywords: apoptosis, arsenite, ID1, microarray, mitotic arrest, p53
Arsenic is both a human carcinogen causing cancer in multiple tissues [National Research Council (NRC) 1999, 2001] and a chemo-therapeutic agent used in the treatment of acute promyelocytic leukemia (Cohen et al. 2001). Arsenic trioxide shows promise for chemotherapy in treatment of solid tumors and inhibits tumor growth in an orthotopic prostate cancer model (Maeda et al. 2001). However, the mechanism by which arsenic selectively induces cell death in tumor cells is not understood.
Clinically achievable concentrations of arsenite induce cell death in a variety of cancer cell lines and virally immortalized cell lines that lack normal cell cycle control. Arsenic trioxide (As2O3) induces apoptosis in several prostate and ovarian cancer cell lines (Du and Ho 2001; Uslu et al. 2000). Arsenite induces mitotic arrest and apoptosis in SV40-transformed human fibroblasts (States et al. 2002), spontaneously immortalized p53-deficient Li-Fraumeni human fibroblasts (Taylor et al. 2006), and in HeLa S3 and KB cells (Huang and Lee 1998). In contrast, these moderate concentrations of arsenite slow the growth of diploid human fibroblasts but do not induce cell death (States et al. 2002; Yih et al. 1997). However, arsenite disrupts normal mitotic progression and induces aneuploidy in human diploid fibroblasts (Yih et al. 1997) and peripheral blood lymphocytes (Vega et al. 1995).
A common feature of the cell lines in which arsenite induces apoptosis is a p53-deficient phenotype, either by mutation or viral oncogene inactivation of p53. p53 regulates cell cycle progression and apoptosis in response to genetic damage. The molecular mechanisms by which p53 arrests cell cycle progression in G1 and G2 phases in response to DNA damage are reasonably well understood (Jin and Levine 2001). p53 is activated in response to mitotic disruption by arsenic (Yih and Lee 2000). However, the signals for p53 activation and the role that p53 plays in the cellular response to mitotic disruption are not clear.
In asynchronous TR9-7 cells (human fibroblasts with tetracycline-regulated p53 expression), arsenite treatment induces a p53-independent accumulation of cells with G2/M phase DNA content (Taylor et al. 2006). However, accumulation of mitotic cells occurs in p53-deficient [p53(−)] but not in p53-expressing [p53( + )] TR9-7 cells. Furthermore, the mitotically arrested p53(−) cells undergo apoptosis, whereas most p53(+) cells exit G2/M phase and arrest in G1 phase. Therefore, we hypothesized that p53 did not prevent delayed G2 transit but did protect against apoptosis subsequent to the arsenite-induced mitotic arrest.
To investigate the role of p53 in the arsenite-induced disruption of G2/M phase progression, we examined the response of p53(+) and p53(−) TR9-7 cells to arsenite treatment after release from G2 phase synchrony. We show that in synchronized TR9-7 cells arsenite delayed entry into mitosis independently of p53 expression but that the exit from mitosis in arsenite-treated cells was p53 dependent. Furthermore, we show that the apoptotic process occurred while the cells attempted cytokinesis. We hypothesized the difference in exit kinetics and resistance to apoptosis between p53(+) and p53(−) cells was due to p53-mediated alterations of gene expression profiles. Microarray analysis of G2 phase–synchronized p53(+) and p53(−) cells treated with arsenite for 3 hr revealed p53-dependent induction of inhibitor of DNA binding-1 (ID1). Negative transcriptional regulation of cyclin-dependent kinase inhibitors by ID1 may have implications for cell cycle progression in p53(+) cells surviving arsenite-induced mitotic perturbation and may play a role in arsenic-induced carcinogenesis.
Materials and Methods
Cell culture and synchronization
TR9-7 cells were the gift of M.A. Tainsky (Wayne State University, Detroit, MI). Cells were cultured at 37ºC at 5% CO2 in a humidified incubator in alpha modification of minimal essential media (GIBCO/BRL, Gaithersburg, MD) supplemented with 10% fetal bovine serum (HyClone, Logan, UT), 10 mM HEPES (pH 7.0), 200 μg/mL geneticin (Invitrogen Corp., Carlsbad, CA), 40 μg/mL hygromycin B (Sigma Chemical Co., St. Louis, MO), 100 U/mL penicillin, and 10 U/mL streptomycin.
Cells were synchronized in G2 phase of the cell cycle according to adaptation of published procedures (Kuhholzer and Prather 2001; Tobey et al. 1990). Briefly, we plated cells in the presence of 15 ng/mL tetracycline (Fisher Scientific, Pittsburgh, PA) to allow moderate p53 expression. We replaced media with media containing 5 μg/mL aphidicolin (Sigma) and incubated the cells for 24 hr. We then washed the cells with phosphate-buffered saline (PBS) and incubated them in media containing 0.1 μg/mL Hoechst 33342 (Sigma) for 12 hr. After 6 hr of incubation with Hoechst 33342, p53 expression was suppressed in half the cultures by direct addition of tetracycline (1 mg/mL) to the media to a final concentration of 1,500 ng/mL. Cells were released from G2 phase by washing with PBS and refeeding with fresh drug-free media.
Arsenic compounds
We prepared fresh working aqueous solutions of sodium arsenite (NaAsO2) (Sigma) on the day of treatment and filter sterilized prior to use.
Flow cytometry
For DNA content analysis, cells were harvested by trypsinization and fixed in 1 mL ice-cold 70% ethanol. Following fixation overnight at 4ºC, we centrifuged the samples at 1,500 × g and resuspended the resulting cell pellets in PBS containing propidium iodide and 100 U/mL RNase A (Sigma) for 30 min at room temperature. We analyzed propidium iodide–stained samples on a FACSCalibur (Becton Dickinson and Co., San Jose, CA) using doublet discrimination. Propidium iodide fluorescence was collected on channel two (FL2) (585/42 nm) using linear amplification. A minimum of 20,000 events/sample was analyzed. Data were collected using CellQuest software (Becton Dickinson) and analyzed for cell cycle distribution using Modfit software (Verity Software House, Inc., Topsham, ME).
Mitotic indices
Cells were harvested for mitotic index analysis by trypsinization. Media, wash, and cells were collected together and centrifuged. We resuspended cell pellets in 150 μL serum free media and added 2.5 mL of 0.4% KCl. We incubated the suspension for 10 min at 37ºC, then added methanol:acetic acid fixative solution (3:1, vol/vol) to 2% (vol/vol) and collected cells by centrifugation. Cells were resuspended in 2.5 mL fixative solution and fixed at room temperature for 20 min. Samples were centrifuged; cell pellets were resuspended in 0.5 mL fixative and chilled on ice for 1 hr. Aliquots of the suspensions were dropped onto glass slides, air dried, and stained with Wright Giemsa solution (Fisher). We examined slides under a microscope and counted a minimum of 200 cells to determine mitotic index.
Western blot analyses
Media from culture dishes were removed and collected along with one PBS wash. Floating cells were pelleted via centrifugation, washed once with PBS, and lysed with lysis buffer [10 mM Tris–HCl (pH 7.4), 1 mM Na2–EDTA, 0.1% SDS, 180 μg/mL phenylmethylsulfonyl fluoride]. Adherent cells were lysed directly in the plates. Lysates from floating and adherent fractions were then combined. Protein concentration in the lysates was determined by Bradford assay (BioRad Laboratories, Inc., Hercules, CA). Proteins were resolved by SDS–PAGE in 12% polyacrylamide gels and transferred to supported nitrocellulose membranes by electro-blotting. p53, β-actin, and ID1 were detected by probing membranes with antibodies for p53 (LabVision, Fremont, CA), β-actin (Sigma), or ID1 (Santa Cruz Biotechnologies, Santa Cruz, CA), respectively. Blots were incubated with secondary anti-mouse or anti-rabbit antibody conjugated to horse radish peroxidase (Zymed Laboratories, San Francisco, CA) and visualized with enhanced chemiluminescence (Amersham Biosciences Inc., Piscataway, NJ).
Statistical analyses
Comparison of mitotic indices of arsenite-treated p53(+) and p53(−) cells was performed by Student t-test using SlideWrite software (version 6.10; Advanced Graphics Software, Encinitas, CA) and confirmed by analysis of variance (ANOVA).
Microarray analyses
We harvested cells via trypsinization and prepared poly(A)+ mRNAs using Micro-FastTrack (Invitrogen Corp.). SuperScript II (Invitrogen Corp.) was used with an oligo(dT) primer linked to the T7 RNA polymerase-binding site sequence to synthesize cDNAs. cDNAs were extracted with phenol:chloroforam:isoamyl alcohol (25:24:1), precipitated with NH4OAc and ethanol, and resuspended in RNase-free water. We synthesized biotin-labeled cRNA with an ENZO RNA transcript labeling kit (Enzo Life Sciences, Farmingdale, NY), using 0.7 μg cDNA per reaction. cRNAs were purified with RNeasy purification columns (Qiagen, Valencia, CA). cRNA, 15 μg per sample, was heated at 80ºC for 33 min in fragmentation buffer (Affymetrix, Inc., Santa Clara, CA). Fragmented cRNAs were hybridized to U95Av2 GeneChips (Affymetrix) for 16 hr at 45ºC. GeneChips were stained with streptavidin phycoerythrin stain solution (Affymetrix). Signal was amplified with goat anti-streptavidin antibody and biotinylated goat IgG (Affymetrix). We scanned stained GeneChips on an Agilent GeneArray Scanner (Agilent Technologies, Palo Alto, CA) and performed four biological replicates.
We performed ANOVA of the signal data with Bioconductor/R software using MAS (Microarray Suite software) background correction, quantiles normalization, MAS PM (perfect match) correction, MAS summary, and statistical significance defined as p < 0.05. Data were filtered on p53 status and arsenite exposure. Analysis was conducted with and without application of the Benjamini and Hochberg false discovery rate correction. Venn analysis was conducted on gene lists using GeneSpring software (Agilent Technologies).
Results
To determine the role that p53 plays in preventing arsenite-induced accumulation of mitotic cells and in preventing arsenite-induced apoptosis, TR9-7 cells were synchronized in G2 using Hoechst 33342. Hoechst 33342 is a topoisomerase inhibitor that interferes with chromatin condensation, resulting in G2 phase arrest (Kuhholzer and Prather 2001). The effects of arsenite treatment on the progression from G2 phase and through mitosis of synchronized p53(+)) and p53(−) cells were evaluated after release from Hoechst 33342-induced G2 phase synchronization.
TR9-7 cells are a spontaneously immortalized human fibroblast cell line, derived from a Li-Fraumeni patient, and subsequently stably transfected with a tetracycline-regulated p53 expression vector (Yin et al. 1992). p53 expression in TR9-7 cells is inversely proportional to the tetracycline concentration in the media (tet-off). Absence of tetracycline strongly induces p53 and causes TR9-7 cells to arrest in G1 and G2 phases (Agarwal et al. 1995). Tetracycline at a concentration of 15 ng/mL induces p53 to a modest level that allows the cells to grow at a rate comparable to p53(−) TR9-7 cells (Taylor et al. 2006).
Synchronization of TR9-7 cells in G2 phase using Hoechst 33342
Synchronization of TR9-7 cells in G2 phase required plating cells with moderate p53 expression. Cells with high levels of p53 tended to arrest in G1 or G2 phase and not re-enter the cell cycle. In addition, induction of p53 by lowering the tetracycline concentration proved difficult to time reproducibly. We obtained reproducible changes in p53 expression by repressing p53 by addition of tetracycline (data not shown). The experimental design we developed is presented in Figure 1. Cells were synchronized in G1 phase by addition of aphidicolin after allowing freshly plated cells to attach and to acclimate for 24 hr. Cells were released from G1 phase synchrony after 24-hr aphidicolin exposure by washing the cells and applying fresh media containing Hoechst 33342, which induced G2 phase synchrony. After 6 hr of incubation in Hoechst 33342 media, p53 transcription was suppressed in half the cultures by addition of tetracycline to 1,500 ng/mL. Cells were incubated for an additional 6 hr. At this point, media were changed again and half of each tetracycline group received media containing 0 or 5 μM NaAsO2 with tetracycline appropriate to maintain p53 expression levels. Samples were taken at each change of media and at several time points post-Hoechst 33342 incubation. Samples were analyzed with flow cytometry to determine cell cycle distribution (Figure 1B), by Western blot to determine p53 and ID1 expression (Figure 2), and for mitotic spread analysis to determine mitotic index (Figure 3A). We performed microarray analysis to determine changes in gene expression (Tables 1–3).
Figure 1.

Synchronization of TR9-7 cells expressing or not expressing p53 in G2 phase. Abbreviations: FL2-A, propidium iodide fluorescence on channel two; Tet, tetracycline. (A) Experimental design. (B) Cell cycle distribution of synchronized cells. Samples were harvested after treatments indicated and analyzed by flow cytometry. Positions of cells with G1 and G2/M phase DNA content are indicated.
Figure 2.

p53 and ID1 expression in cells after G2 phase synchronization. Cells were synchronized, treated with 5 μM NaAsO2, and samples were taken at the indicated times. Levels of p53 and ID1 proteins were assessed by Western blot analysis with β-actin as a loading control. 0 hr = release from Hoechst 33342. A representative example is shown.
Figure 3.
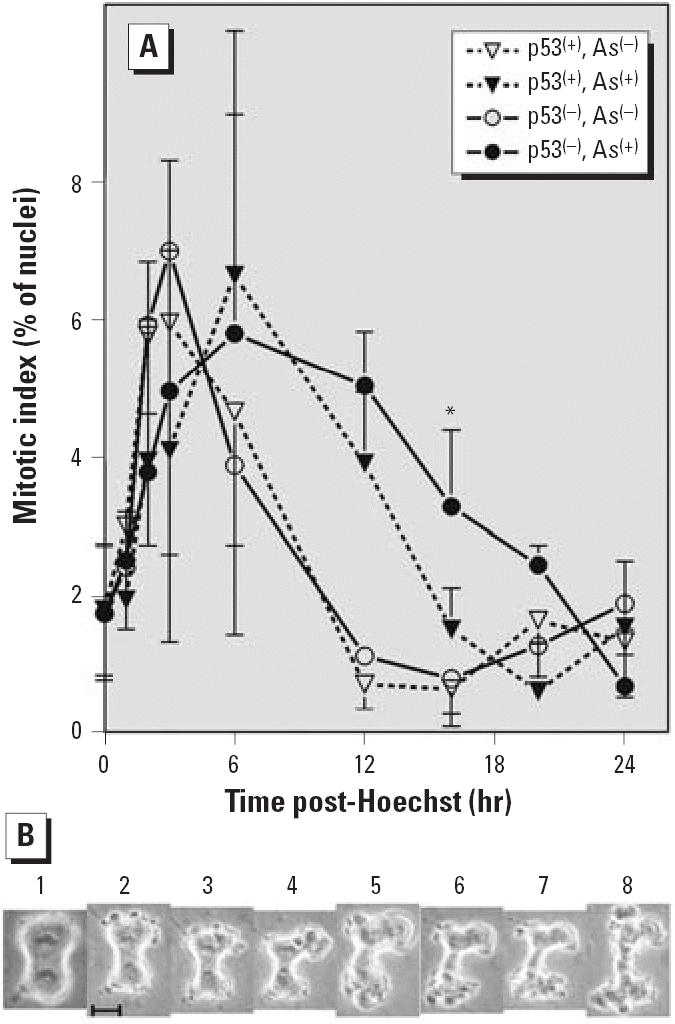
Arsenite treatment delays mitotic entry, and p53 deficiency delays mitotic exit in arsenite-treated cells. Abbreviations: As(+), + 5 μg sodium arsenite; As(−), 0 μg sodium arsenite. (A) Mitotic indices of TR9-7 cells released from G2 phase synchronization. Mean ± SD from four independent experiments performed with triplicate cultures are plotted. Not all time points assayed in every experiment. (B) Arsenite-treated TR9-7 cells in late anaphase/early telophase undergoing apoptosis. Cells were released from G2 phase by feeding with fresh media containing 5 μM NaAsO2 and monitored by phase microscopy. Photographs in panels 1–8 were taken in order over a 30-min period. Bar, 5 μm. *Significant difference between arsenite-exposed p53(+) and p53(−) cells.
Table 1.
Arsenite-dependent gene induction detected by microarray analysis at 0 and 3 hr after release from G2 phase synchrony with FDR applied.a
| Fold-change
|
|||||
|---|---|---|---|---|---|
| UniGene ID | Probe set ID | Gene title | Gene symbol | p53(+) | p53(−) |
| 517581 | 33802_at | heme oxygenase (decycling) 1 | HMOX1 | 27.2 | 26.1 |
| 504609 | 36618_g_at | inhibitor of DNA binding 1 | ID1 | 6.9 | 4.3 |
| 418241 | 39081_at | metallothionein 2A | MT2A | 3.9 | 3.6 |
| 519469 | 34759_at | solute carrier family 30 (zinc transporter), member 1 | SLC30A1 | 3.2 | 3.1 |
| 504609 | 36619_r_at | inhibitor of DNA binding 1 | ID1 | 3.8 | 2.7 |
| 504609 | 36617_at | inhibitor of DNA binding 1 | ID1 | 3.7 | 2.5 |
| 534330 | 36130_f_at | metallothionein 1E (functional) | MT1E | 1.9 | 1.9 |
| 513626 | 31622_f_at | metallothionein 2A | MT2A | 1.9 | 1.8 |
| 284141 | 33835_at | TSPY-like 4 | TSPYL4 | 1.7 | 1.9 |
| 282326 | 32168_s_at | Down syndrome critical region gene 1 | DSCR1 | 1.6 | 1.8 |
| 37055 | 1732_at | fibroblast growth factor 5 | FGF5 | 1.7 | 1.6 |
| 440939 | 31623_f_at | metallothionein 2A | MT2A | 1.6 | 1.5 |
| 105269 | 33369_at | sterol-C4-methyl oxidase-like | SC4MOL | 1.4 | 1.5 |
| 467020 | 1700_at | BCL2 binding component 3 | BBC3 | 0.7 | 0.6 |
| 503093 | 32588_s_at | zinc finger protein 36, C3H type-like 2 | ZFP36L2 | 0.4 | 0.3 |
Gene annotations from NetAffx (http://www.affymetrix.com/analysis/index.affx).
Table 3.
p53-dependent gene induction by arsenite determined by Venn analysis (FDR not applied).a
| Unigene ID | Probe set ID | Gene title | Gene symbol | Fold-change |
|---|---|---|---|---|
| P53(+) cells | ||||
| 520028 | 31692_at | heat shock 70kDA protein 1A | HSPA1A | 4.7 |
| 419 | 34585_at | distal-less homeo box 2 | DLX2 | 2.3 |
| 171695 | 1005_at | dual specificity phosphatase 1 (MKP-1) | DUSP1 | 2.1 |
| P53(−) cells | ||||
| 142912 | 36799_at | frizzled homolog 2 (Drosophila) | FZD2 | 4.2 |
| 50308 | 40121_at | huntingtin interacting protein 2 | HIP2 | 2.5 |
| 93002 | 1651_at | ubiquitin-conjugating enzyme E2C | UBE2C | 2.2 |
| 524630 | 36604_at | ubiquitin-conjugating enzyme E2N | UBE2N | 2.2 |
Gene annotations from NetAffx (http://www.affymetrix.com/analysis/index.affx).
Cell cycle distribution of synchronized cells
Using the procedure depicted in Figure 1A, we were able to achieve reasonably good synchronization. Unsynchronized TR9-7 cells show a typical cell cycle distribution with a majority of the cells in G1 phase and a distinct but small G2/M phase peak (Figure 1B). After aphidicolin treatment, > 90% of cells were in the G1 phase (Figure 1B). After incubation with Hoechst 33342, approximately 60% of the cells were in G2 phase, as characterized using ModFit software to analyze the flow cytometry data (Figure 1B). The approximately 40% of cells remaining in G1 phase appeared irreversibly arrested in G1.
p53 expression in cells after G2 synchronization
p53 levels were assessed in cells during synchronization and after release from G2 phase. Generally, p53 expression was maintained consistently in cultures with low tetra-cycline and suppressed by tetracycline at 1,500 ng/mL throughout the experiment (Figure 2). The achievement of a high level of synchrony in the cultures required the moderate expression of p53 that was suppressed by addition of tetracycline at 1,500 ng/mL 6 hr prior to release from G2 phase blockade. A modest amount of residual p53 was detectable in 1,500 ng/mL tetracylcline cultures [p53(−)] at 0 and 3 hr after release, although p53 expression was much lower than that observed in cultures with only 15 ng/mL tetracycline [p53(+)].
Arsenite delays mitotic exit selectively in p53(−) cells
We examined arsenite effects on entry into and exit from mitosis in synchronized cells. Mitotic indices and p53 expression were determined in triplicate cultures exposed or not exposed to 5 μM NaAsO2 and harvested 0, 1, 3, 6, 12, 16, 20 and 24 hr after release from G2 phase synchrony induced by Hoechst 33342 exposure. Mitotic indices were determined only in experiments in which the p53 expression was maintained as expected. The mitotic index of cells not exposed to arsenite peaked 3 hr after release from G2 phase (Figure 3A). There was no difference between p53(+) and p53(−) cells. The mitotic index dropped sharply after 3 hr and was < 1% by 12 hr. The mitotic index started to increase again 24 hr after release from G2 phase, indicating that the cells had started to cycle again. Arsenite treatment delayed the peak in mitotic index to 6 hr after release from G2 phase in both p53(+) and p53(−) cells. The mitotic index of p53(+) cells treated with arsenite decreased after 6 hr and returned to baseline by 16 hr. The slope of the decrease in mitotic index of arsenite-treated p53(+) cells is similar to that of the decline in cells not exposed to arsenite. These results suggest that entry into mitosis is delayed in p53(+) cells but that mitotic exit is normal. In contrast, the mitotic index of p53(−) cells treated with arsenite dropped slowly and was still significantly elevated 16 hr (p < 0.001, n = four experiments) after release from G2 phase arrest. The mitotic index dropped to nearly zero by 24 hr after release from G2 phase.
Arsenite induces alteration of expression profiles
Microarray analysis was conducted on mRNAs prepared from p53(+) and p53(−) cells at 0 and 3 hr after release from G2 phase synchrony with or without arsenite exposure. ANOVA of four biological replicates using a p < 0.05 was performed with Bioconductor/R software using MAS background correction, quantiles normalization, MAS PM correction, and MAS summary. Analysis was conducted with (Table 1) and without (Tables 2, 3) application of the Benjamini and Hochberg false discovery rate (FDR) correction. Table 1 is all inclusive, whereas Table 2 includes only those genes showing ≥2-fold increase in either p53(+) or p53(−) samples. Arsenite induced several genes independently of p53 status. Heme oxygenase 1 (HMOX1) showed a dramatic > 25-fold increase in expression. Metallothioneines 2A and 1X and the zinc transporter SLC30A1 were also induced. Expression of the stress-responsive heat shock protein HSP70B was elevated > 7-fold. ID1 was induced 3.7- to 6.9-fold in p53(+) cells and 2.5- to 4.3-fold in p53(−) cells depending on which of three probe sets was evaluated. Although two-way ANOVA did not indicate that the difference in fold-change between p53(+) and p53(−) cells was statistically significant, Western blot analysis did show ID1 induction that was more extensive and sustained in p53(+) cells (Figure 2). Two-way ANOVA did not identify any genes or which expression was dependent on p53; therefore, Venn analysis was performed with GeneSpring software (Agilent Technologies) on lists of genes altered in p53(+) and p53(−) samples by arsenite treatment. The false discovery rate correction was not applied. This approach identified several genes altered by arsenite in only p53(+) or p53(−) (Table 3). For example, MAP kinase phosphatase 1 (MKP-1) was induced only in p53(+) cells and ubiquitin-conjugating enzyme E2N was induced only in p53(−) cells.
Table 2.
Arsenite-dependent gene induction detected by microarray analysis at 0 and 3 hr after release from G2 phase synchrony with FDR not applied. a
| Fold-change
|
|||||
|---|---|---|---|---|---|
| UniGene ID | Probe set ID | Gene title | Gene symbol | p53(+) | p53(−) |
| 517581 | 33802_at | heme oxygenase (decycling) 1 | HMOX1 | 27.2 | 26.1 |
| 3268 | 35965_at | heat shock 70k protein 6 (HSP70B’) | HSPA6 | 8.5 | 6.9 |
| 504609 | 36618_g_at | inhibitor of DNA binding 1 | ID1 | 6.9 | 4.3 |
| 520028 | 1104_s_at | heat shock 70kDa protein 1A | HSPA1A | 5.8 | 4.9 |
| 418241 | 39081_at | metallothionein 2A | MT2A | 3.9 | 3.6 |
| 609521 | 34759_at | solute carrier family 30 (zinc transporter), member 1 | SLC30A1 | 3.2 | 3.1 |
| 504609 | 36619_r_at | inhibitor of DNA binding 1 | ID1 | 3.8 | 2.7 |
| 504609 | 36617_at | inhibitor of DNA binding 1 | ID1 | 3.7 | 2.5 |
| 374950 | 39120_at | metallothionein 1X | MT1X | 3.3 | 4.5 |
| 148340 | 41215_s_at | inhibitor of DNA binding 2 | ID2 | 3.1 | 2.8 |
| 799 | 38037_at | diphtheria toxin receptor | DTR | 2.8 | 1.9 |
| 153863 | 1955_s_at | SMAD, mothers against DPP homolog 6 | SMAD6 | 2.4 | 2.0 |
| 76884 | 37043_at | inhibitor of DNA binding 3 | ID3 | 2.4 | 2.5 |
| 491611 | 1137_at | solute carrier family 20 member 2 | SLC20A2 | 2.2 | 2.3 |
| 517617 | 36711_at | v-maf musculoaponeurotic fibrosarcoma oncogene | MAFF | 2.2 | 1.9 |
| 520819 | 35303_at | insulin induced gene 1 | INSIG1 | 2.1 | 1.8 |
| 196054 | 37483_at | histone deacetylase 9 | HDAC9 | 2.1 | 1.7 |
Gene annotations from NetAffx (http://www.affymetrix.com/analysis/index.affx).
Arsenite induces cytokinesis failure and apoptosis
An example of a mitotic cell undergoing apoptosis in an arsenite-treated culture is shown in Figure 3B. Mitosis, from the start of prophase to the completion of cytokinesis, normally takes < 30 min in these cells. As can be observed in Figure 3B, 30 min elapsed from the time a cell was in late anaphase/early telophase (panel 1) to when the cell collapsed (panel 8). The metaphase plates holding the chromosomes did not change position while the membrane developed multiple blebs (panels 1–4). The chromosomes appeared to partially decon-dense (panel 5). The cell body collapsed between the plates but the membranes did not fuse to complete cytokinesis (panels 6–8).
Discussion
Arsenite (as either NaAsO2 or As2O3) is known to activate the G2 phase checkpoint (Chen et al. 2002) and to induce mitotic arrest and apoptosis in a variety of cell lines including HeLa (Huang and Lee 1998), U937 (Halicka et al. 2002; McCabe et al. 2000), SV40-transformed human fibroblasts (Halicka et al. 2002; McCabe et al. 2000; States et al. 2002), and several prostate and ovarian carcinoma cells (Uslu et al. 2000). A common feature of the cells in which arsenite induces both mitotic arrest and apoptosis is a functional loss of p53. We have shown that arsenite-induced mitotic arrest occurs in asynchronous cells lacking p53 expression but not in cells expressing p53 (Taylor et al. 2006). In the present study, we have determined whether p53 expression affects the progression of arsenite-treated cells from G2 phase into M phase and/or the exit from mitosis.
TR9-7 cells are a well-characterized model to study p53-dependent processes, taking advantage of tetracycline-regulated p53 expression (Adimoolam and Ford 2002; Agarwal et al. 1995; Ford and Hanawalt 1997; Lloyd and Hanawalt 2002; Robles et al. 2001; Taylor et al. 2001) We synchronized these human fibroblasts in G2 phase using a multi-step protocol developed by others (Kuhholzer and Prather 2001; Tobey et al. 1990) and adapted to our experiments. The fraction of the cells in G2 phase was not as high as some investigators have achieved with normal diploid fibroblasts (Tobey et al. 1990) but higher than that achieved by another group (Kuhholzer and Prather 2001).
Our results indicate that arsenite exposure delays progression from G2 phase to M phase in both p53(−) and p53(+) cells. This result is consistent with reported arsenite-induced G2 phase delay (McCollum et al. 2005; Park et al. 2001). However, the exit from mitosis was delayed only in p53(−) cells. The mitotic index in arsenite-treated p53(+) cells declined with the same kinetics as in untreated cells. The mitotic index declined more slowly in arsenite-treated p53(−) cells, suggesting that these cells exit mitosis more slowly. The arsenite-treated cells entered apoptosis while futilely attempting cytokinesis. These data indicate that arsenite delays entry into mitosis and progression to anaphase in TR9-7 cells regardless of p53 expression but that exit from mitosis is p53 dependent in arsen-itetreated cells. Thus, both entry into and exit from mitosis is delayed in p53(−) cells. Furthermore, the p53(−) cells undergo apoptosis as a result of failed or abnormal mitotic exit.
As p53 is a transcription factor, we hypothesized that expression profile changes could contribute to delayed mitotic exit kinetics seen in p53(−) cells. To test this, we performed microarray analysis of RNAs prepared from G2 phase synchronized p53(+) and p53(−) cells harvested at 0-hr and after 3-hr exposure to 5 μM sodium arsenite. HMOX1 was highly and rapidly induced, independently of p53. HMOX1 induction is a well-documented response to arsenite (Andrew et al. 2003; Liu et al. 2001). HMOX1 induction is associated with cellular response to oxidative damage, as the degradation products of heme are known to scavenge free radicals (Abraham 2003).
The induction of metallothioneins 2A and 3 may also serve a protective role, as metallothionein has been shown to bind arsenicals (Jiang et al. 2003) and metallothionein null mice are hypersensitive to arsenite toxicity (Liu et al. 2000). MKP1, a phosphatase, was induced in p53(+) cells and may serve a protective role as it opposes the activation of SAPK/JNK (Li et al. 2003), a kinase involved in the induction of apoptosis by arsenite (Davison et al. 2004), MKP1 is a transcriptional target of p53 (Li et al. 2003) and has been shown to be arsenite-induced (Li et al. 2001). The expression of ID1 was elevated by arsenite and Western blot analysis showed the extent of induction was greater and more sustained in p53(+) cells. ID1 is a dominant negative inhibitor of transcription that forms inactive heterodimers with basic helix–loop–helix transcription factors. Genes identified as targets of repression by ID1 include those encoding the cyclin-dependent kinase inhibitory proteins p15INK4B, p16INK4A, and p21CIP1/WAF1 (Sikder et al. 2003). Our previous results indicate that the increased sensitivity to mitotic arrest associated apoptosis of p53-deficient cells is due to the lack of p21CIP1/WAF1, a transcriptional target of p53 (Taylor et al. 2006). p21CIP1/WAF1 may prevent mitotic arrest associated apoptosis in p53(+) cells by allowing for mitotic exit of cells arrested in mitosis. Mitotic exit requires the destruction of cyclin B or inhibition of the cyclin B/CDC2 complex. The promotion of mitotic exit by p21CIP1/WAF1 would involve its direct inhibition of cyclin B/CDC2 (Taylor and Stark 2001). However, once p53(+) cells exit mitosis, cell cycle progression would require the inhibition of p21CIP1/WAF1. The induction of ID1 by arsenite may serve to repress p21CIP1/WAF1 and allow for continued cell cycle progression (Figure 4).
Figure 4.
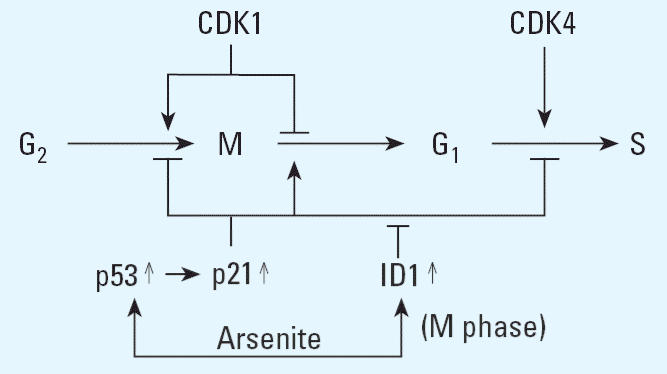
Model for exit from mitotic arrest induced by arsenite and subsequent cell cycle progression dependent on p21CIP1/WAF1 and ID1 induction in p53-expressing cells.
Ectopic overexpression of ID1 immortalizes primary keratinocytes (Alani et al. 1999) and a portion of the ID1 localizes to centrosomes, causing abnormal centrosome number (Hasskarl et al. 2004). Centrosomes serve as the organizational units for the mitotic spindle apparatus. Therefore, an aberrant number of centrosomes may be aneuploidogenic by preventing proper chromosomal segregation. The induction of aneuploidy was posited as the most likely mechanism of arsenic carcinogenesis (NRC 1999, 2001). ID1 also protects against apoptosis through activation of the nuclear factor kappa B signaling pathway (Ling et al. 2003). Higher ID1 levels in p53(+) cells may therefore promote survival. ID1 basal levels are elevated in a variety of cancers, and ID1 is induced during malignant transformation of rat liver cells by arsenic (Chen et al. 2001). Consequently, ID1 induction by arsenite may play an important role in the mechanism of arsenic carcinogenesis by allowing for survival of cells progressing through aberrant mitosis and inducing abnormal chromosome numbers.
Loss of p53 function as seen in many tumors has been suggested to allow cells to bypass cell cycle checkpoints after DNA damage and to enter a nonmitotic state (Erenpreisa and Cragg 2001). Our results suggest p53 plays an important role in prevention of arsenite-induced mitotic arrest and also in apoptosis consequent to the arrest. The prevention of mitotic arrest–associated apoptosis is likely a key event in arsenic-induced carcinogenesis because it predisposes cells to aneuploidy. Understanding the link between p53 and prevention of apoptosis associated with arsenite-induced mitotic arrest will provide essential information regarding the mechanism of arsenic carcinogenesis and also help identify a key target for cancer chemotherapeutics.
Footnotes
This work was supported in part by National Institutes of Health grants R01 ES06460, R01 ES11314, F30 ES013372, T32 ES011564, and P30 ES01247, and the James Graham Brown Cancer Center.
References
- Abraham NG. Therapeutic applications of human heme oxygenase gene transfer and gene therapy. Curr Pharm Des. 2003;9:2513–2524. doi: 10.2174/1381612033453758. [DOI] [PubMed] [Google Scholar]
- Adimoolam S, Ford JM. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc Natl Acad Sci USA. 2002;99:12985–12990. doi: 10.1073/pnas.202485699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal ML, Agarwal A, Taylor WR, Stark GR. p53 controls both the G2/M and the G1 cell cycle checkpoints and mediates reversible growth arrest in human fibroblasts. Proc Natl Acad Sci USA. 1995;92:8493–8497. doi: 10.1073/pnas.92.18.8493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alani RM, Hasskarl J, Grace M, Hernandez MC, Israel MA, Munger K. Immortalization of primary human keratinocytes by the helix-loop-helix protein, Id-1. Proc Natl Acad Sci USA. 1999;96:9637–9641. doi: 10.1073/pnas.96.17.9637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrew AS, Warren AJ, Barchowsky A, Temple KA, Klei L, Soucy NV, et al. Genomic and proteomic profiling of responses to toxic metals in human lung cells. Environ Health Perspect. 2003;111:825–835. doi: 10.1289/ehp.111-1241504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Zhang Z, Bower J, Lu Y, Leonard SS, Ding M, et al. Arsenite-induced Cdc25C degradation is through the KEN-box and ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 2002;99:1990–1995. doi: 10.1073/pnas.032428899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Liu J, Merrick BA, Waalkes MP. Genetic events associated with arsenic-induced malignant transformation: applications of cDNA microarray technology. Mol Carcinog. 2001;30:79–87. doi: 10.1002/1098-2744(200102)30:2<79::aid-mc1016>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- Cohen MH, Hirschfeld S, Flamm HS, Ibrahim A, Johnson JR, O’Leary JJ, et al. Drug approval summaries: arsenic trioxide, tamoxifen citrate, anastrazole, paclitaxel, bexarotene. Oncologist. 2001;6:4–11. doi: 10.1634/theoncologist.6-1-4. [DOI] [PubMed] [Google Scholar]
- Davison K, Mann KK, Waxman S, Miller WH., Jr JNK activation is a mediator of arsenic trioxide-induced apoptosis in acute promyelocytic leukemia cells. Blood. 2004;103:3496–3502. doi: 10.1182/blood-2003-05-1412. [DOI] [PubMed] [Google Scholar]
- Du YH, Ho PC. Arsenic compounds induce cytotoxicity and apoptosis in cisplatin-sensitive and -resistant gynecological cancer cell lines. Cancer Chemother Pharmacol. 2001;47:481–490. doi: 10.1007/s002800100278. [DOI] [PubMed] [Google Scholar]
- Erenpreisa J, Cragg MS. 2001. Mitotic death: a mechanism of survival? A review. Cancer Cell Int 1:1. Available: http://www.cancerci.com/content/1/1/1 [accessed 9 August 2006]. [DOI] [PMC free article] [PubMed]
- Ford JM, Hanawalt PC. Expression of wild-type p53 is required for efficient global genomic nucleotide excision repair in UV-irradiated human fibroblasts. J Biol Chem. 1997;272:28073–28080. doi: 10.1074/jbc.272.44.28073. [DOI] [PubMed] [Google Scholar]
- Halicka HD, Smolewski P, Darzynkiewcz Z, Dai W, Traganos F. Arsenic trioxide arrests cells early in mitosis leading to apoptosis. Cell Cycle. 2002;1:201–209. [PubMed] [Google Scholar]
- Hasskarl J, Duensing S, Manuel E, Munger K. The helix-loop-helix protein ID1 localizes to centrosomes and rapidly induces abnormal centrosome numbers. Oncogene. 2004;23:1930–1938. doi: 10.1038/sj.onc.1207310. [DOI] [PubMed] [Google Scholar]
- Huang SC, Lee TC. Arsenite inhibits mitotic division and perturbs spindle dynamics in HeLa S3 cells. Carcinogenesis. 1998;19:889–896. doi: 10.1093/carcin/19.5.889. [DOI] [PubMed] [Google Scholar]
- Jiang G, Gong Z, Li XF, Cullen WR, Le XC. Interaction of trivalent arsenicals with metallothionein. Chem Res Toxicol. 2003;16:873–880. doi: 10.1021/tx034053g. [DOI] [PubMed] [Google Scholar]
- Jin S, Levine AJ. The p53 functional circuit. J Cell Sci. 2001;114:4139–4140. doi: 10.1242/jcs.114.23.4139. [DOI] [PubMed] [Google Scholar]
- Kuhholzer B, Prather RS. Synchronization of porcine fetal fibroblast cells with topoisomerase-inhibitor Hoechst 33342. Anim Reprod Sci. 2001;66:109–116. doi: 10.1016/s0378-4320(01)00088-4. [DOI] [PubMed] [Google Scholar]
- Li J, Gorospe M, Hutter D, Barnes J, Keyse SM, Liu Y. Transcriptional induction of MKP-1 in response to stress is associated with histone H3 phosphorylation-acetylation. Mol Cell Biol. 2001;21:8213–8224. doi: 10.1128/MCB.21.23.8213-8224.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Zhou JY, Ge Y, Matherly LH, Wu GS. The phosphatase MKP1 is a transcriptional target of p53 involved in cell cycle regulation. J Biol Chem. 2003;278:41059–41068. doi: 10.1074/jbc.M307149200. [DOI] [PubMed] [Google Scholar]
- Ling MT, Wang X, Ouyang XS, Xu K, Tsao SW, Wong YC. Id-1 expression promotes cell survival through activation of NF-kappaB signalling pathway in prostate cancer cells. Oncogene. 2003;22:4498–4508. doi: 10.1038/sj.onc.1206693. [DOI] [PubMed] [Google Scholar]
- Liu J, Kadiiska MB, Liu Y, Lu T, Qu W, Waalkes MP. Stress-related gene expression in mice treated with inorganic arsenicals. Toxicol Sci. 2001;61:314–320. doi: 10.1093/toxsci/61.2.314. [DOI] [PubMed] [Google Scholar]
- Liu J, Liu Y, Goyer RA, Achanzar W, Waalkes MP. Metallothionein-I/II null mice are more sensitive than wild-type mice to the hepatotoxic and nephrotoxic effects of chronic oral or injected inorganic arsenicals. Toxicol Sci. 2000;55:460–467. doi: 10.1093/toxsci/55.2.460. [DOI] [PubMed] [Google Scholar]
- Lloyd DR, Hanawalt PC. p53 controls global nucleotide excision repair of low levels of structurally diverse benzo(g)chrysene-DNA adducts in human fibroblasts. Cancer Res. 2002;62:5288–5294. [PubMed] [Google Scholar]
- Maeda H, Hori S, Nishitoh H, Ichijo H, Ogawa O, Kakehi Y, et al. Tumor growth inhibition by arsenic trioxide (As2O3) in the orthotopic metastasis model of androgen-independent prostate cancer. Cancer Res. 2001;61:5432–5440. [PubMed] [Google Scholar]
- McCabe MJ, Jr, Singh KP, Reddy SA, Chelladurai B, Pounds JG, Reiners JJ, Jr, et al. Sensitivity of myelomonocytic leukemia cells to arsenite-induced cell cycle disruption, apoptosis, and enhanced differentiation is dependent on the inter-relationship between arsenic concentration, duration of treatment, and cell cycle phase. J Pharmacol Exp Ther. 2000;295:724–733. [PubMed] [Google Scholar]
- McCollum G, Keng PC, States JC, McCabe MJ. Arsenite delays progression through each cell cycle phase and induces apoptosis following G2/M arrest in U937 myeloid leukemia cells. J Pharmacol Exp Ther. 2005;313:877–887. doi: 10.1124/jpet.104.080713. [DOI] [PubMed] [Google Scholar]
- NRC (National Research Council) 1999. Arsenic in Drinking Water. Washington DC:National Academy Press.
- NRC (National Research Council) 2001. Arsenic in Drinking Water:2001 Update. Washington DC:National Academy Press.
- Park JW, Choi YJ, Jang MA, Baek SH, Lim JH, Passaniti T, et al. Arsenic trioxide induces G2/M growth arrest and apoptosis after caspase-3 activation and bcl-2 phosphorylation in promonocytic U937 cells. Biochem Biophys Res Commun. 2001;286:726–734. doi: 10.1006/bbrc.2001.5416. [DOI] [PubMed] [Google Scholar]
- Robles AI, Bemmels NA, Foraker AB, Harris CC. APAF-1 is a transcriptional target of p53 in DNA damage-induced apoptosis. Cancer Res. 2001;61:6660–6664. [PubMed] [Google Scholar]
- Sikder HA, Devlin MK, Dunlap S, Ryu B, Alani RM. Id proteins in cell growth and tumorigenesis. Cancer Cell. 2003;3:525–530. doi: 10.1016/s1535-6108(03)00141-7. [DOI] [PubMed] [Google Scholar]
- States JC, Reiners JJ, Jr, Pounds JG, Kaplan DJ, Beauerle BD, McNeely SC, et al. Arsenite disrupts mitosis and induces apoptosis in SV40-transformed human skin fibro-blasts. Toxicol Appl Pharmacol. 2002;180:83–91. doi: 10.1006/taap.2002.9376. [DOI] [PubMed] [Google Scholar]
- Taylor BF, McNeely SC, Miller HL, Lehmann GM, McCabe MJ, Jr, States JC. p53 suppression of arsenite-induced mitotic catastrophe is mediated by p21CIP1/WAF1. J Pharmacol Exp Ther. 2006;318:142–151. doi: 10.1124/jpet.106.103077. [DOI] [PubMed] [Google Scholar]
- Taylor WR, Schonthal AH, Galante J, Stark GR. p130/E2F4 binds to and represses the cdc2 promoter in response to p53. J Biol Chem. 2001;276:1998–2006. doi: 10.1074/jbc.M005101200. [DOI] [PubMed] [Google Scholar]
- Taylor WR, Stark GR. Regulation of the G2/M transition by p53. Oncogene. 2001;20:1803–1815. doi: 10.1038/sj.onc.1204252. [DOI] [PubMed] [Google Scholar]
- Tobey RA, Oishi N, Crissman HA. Cell cycle synchronization: reversible induction of G2 synchrony in cultured rodent and human diploid fibroblasts. Proc Natl Acad Sci USA. 1990;87:5104–5108. doi: 10.1073/pnas.87.13.5104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uslu R, Sanli UA, Sezgin C, Karabulut B, Terzioglu E, Omay SB, et al. Arsenic trioxide-mediated cytotoxicity and apoptosis in prostate and ovarian carcinoma cell lines. Clin Cancer Res. 2000;6:4957–4964. [PubMed] [Google Scholar]
- Vega L, Gonsebatt ME, Ostrosky-Wegman P. Aneugenic effect of sodium arsenite on human lymphocytes in vitro: an individual susceptibility effect detected. Mutat Res. 1995;334:365–373. doi: 10.1016/0165-1161(95)90074-8. [DOI] [PubMed] [Google Scholar]
- Yih LH, Ho IC, Lee TC. Sodium arsenite disturbs mitosis and induces chromosome loss in human fibroblasts. Cancer Res. 1997;57:5051–5059. [PubMed] [Google Scholar]
- Yih LH, Lee TC. Arsenite induces p53 accumulation through an ATM-dependent pathway in human fibroblasts. Cancer Res. 2000;60:6346–6352. [PubMed] [Google Scholar]
- Yin Y, Tainsky MA, Bischoff FZ, Strong LC, Wahl GM. Wild-type p53 restores cell cycle control and inhibits gene amplification in cells with mutant p53 alleles. Cell. 1992;70:937–948. doi: 10.1016/0092-8674(92)90244-7. [DOI] [PubMed] [Google Scholar]