

Abstract
Histone methylation at specific lysine residues brings about various downstream events that are mediated by different effector proteins. The WD40 domain of WDR5 represents a new class of histone methyl-lysine recognition domains that is important for recruiting H3K4 methyltransferases to K4-dimethylated histone H3 tail as well as for global and gene-specific K4 trimethylation. Here we report the crystal structures of full-length WDR5, WDR5Δ23 and its complexes with unmodified, mono-, di- and trimethylated histone H3K4 peptides. The structures reveal that WDR5 is able to bind all of these histone H3 peptides, but only H3K4me2 peptide forms extra interactions with WDR5 by use of both water-mediated hydrogen bonding and the altered hydrophilicity of the modified lysine 4. We propose a mechanism for the involvement of WDR5 in binding and presenting histone H3K4 for further methylation as a component of MLL complexes.
Keywords: chromatin, methyl-lysine recognition, WDR5
Introduction
The regulation of higher order chromatin structure is closely linked to covalent histone post-translational modifications, which includes acetylation, methylation, phosphorylation and ubiquitination (Margueron et al, 2005). In the past decade, tremendous progress has been made in both the functional and structural studies of histone-modifying enzymes, but little progress has been made in identifying the effector proteins responsible for deciphering these modifications. So far, we only know that bromodomains recognize acetylated histones (Zeng and Zhou, 2002), and chromodomains from HP1 and Polycomb recognize histone H3 methylated at K9 and K27, respectively (Jacobs and Khorasanizadeh, 2002; Nielsen et al, 2002; Fischle et al, 2003; Min et al, 2003). Recently, it has been shown that the double chromodomain of CHD1 and the double tudor domain of JMJD2A recognize histone H3 methylated at K4 (Flanagan et al, 2005; Huang et al, 2006).
The WD40-repeat proteins are a large family of proteins found in all eukaryotes and implicated in very diverse functions such as signal transduction, vesicular trafficking, cytoskeletal assembly, cell cycle control, apoptosis and transcription regulation (Smith et al, 1999). The common function of these WD40-repeat proteins is to serve as a rigid scaffold for protein–protein interaction, and to coordinate downstream events, such as ubiquitination (Nash et al, 2001). Recently, it was shown that a common component of MLL1, MLL2, and hSet1 H3K4 methyltransferase complexes, the WD40-repeat protein WDR5, directly associates with K4-dimethylated H3 in vitro and in vivo (Dou et al, 2005; Wysocka et al, 2005). WDR5 is required for recruiting the methyltransferase complexes to the K4-dimethylated H3 tail and is involved in di- to trimethylation conversion of histone H3 at K4. Han et al (2006) recently reported the structure of WDR5 bound to a K4-dimethylated H3 peptide, revealing a novel mode of dimethyl-lysine recognition that differs significantly from that of chromodomain and tudor domain proteins, which use an aromatic cage to recognize methyl-lysine. Instead, H3K4me2 appeared to interact with WDR5 via nonconventional hydrogen bonds between the two ζ-methyl groups of the dimethylated K4 and the carboxylate oxygen of Glu322 in WDR5 (Han et al, 2006). During revision of this manuscript, two additional reports of WDR5 structure were published (Couture et al, 2006b; Ruthenburg et al, 2006). Ruthenburg et al (2006) reported that dimethylation of H3K4 is modestly recognized by WDR5, while Couture et al (2006b) suggested that the methylation state of K4 does not affect histone H3 binding.
We report here the crystal structures of full-length WDR5, N-terminally truncated WDR5 and its complexes with unmodified, mono-, di- and trimethylated histone H3K4 peptides. These structures together with three different types of peptide binding measurements reveal that WDR5 is able to bind a variety of arginine-containing sequences with similar apparent binding constants. Our combined data reveal how WDR5 can discriminate H3K4me2 from other states of H3K4, and present it for subsequent methylation by use of both water-mediated hydrogen bonding and the altered hydrophilicity of the modified K4.
Results and discussion
WDR5 is a highly conserved protein that shares over 90% sequence identity among all vertebrates, and 68 and 34% sequence identity with Caenorhabditis elegans and Saccharomyces cerevisiae WDR5 homologs, respectively (Figure 1). WDR5 contains seven WD40 repeats, forming a seven-blade β-propeller structure. To elucidate the mechanism of selective binding of H3K4me2 to WDR5, we determined the crystal structures of full-length WDR5, WDR5Δ23 (lacking residues 1–23) and its complexes with mono-, di- and trimethylated and unmodified H3K4 peptides.
Figure 1.
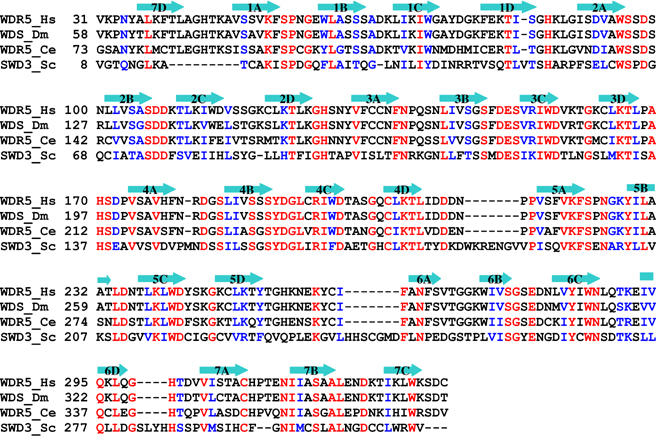
Structure-based sequence alignment of WDR5 homologs. Identical residues are colored in red and conserved residues are colored in blue. The secondary structural elements of human WDR5 are shown above the sequence. Hs: Homo sapiens; Dm: Drosophila melanogaster; Ce: Caenorhabditis elegans; Sc: Saccharomyces cerevisiae.
As anticipated from previous structural studies, full-length WDR5 folds into a seven-blade β-propeller structure preceded by a ∼30 residue mostly unstructured loop (Figure 2). The seven blades are symmetrically arranged around a pseudosymmetry axis and a narrow channel runs through the middle of the β-propeller structure. The β-propeller core domains of our six structures are very similar with an RMSD of 0.3–0.4 Å for all aligned Cα atoms, and the mode of peptide binding is very similar to that recently reported by Han et al (2006). Unexpectedly, a stretch of N-terminal residues (aa11–18) from full-length WDR5 and part of the His-tag from the WDR5Δ23 bind to the top face of the β-propeller structure of their corresponding symmetry-related molecule (Figure 2A), mimicking the binding of the histone H3 peptide in the same cleft of the WDR5 core domain (Figure 2B). The histone H3 and the mimic peptides make contacts to all of the seven blades. All bound peptides share a common residue, arginine, which dips into the central channel (Figure 3). Most of the residues of the β-propeller core domain that interact with the peptides are conserved, and are located in the loop immediately N-terminal to the A strand and the N-terminus of the A strand, and the BC loop.
Figure 2.
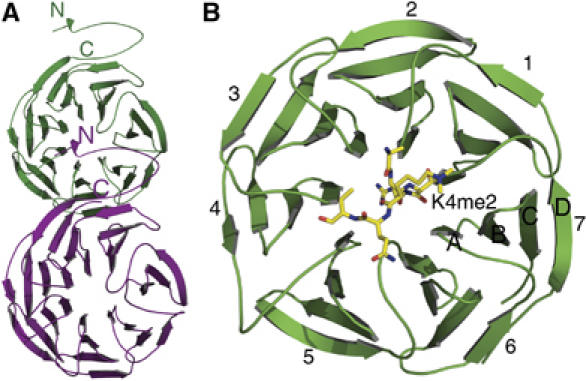
Overall structures of His-tagged WDR5Δ23 alone and in complex with H3K4me2. (A) The N-terminal tail of His-tagged WDR5Δ23 binds to the top face of the β-propeller core domain of a symmetry-related molecule. The structure of full-length WDR5 is similar to that of His-tagged WDR5Δ23. (B) The K4-dimethylated H3 peptide binds to the top face of WDR5 with Arg2 inserting into the central channel of the β-propeller structure. The seven blades of the WD40 domain are numbered from 1 to 7, and the strands of each blade are numbered A–D from the innermost strand to the outermost strand.
Figure 3.
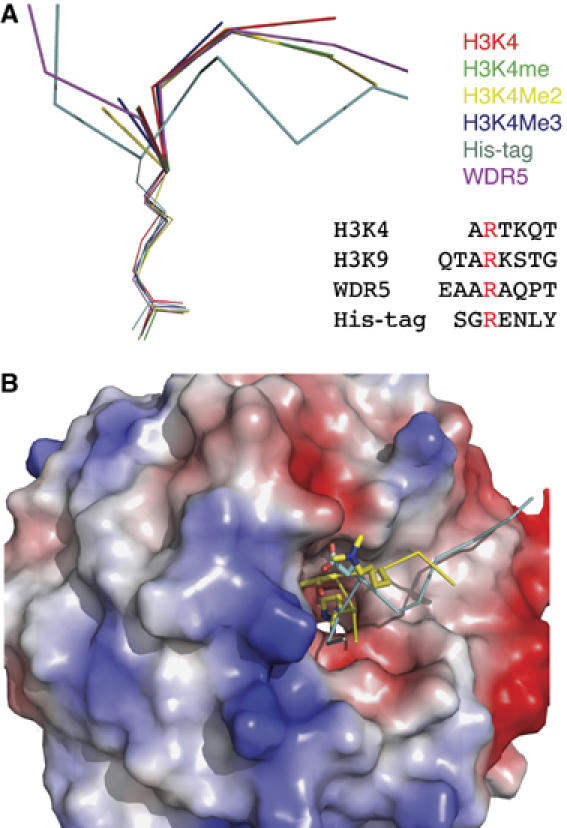
Structures of WDR5 bound to different peptides. (A) Superimposition of the six peptides that are bound to WDR5 core domain. Unmodified histone H3 is colored in red, H3K4me is colored in green, H3K4me2 is colored in yellow, H3K4me3 is colored in blue, a stretch of residues from the His-tag of WDR5Δ23 is colored in cyan and the N-terminal tail (aa11–18) of WDR5 is colored in magenta. The arginine-containing sequences of histone H3K4, H3K9, WDR5 N-terminal tail (aa11–18) and part of the His-tag from WDR5Δ23 that binds to WDR5 core domain are also shown in this panel. (B) Surface representation of WDR5 core domain. H3K4me2 peptide is colored in yellow, of which Thr3 and K4me2 are shown in a stick model. His-tag fragment of WDR5Δ23 is colored in cyan and the glutamic acid corresponding to Thr3 in H3 is shown in a stick model.
In the structure of WDR5 bound to histone H3 dimethylated at K4, there are two complexes per asymmetric unit. The structures of these two complexes are very similar, except that in one complex the first six residues of the histone peptide have clear electron density, while in the other complex only the first five residues are visible. The side chain of the dimethylated lysine interacts with Glu322 of WDR5 via a conserved water molecule found in both complexes of the asymmetric unit (Figure 4). In the structure of WDR5 bound to histone H3 trimethylated at K4, only the first four residues of the peptide are visible and the side chain of the trimethylated lysine is disordered. In the structure of WDR5 bound to histone H3 monomethylated at K4, only the first five residues of the peptide are visible and the side chain of the monomethylated lysine is disordered. In the structure of WDR5 bound to unmodified histone H3, the first six residues are ordered, and K4 is stabilized by crystal contacts.
Figure 4.
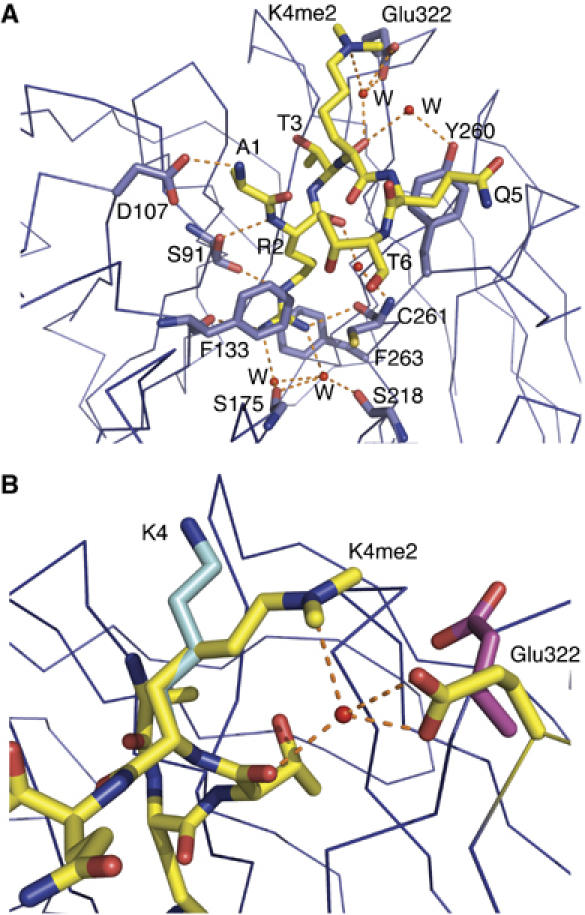
Interaction between WDR5 and histone peptides. (A) H3K4me2 peptide is shown in a stick model colored in yellow, and residues in WDR5 that make hydrogen bonds with H3K4me2 are also shown in a stick model and colored in blue. Hydrogen bonds are denoted as orange dotted lines. (B) Interaction of different states of H3K4 with Glu322 in WDR5. Dimethylated K4 is shown as a stick model colored in yellow, which interacts with Glu322 in WDR5, colored in yellow, via a water molecule. Unmodified K4 in the WDR5–H3K4 complex is colored in cyan, which is stabilized by crystal contacts. The side chain of Glu322 in this complex is either disordered or points to solvent. In the WDR5Δ23–H3K4me complex structure, the side chain of K4me is disordered. The side chain of Glu322 is either disordered or adopts the conformation of Glu322 in the WDR5Δ23–H3K4me2 complex. K4me3 in the WDR5–H3K4me3 complex is disordered, and Glu322, colored in magenta, points to solvent.
The first three residues of the histone H3 peptide form a 310 helix-like conformation, which is also found in the mimic peptides (Figure 4). Arg2 in H3 inserts into the central pore of the β-propeller and forms the largest number of interactions with WDR5 through a network of hydrogen bonds and cation–π interactions (Figure 4A). Arg2 acts like a pin, which anchors the peptide into a shallow groove at the top of the β-propeller. Presumably, it contributes to most of the binding affinity, which is confirmed by our mutant peptide binding studies. Mutation of Arg2 to alanine results in a severe decrease in binding (Figure 8).
Figure 8.
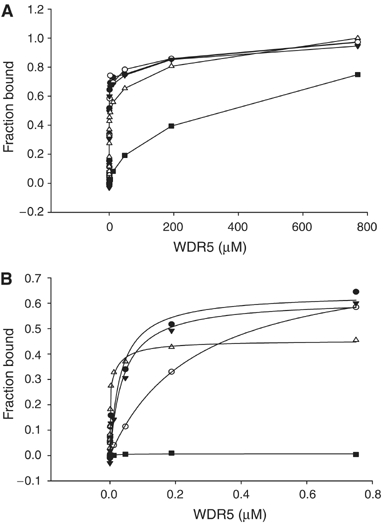
Fluorescence polarization measurements of peptide binding to WDR5. (A) Different concentrations of WDR5 were added to 30 nM of fluorescein-labeled peptide, H3K4me3 (aa1–11) (•), H3K4me2 (aa1–11) (○), H3K4me1 (aa1–8) (▾), unmodified H3K4 (aa1–11) (▵) and R2A H3 (aa1–8) (▪). Points are connected by straight lines. (B) The data measured up to 0.8 μM WDR5 fit to a one-site binding model.
In addition to the histone H3K4 peptides, we also found that a stretch of N-terminal residues (aa11–18) from WDR5 and part of the His-tag from the WDR5Δ23 bind the top face of the β-propeller structure. As mentioned above, their binding mode is conserved with a short consensus sequence ϕδRϕ in each peptide (where δ is a small residue, and ϕ is a residue for which a bulky side-chain residue is disfavored). Steric restrictions in the δ residue pocket allow only a short side-chain residue in this position. In the structure of His-tagged WDR5Δ23, a glutamic acid succeeds the arginine residue, and corresponds to threonine in histone H3 and alanine in WDR5 N-terminal tail peptide. The longer side chain of this glutamic acid prevents the His-tag fragment from taking the main chain conformation as seen in the other peptides (Figure 3B). Our structures demonstrate that WDR5 exhibits plasticity in recognizing different peptides with the consensus sequence, and suggest that WDR5 may be able to recognize other substrates and facilitate their methylation. This is not unusual. For example, Set7/9 methylates both histone H3K4 as well as other transcription factors, such as p53 and TAF10 (Chuikov et al, 2004; Couture et al, 2006a). It is tempting to speculate that MLL complexes may methylate different substrates that require WDR5 as an effector component. It was found that trimethylated H3K9 peptide has a weak binding affinity with WDR5 based on our peptide binding assay (Figure 4) and pull-down assay by Wysocka et al (2005). From our structural analysis, it is permissible for H3K9 to bind WDR5 in a manner similar to that of the His-tag fragment. However, its biological significance is unclear and no association of WDR5- with H3K9-methylated nucleosome was detected (Wysocka et al, 2005). Furthermore, H3K9 methylation is generally implicated in transcriptional repression, whereas H3K4 methylation by MLL complexes is linked to transcriptional activation, suggesting other WD40-repeat containing complexes may exist, which are the physiologically relevant enzymes for modifying other lysine sites. In such cases, the relevant WD40-repeat protein components would likely act as a recognition and presentation module.
Figure 4B shows the interaction of Glu322 in WDR5 with differently modified K4 in histone H3 peptides. Although dimethylated K4 is exposed to solvent in the WDR5Δ23–H3K4me2 complex, it has a well-defined conformation, whose side chain interacts with Glu322 via a conserved water molecule found in both complexes of the asymmetric unit (Figures 4B and 5A). The Nɛ atom of K4me2 forms a hydrogen bond with the conserved water molecule, which is confirmed by the observed polarization of the electron density of the methyl-ammonium group directed towards the water molecule. The water molecule in turn forms two hydrogen bonds with either the two carboxylate oxygens of Glu322 in WDR5 or with one of the two oxygens in the carboxylate group of Glu322 and the backbone carbonyl oxygen of Thr3 in the H3K4me2 peptide (Figure 5B). The K4me2–Glu322 interaction is further reinforced by the electrostatic interaction between the positively charged methyl-ammonium group of K4me2 and the negatively charged carboxylate group of Glu322 as a salt bridge. The importance of Glu322 in K4me2 recognition is supported by the mutagenesis data of Han et al (2006), in which mutation of Glu322 to Ala diminished binding of the H3K4me2 peptide to WDR5.
Figure 5.
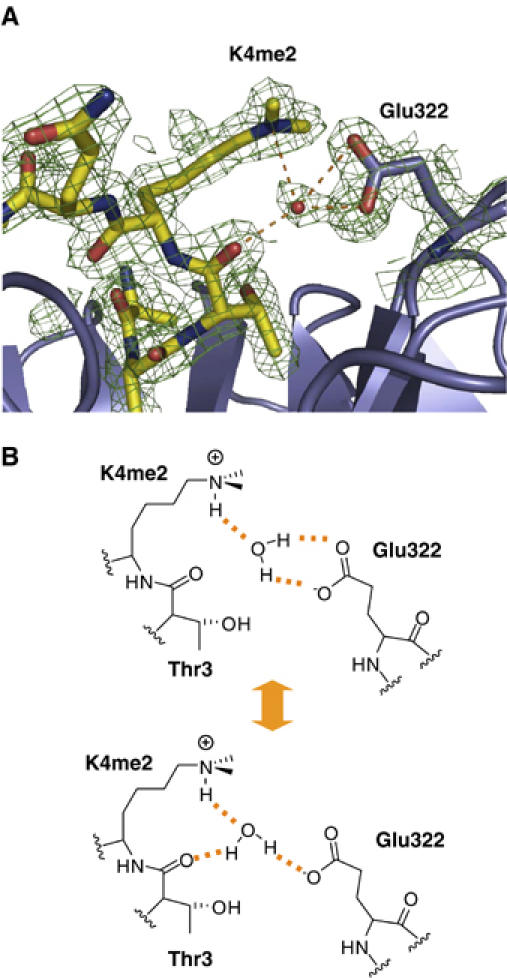
Detailed interaction between me2K4 in H3K4me2 and Glu322 in WDR5. (A) The H3K4me2 peptide and WDR5 are colored in yellow and blue, respectively. The electron density map is contoured at 1σ. Potential water-mediated hydrogen bonds are shown as dashed orange lines. (B) The lower panel shows schematics of two alternative hydrogen binding networks involving the water molecule.
In the WDR5Δ23–H3K4me3 complex structure, the side chain of K4me3 is disordered and Glu322 points to solvent (Figure 4B). The carboxylate oxygen Oɛ1 of Glu322 forms a hydrogen bond with the backbone amine group of Ala47. In the WDR5Δ23–H3K4me1 complex structure, the side chain of K4me1 is disordered. The side chain of Glu322 is either disordered or adopts the conformation of Glu322 in the WDR5Δ23–H3K4me2 complex (Figure 4B). In the WDR5Δ23-unmodified H3K4 complex structure, K4 points to solvent and is stabilized by hydrogen bonding with Glu151 and His170 from another independent WDR5 molecule (Figure 4B). The side chain of Glu322 is either disordered or points to solvent. Therefore, only K4me2 of H3 forms extra interactions with WDR5, and this specificity is conferred by its degree of methylation.
Trimethylation of K4 abolishes its ability to interact with Glu322 in WDR5 via a water molecule, although its stronger hydrophobicity favors its association with the protein. Monomethylated K4 has two hydrogen bond donors at its ammonium moiety, and potentially could form two hydrogen bonds with two water molecules, one water molecule mediating the interaction between K4 and Glu322, and the other one from the solvent. However, such interactions are not observed likely due to the greater hydrophilicity of the monomethylated lysine and a consequent higher propensity to interact with the solvent. A similar argument can be made for K4 in the unmodified H3 peptide.
This mechanism of specificity differs from that recently suggested by Han et al (2006), who invoked the presence of ‘unconventional hydrogen bonds' between the methyl groups of K4me2 and Glu322 as the key element of recognition. The additional data presented here leads us to an alternative interpretation. Our structures of all four methylation states of H3K4 do not support a role for unconventional hydrogen bonds from the methyl groups. Instead, WDR5 uses the subtle changes in the electronic and electrostatic properties of modified K4 in H3 to discriminate the dimethylated lysine from other states of lysine.
Based on biochemical studies by Wysocka et al (2005), WDR5 preferentially binds K4-dimethylated histone H3. However, from the complex structures we solved, the only major difference among these complex structures is that K4me2 of H3 forms extra interactions with WDR5 via Glu322. To investigate the binding specificity of WDR5 for H3K4me2, we carried out an initial assay of relative binding affinities by differential static light scattering, which measures the increase in thermostability of the protein in the presence of peptides (Murphy, 2001; Matulis et al, 2005). Although all the peptides stabilized WDR5, the stabilizing effect was most pronounced with H3K4me2 peptide (Figure 6).
Figure 6.
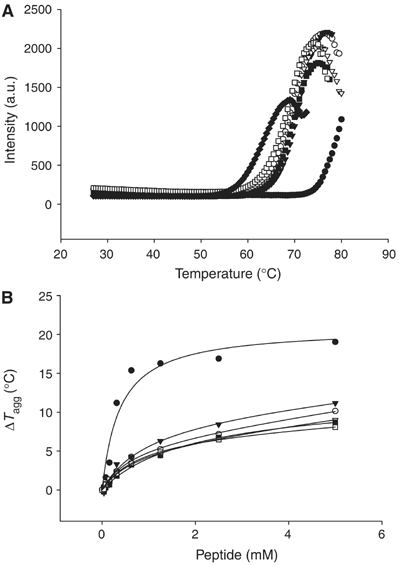
Effects of histone peptides on the thermostability of WDR5Δ23. (A) Thermostability of WDR5Δ23 alone (♦), and in the presence of 2.5 mM of H3K4me1 (aa1–11) (□), H3K4me2 (aa1–11) (•), H3K4me3 (aa1–11) (○), H3K4 (aa1–11) (▵), WDR5 (aa11–18) (▾) and H3K9me3 (▪). (B) Thermostability of WDR5Δ23 as a function of peptide concentration.
To further analyze the binding affinities of the different peptides to WDR5, isothermal titration calorimetry measurements were performed in duplicate. The data were fit to a one-site binding model with KD values of 35±8, 115±13, 77±16 and 192±26 μM for H3K4, H3K4me1, H3K4me2 and H3K4me3 peptides, respectively (Figure 7). Although the data fit well for unmodified and monomethylated H3K4 peptides, the fitting of the data for di- and trimethylated H3K4 peptides into a one-site binding model is not as good as that for the unmodified and monomethylated H3 peptides, suggesting a more complicated binding situation for di- and trimethylated H3K4 peptides (Figure 7). Therefore, additional peptide binding assays were performed using fluorescence polarization method. Again, very similar apparent equilibrium binding constants were obtained for all four H3K4 peptides; however, the data could not be reliably fit to a single-site binding model over the full range of concentrations (Figure 8).
Figure 7.
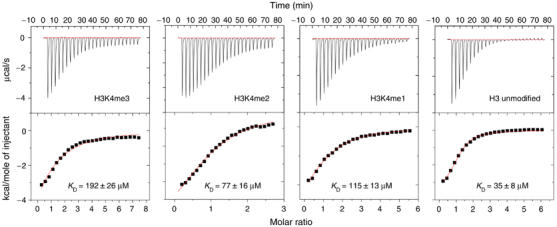
Isothermal titration calorimetry data for binding of methylated and unmodified H3 peptides to WDR5 at 25°C. The upper panels show the sequential heat pulses for peptide–WDR5 binding and the lower panels show the integrated heat data, corrected for the heat of dilution and fit to a single-site binding model using Origin Software. The error values are the s.d. of the values of two titration experiments.
These ITC and FP binding measurements are in apparent conflict with the ability of WDR5 to preferentially bind to H3K4me2 as observed by Wysocka et al (2005), but are consistent with very recent binding data reported by Couture et al (2006b) and Ruthenburg et al (2006). Ruthenburg et al (2006) suggested that this conflict might be explained by slower kon and koff rates for the H3K4me2 peptide. This proposal is consistent with our fluorescence polarization data at low nM concentrations of WDR5 (<100 nM). In this concentration range, the data fits well into a one-site binding model (Figure 8B), with apparent dissociation constants for unmodified H3K4, H3K4me1, H3K4me2 and H3K4me3 peptides being 0.9±0.4, 40±11, 264±11 and 32.8±19 nM, respectively. At these low concentrations of WDR5, the apparent dissociation constant is more a reflection of kon rather than KD. The much higher apparent dissociation constant for H3K4me2 is consistent with Ruthenburg et al's (2006) observation of slower kon and koff rates for H3K4me2 using surface plasmon resonance. They claimed that the binding preference of H3K4me2 for WDR5 might be due to a modest kinetic discrimination for H3K4me2, which displays slower kon and koff rates for H3K4me2. On the other hand, fluorescence polarization experiments were carried out for a mutant H3 peptide with Arg2 substituted with alanine; the data fits perfectly to a single-site binding model (KD=247 μM) over the entire concentration range. The biphasic H3 binding thus seems to be related to the arginine residue, which binds deeply in the central pore and contributes substantial binding affinity to the interaction. The strong arginine contribution to the interaction is likely to mask any weaker but discriminatory binding of WDR5 to the differently methylated peptides. Taken together, the binding specificity of WDR5 for H3K4me2 is not as pronounced as that measured by peptide pull-down assays (Wysocka et al, 2005). Nevertheless, our differential static light scattering results, which monitor protein stability, clearly show the binding preference of WDR5 to H3K4me2 over other modified states of H3K4 peptides (ΔT=∼18°C for H3K4Me2 versus ∼8°C for H3K4, H3K4me1 and H3K4me3) (Figure 6).
WDR5 is a highly conserved protein from yeast to humans, raising the possibility that WDR5 is also important for H3K4 trimethylation in other organisms. The yeast Set1 complex contains a WDR5 homolog, Swd3, which is essential for the H3K4 methylation (Miller et al, 2001; Roguev et al, 2001; Nagy et al, 2002). However, the residue corresponding to Glu322 is an asparagine in yeast Swd3 (Figure 1). We modeled an Asn at residue 322 instead of Glu into the human WDR5 structure, and found that the asparagine could potentially form hydrogen bonds with dimethylated lysine via the conserved water molecule. This suggests that Swd3, like human WDR5 in hSET1, MLL1 and MLL2 complexes, may be directly involved in H3K4 trimethylation as a component of the yeast Set1 complex.
Based on our structural and binding studies, and in combination with previous studies by Wysocka et al (2005), we propose a possible mechanism for the involvement of WDR5 in binding and presenting the H3K4 peptide as a component of MLL complexes for gene activation. MLL complexes first dimethylate K4 of histone H3 in transcriptionally active chromatin domains without assistance of WDR5 (Wysocka et al, 2005). To facilitate MLL complexes to further deprotonate dimethylated K4, WDR5 binds K4-dimethylated histone H3 as an essential component of MLL complexes. Binding of WDR5 positions K4me2 (via Glu322/H2O interactions) and presents it in an ideal conformation and electronic state for the addition of a third methyl group by the catalytic domain (SET domain) of MLL. Glu322 may play a role in deprotonation of dimethylated K4.
Materials and methods
Protein expression and purification
The full-length human WDR5 was subcloned into a modified pET28a-LIC vector, and the N-terminal 23-residue deletion construct WDR5Δ23 was subcloned into pET28a-MHL vector, respectively. The recombinant proteins were overexpressed at 12°C as an N-terminal His6-tagged protein in Escherichia coli BL21 (DE3) Codon plus RIL (Stratagene) and were purified by HiTrap Ni column and Superdex 200 gel-filtration column. The protein was concentrated to 10 mg/ml in a buffer containing 20 mM HEPES–NaOH, pH 7.5, and 0.25 M NaCl.
Binding studies by differential static light scattering
Binding affinity and thermostability of WDR5 was studied using differential static light scattering technology (Reinking et al, 2005) that monitors protein stability by its aggregation properties. Protein samples at 0.4 mg/ml were heated from 27 to 80°C at the rate of 0.5°C/min in clear-bottom 384-well plates (Nunc) in 50 μl of buffer (100 mM HEPES, pH 7.5, 150 mM NaCl). Protein aggregation was monitored by capturing images of scattered light every 30 s with a CCD camera. The pixel intensities in a preselected region of each well were integrated using proprietary software to generate a value representative of the total amount of scattered light in that region. These total intensities were then plotted against temperature for each sample well and fitted to the Boltzman equation by nonlinear regression. The resulting point of inflection of each resulting curve was identified as Tagg. Peptide binding was detected by monitoring the increase in Tagg in the presence of different peptide concentrations ranging from 39 μM to 5 mM in 100 mM HEPES, pH 7.5, and 150 mM NaCl. ΔTagg (Tagg (protein)−Tagg (protein+peptide)) was calculated and plotted against peptide concentration and was fitted to a hyperbolic function to assess the binding ability of each peptide.
Isothermal titration calorimetry
Isothermal titration calorimetry measurements were performed in duplicate at 25°C, using a VP-ITC microcalorimeter (MicroCal Inc.). Experiments were performed by injecting 10 μl of peptide solution (2–4 mM) into a sample cell containing 130 μM WDR5Δ23 in 25mM Tris–HCl, pH 7.5, 200 mM NaCl and 2 mM β-mercaptoethanol. Unmodified H3, H3K4me1, H3K4me2 and H3K4me3 were dissolved and dialyzed into the same buffer as WDR5. Peptide concentrations were estimated from the weighted sample. A total of 25 injections were performed with a spacing of 180 s and a reference power of 15 μcal/s. Binding isotherms were plotted and analyzed using Origin Software (MicroCal Inc.).
Fluorescence polarization
Fluorescence polarization assays were performed with a Beacon 2000 fluorescence polarization system (PanVera) (Jacobs et al, 2001). All peptides were N-terminally labeled with fluorescein succinimidyl ester (Invitrogen). Peptide (0.2 mg) was mixed with 1.2 mM fluorescein in 100 mM potassium phosphate, pH 7 (100 μl), incubated for 1 h at 37°C and quenched by adding 10 μl of 1 M Tris–HCl, pH 8.0. Labeled peptides were separated from free fluorescein and hydroxyl succinamide using a 1 ml G10 Sephadex column. Binding assays were performed at a constant labeled peptide concentration (30 nM), by titrating WDR5Δ23 (up to 770 μM) into 50 mM sodium phosphate, pH 6.0, and 25 mM NaCl. A maximum fluorescence polarization of about 280 mP units was detected upon binding of WDR5 to the labeled peptides, while typically about 40 mP was detected for free labeled peptides. To determine KD values, the data were fit to a hyperbolic function using Sigma Plot software.
Protein crystallization
For cocrystallization, purified proteins were mixed with histone H3K4 peptides (aa1–11) by directly adding a four-fold molar excess of peptide to the protein solution, and crystallized using the sitting drop vapor diffusion method at 18°C. His-tag cleaved full-length WDR5 and the complex of His-tag cleaved WDR5Δ23 and H3K4me2 peptide were crystallized in a buffer containing 20% PEG5000 MME and 0.1 M Bis–Tris, pH 6.5. His-tagged WDR5Δ23 was crystallized in a buffer containing 28% PEG400, 0.2 M ammonium sulfate and 0.1 M Bis–Tris, pH 6.5. The complex of His-tag cleaved WDR5Δ23 with unmodified H3K4 peptide was crystallized in a buffer containing 20% PEG3350 and 0.2 M di-Na tartrate. The complex of His-tag cleaved WDR5Δ23 with H3K4me peptide was crystallized in a buffer containing 20% PEG3350 and 0.2 M Na3PO4. The complex of His-tagged WDR5Δ23 and H3K4me3 peptide (aa1–11) was crystallized in a buffer containing 30% PEG4000, 0.2 M ammonium acetate and 0.1 M Na citrate, pH 5.6. Before flash-freezing crystals in liquid nitrogen, crystals were soaked in a cryoprotectant consisting of 80% reservoir solution and 20% ethyleneglycol.
Structure determination
X-ray diffraction data were collected at 100K using Rigaku FRE High Brilliance X-Ray Generator with R-AXIS IV detector. Data were processed using the HKL software package (Otwinowski and Minor, 1997). The structure of full-length WDR5 was solved by molecular replacement using the program MOLREP (Vagin and Teplyakov, 1997). The crystal structure of LIS1 (PDB 1VYH; Tarricone et al, 2004) was used as the search model. ARP/wARP was used for automatic model building (Perrakis et al, 2001). The other structures were solved by molecular replacement using the full-length WDR5 model as the search model. Graphics program COOT (Emsley and Cowtan, 2004) was used for model building and visualization. Crystal diffraction data and refinement statistics for these six structures are displayed in Table I.
Table 1.
Crystallography data and refinement statistics
| WDR5 | WDR5Δ23 | H3K4 | H3K4me | H3K4me2 | H3K4me3 | |
|---|---|---|---|---|---|---|
| Data collection | ||||||
| Space group | P212121 | P212121 | P1 | P1 | C2 | C2221 |
| Cell dimensions | ||||||
| a, b, c (Å) | 50.05, 55.32, 124.31 | 50.09, 55.54, 123.93 | 46.95, 61.38, 64.73 | 46.74, 56.94, 64.80 | 134.05, 46.98, 112.24 | 78.79, 98.73, 80.21 |
| α, β, γ (deg.) | 90, 90, 90 | 90, 90, 90 | 110.34, 91.23, 112.66 | 71.15, 89.31, 66.05 | 90, 117.26, 90 | 90, 90, 90 |
| Resolution (Å) | 1.8 | 1.75 | 1.9 | 2.0 | 1.9 | 1.91 |
| Rmerge | 3.9 (11.4) | 4.1 (22.4) | 6.7 (21.8) | 12.0 (35.3) | 8.6 (42.1) | 8.9 (46.6) |
| I/σI | 26.1 (10.4) | 18.3 (3.9) | 12.4 (2.7) | 8.4 (2.5) | 10.6 (2.1) | 13.3 (3.8) |
| Completeness (%) | 93.0 (62.7) | 94.5 (67.1) | 91.1 (73.1) | 94.6 (88.2) | 96.4 (76.0) | 95.2 (72.9) |
| Redundancy | 5.8 (4.3) | 4.6 (3.0) | 2.5 (2.1) | 3.6 (3.4) | 3.5 (2.6) | 6.2 (4.9) |
| Refinement | ||||||
| No. of reflections | 30 580 | 33 771 | 44 230 | 36 592 | 47 729 | 23 620 |
| Rwork/Rfree | 14.9/19.2 | 16.7/20.1 | 20.4/25.7 | 22.0/27.9 | 17.4/22.6 | 16.4/20.3 |
| No. of atoms | ||||||
| Protein | 2488 | 2536 | 4727 | 4721 | 4724 | 2370 |
| Peptide | NA | NA | 89 | 84 | 83 | 35 |
| Water | 484 | 429 | 477 | 264 | 627 | 267 |
| R.m.s. deviations | ||||||
| Bond lengths (Å) | 0.011 | 0.012 | 0.008 | 0.012 | 0.013 | 0.016 |
| Bond angles (deg.) | 1.28 | 1.34 | 1.16 | 1.38 | 1.35 | 1.53 |
| PDB code | 2GNQ | 2H9L | 2H9M | 2H9N | 2H9O | 2H9P |
| NA=not applicable. | ||||||
Structural data deposition
The atomic coordinates and structural factor amplitudes have been deposited in the Protein Data Bank for structures of full-length WDR5 (2GNQ), His-tagged WDR5Δ23 (2H9L), WDR5Δ23–H3K4 complex (2H9M), WDR5Δ23–H3K4me complex (2H9N), WDR5Δ23–H3K4me2 complex (2H9O) and WDR5Δ23–H3K4me3 complex (2H9P).
Acknowledgments
This research was supported by the Structural Genomics Consortium with funds from Genome Canada through the Ontario Genomics Institute, the Canadian Institutes for Health Research, the Canada Foundation for Innovation, the Ontario Research and Development Challenge Fund, the Ontario Innovation Trust, the Wellcome Trust, GlaxoSmithKline, the Knut and Alice Wallenberg Foundation, Vinnova and Swedish Foundation for Strategic Research.
References
- Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst P, Prives C, Gamblin SJ, Barlev NA, Reinberg D (2004) Regulation of p53 activity through lysine methylation. Nature 432: 353–360 [DOI] [PubMed] [Google Scholar]
- Couture JF, Collazo E, Hauk G, Trievel RC (2006a) Structural basis for the methylation site specificity of SET7/9. Nat Struct Mol Biol 13: 140–146 [DOI] [PubMed] [Google Scholar]
- Couture JF, Collazo E, Hauk G, Trievel RC (2006b) Molecular recognition of histone H3 by the WD40 protein. Nat Struct Mol Biol 13: 698–703 [DOI] [PubMed] [Google Scholar]
- Dou Y, Milne TA, Tackett AJ, Smith ER, Fukuda A, Wysocka J, Allis CD, Chait BT, Hess JL, Roeder RG (2005) Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell 121: 873–885 [DOI] [PubMed] [Google Scholar]
- Emsley P, Cowtan K (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr D 60: 2126–2132 [DOI] [PubMed] [Google Scholar]
- Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S (2003) Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev 17: 1870–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan JF, Mi LZ, Chruszcz M, Cymborowski M, Clines KL, Kim Y, Minor W, Rastinejad F, Khorasanizadeh S (2005) Double chromodomains cooperate to recognize the methylated histone H3 tail. Nature 438: 1181–1185 [DOI] [PubMed] [Google Scholar]
- Han Z, Guo L, Wang H, Shen Y, Deng XW, Chai J (2006) Structural basis for the specific recognition of methylated histone H3 lysine 4 by the WD-40 protein WDR5. Mol Cell 22: 137–144 [DOI] [PubMed] [Google Scholar]
- Huang Y, Fang J, Bedford MT, Zhang Y, Xu RM (2006) Recognition of histone H3 lysine-4 methylation by the double tudor domain of JMJD2A. Science 312: 748–751 [DOI] [PubMed] [Google Scholar]
- Jacobs SA, Khorasanizadeh S (2002) Structure of HP1 chromodomain bound to a lysine 9-methylated histone H3 tail. Science 295: 2080–2083 [DOI] [PubMed] [Google Scholar]
- Jacobs SA, Taverna SD, Zhang Y, Briggs SD, Li J, Eissenberg JC, Allis CD, Khorasanizadeh S (2001) Specificity of the HP1 chromo domain for the methylated N-terminus of histone H3. EMBO J 20: 5232–5241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margueron R, Trojer P, Reinberg D (2005) The key to development: interpreting the histone code? Curr Opin Genet Dev 15: 163–176 [DOI] [PubMed] [Google Scholar]
- Matulis D, Kranz JK, Salemme FR, Todd MJ (2005) Thermodynamic stability of carbonic anhydrase: measurements of binding affinity and stoichiometry using ThermoFluor. Biochemistry 44: 5258–5266 [DOI] [PubMed] [Google Scholar]
- Miller T, Krogan NJ, Dover J, Erdjument-Bromage H, Tempst P, Johnston M, Greenblatt JF, Shilatifard A (2001) COMPASS: a complex of proteins associated with a trithorax-related SET domain protein. Proc Natl Acad Sci USA 98: 12902–12907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min J, Zhang Y, Xu RM (2003) Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev 17: 1823–1828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy KP (2001) Stabilization of protein structure. Methods Mol Biol 168: 1–16 [DOI] [PubMed] [Google Scholar]
- Nagy PL, Griesenbeck J, Kornberg RD, Cleary ML (2002) A trithorax-group complex purified from Saccharomyces cerevisiae is required for methylation of histone H3. Proc Natl Acad Sci USA 99: 90–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash P, Tang X, Orlicky S, Chen Q, Gertler FB, Mendenhall MD, Sicheri F, Pawson T, Tyers M (2001) Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 414: 514–521 [DOI] [PubMed] [Google Scholar]
- Nielsen PR, Nietlispach D, Mott HR, Callaghan J, Bannister A, Kouzarides T, Murzin AG, Murzina NV, Laue ED (2002) Structure of the chromo barrel domain from the MOF acetyltransferase. Nature 416: 103–10711882902 [Google Scholar]
- Otwinowski Z, Minor W (1997) Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol 276: 307–326 [DOI] [PubMed] [Google Scholar]
- Perrakis A, Harkiolaki M, Wilson KS, Lamzin VS (2001) ARP/wARP and molecular replacement. Acta Crystallogr D 57: 1445–1450 [DOI] [PubMed] [Google Scholar]
- Reinking J, Lam MM, Pardee K, Sampson HM, Liu S, Yang P, Williams S, White W, Lajoie G, Edwards A, Krause HM (2005) The Drosophila nuclear receptor e75 contains heme and is gas responsive. Cell 122: 195–207 [DOI] [PubMed] [Google Scholar]
- Roguev A, Schaft D, Shevchenko A, Pijnappel WW, Wilm M, Aasland R, Stewart AF (2001) The Saccharomyces cerevisiae Set1 complex includes an Ash2 homologue and methylates histone 3 lysine 4. EMBO J 20: 7137–7148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruthenburg AJ, Wang W, Graybosch DM, Li H, Allis CD, Patel DJ, Verdine GL (2006) Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nat Struct Mol Biol 13: 704–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TF, Gaitatzes C, Saxena K, Neer EJ (1999) The WD repeat: a common architecture for diverse functions. Trends Biochem Sci 24: 181–185 [DOI] [PubMed] [Google Scholar]
- Tarricone C, Perrina F, Monzani S, Massimiliano L, Kim MH, Derewenda ZS, Knapp S, Tsai LH, Musacchio A (2004) Coupling PAF signaling to dynein regulation: structure of LIS1 in complex with PAF-acetylhydrolase. Neuron 44: 809–821 [DOI] [PubMed] [Google Scholar]
- Vagin A, Teplyakov A (1997) MOLREP: an Automated Program for Molecular Replacement. J Appl Crystallogr 30: 1022–1025 [Google Scholar]
- Wysocka J, Swigut T, Milne TA, Dou Y, Zhang X, Burlingame AL, Roeder RG, Brivanlou AH, Allis CD (2005) WDR5 associates with histone H3 methylated at K4 and is essential for H3 K4 methylation and vertebrate development. Cell 121: 859–872 [DOI] [PubMed] [Google Scholar]
- Zeng L, Zhou MM (2002) Bromodomain: an acetyl-lysine binding domain. FEBS Lett 513: 124–128 [DOI] [PubMed] [Google Scholar]