

Abstract
Objective:
The phosphatidylinositol 3-kinase (PI3K) pathway promotes cancer cell proliferation and survival. The authors determined the pattern of distribution of PI3K pathway components (ie, the p85α regulatory subunit, p110α catalytic subunit, Akt1, Akt2, and the tumor suppressor PTEN) in human colorectal cancer. In addition, inhibition of in vitro proliferation and in vivo liver metastasis by p85α or p110α siRNA treatment was analyzed.
Summary Background Data:
Small interfering RNA (siRNA) molecules suppress expression of target genes and may have therapeutic applications as target-specific therapies for cancer. Therefore, the purpose of this study was 2-fold: 1) to analyze the distribution pattern of PI3K pathway components in human normal colorectal cancers, and 2) to determine whether targeted inhibition of PI3K inhibits colon cancer growth in vitro and suppresses metastatic growth in vivo.
Methods:
Immunohistochemical analysis was performed on colorectal adenocarcinomas and adjacent normal mucosa for PI3K pathway components, including p85α, p110α, Akt1, Akt2, and the tumor suppressor PTEN, which inhibits PI3K. HT29 and KM20 human colon cancer cells were treated with siRNA directed to p85α or p110α, and cell viability and apoptosis assessed. HT29 cells, transfected with a plasmid containing green fluorescent protein (GFP), were injected into the spleen of athymic nude mice to establish liver metastases; mice were randomized to receive either nontargeting control (NTC), p85α or p110α siRNA.
Results:
PI3K pathway components p85α and Akt2 were highly expressed in glandular elements of colon cancers, with a correlation between staining intensity and clinical stage; PTEN expression was decreased in the colon cancers of all stages. PI3K-specific siRNA treatment decreased cell viability in vitro and suppressed metastatic tumor growth in vivo.
Conclusions:
Selective targeting of PI3K pathway components may enhance the effects of standard chemotherapeutic agents and provide novel adjuvant treatment of selected colorectal cancers.
The pattern of distribution of phosphatidylinositol 3-kinase (PI3K) pathway components in human colorectal cancers was analyzed, and the effects of targeted inhibition on in vitro growth and in vivo metastasis using siRNA to p85α or p110α were assessed.
Phosphatidylinositol 3-kinase (PI3K), a ubiquitous lipid kinase involved in receptor signal transduction by tyrosine kinase receptors, comprises a large and complex family that includes 3 classes with multiple subunits and isoforms.1,2 The class I PI3Ks are composed of a Src homology-2 domain-containing an 85 kDa regulatory subunit (p85) and a 110-kDa catalytic subunit (p110), which catalyze the phosphorylation of phosphoinositol 4-phosphate and phosphoinositol 4,5-phosphate at their D3 position.1,2 The PI3K regulatory subunits include p85α and its truncated splice variants p50α and p55α, as well as p85β and p55γ; the catalytic subunits include p110α, p110β, and p110Δ.2 The regulatory subunits p85α, p50α, and p55α are encoded by the pik3r1 gene; p85α is the most abundantly expressed regulatory isoform of PI3K, and p55α and p50α are 2 additional minor alternative splicing isoforms.3,4
The type I enzymes have been extensively studied and were originally identified in association with tyrosine kinases such as growth factor receptors and products of oncogenes.5 Most studies regarding the type I PI3Ks have focused on the α form. In particular, class IA PI3Ks are strongly expressed in colonic epithelial carcinoma cell lines.6 The gene coding for p110α (pik3cα) is amplified in ovarian and breast tumors,7 implicating pik3cα as a potential oncogene in these cancers. An oncogenic mutated form of p85α has also been described,8 expression of this allele associates with endogenous p110 and increases its activity in a constitutive manner, leading to cell transformation. In addition to the regulation of normal cell processes, the promotion of cell survival by the activation of PI3K occurs by the inhibition of proapoptotic signals and the induction of survival signals, which contribute to the malignant transformation and tumor progression.9 In this regard, there is growing body of evidence to support the notion that the activation of PI3K/Akt is associated with colorectal carcinoma and can convert differentiated human gastric or colonic carcinoma cells to a less differentiated and more malignant phenotype.10 The effects of PI3K on tumor growth and progression are thought to be mediated by Akt, a downstream effector of PI3K.11 The Akt family defines a family of closely related highly conserved cellular homologs of the viral oncoprotein v-akt.12 In humans, there are 3 members of the Akt gene family, designated Akt1, Akt2, and Akt3, which are located on different chromosomes. The Akt gene products, cytoplasmic serine/threonine (ser/thr)-specific protein kinases, are major downstream targets of numerous receptor tyrosine kinases signaling via PI3K.11 Akt is overexpressed in a number of cancers, including colon, pancreatic, ovarian, and some steroid hormone-insensitive breast cancers.13,14 Moreover, it has been reported that Akt phosphorylation in human colon carcinomas correlates with cell proliferation and apoptosis inhibition, as well as with different clinicopathologic parameters such as invasion grade, vessel infiltration, metastasis to lymph nodes, and tumor stage.5,15
Inhibitors of proteins that are involved in PI3K/Akt signaling have been suggested as potential therapeutic agents. These include inhibitors that target both upstream regulators of PI3K/Akt, such as growth factor receptors, PI3K and Akt inhibitors, and downstream effectors, such as the components of the mTOR pathway.6,16 The components of the regulatory system for PI3K/Akt that have proved most amenable to therapeutic intervention are the growth-factor-receptor tyrosine kinases, in particular, the epidermal growth factor receptor (EGFR), its close relative ERBB2, and the fungal metabolite wortmannin, a PI3K inhibitor.17,18 Disadvantages of wortmannin include its short half-life, solubility in organic solvents, and toxicity, which limits its use in clinical trials.19 An alternative approach to the therapeutic targeting of the PI3K/Akt pathway is to specifically inhibit the expression of important pathway proteins by RNA interference (RNAi). RNAi is an evolutionary conserved mechanism that is operative in insects, nematodes, plants, and mammalian cells.20 In this process, sequence-specific posttranscriptional silencing is initiated by the introduction into cells of double-stranded annealed sense and antisense RNAs that are homologous to the sequence of the silenced gene.20 Small interfering RNAs (siRNAs) can be targeted to tumors, and several recent studies indicate the potential for application of this technique in the therapy for various cancers.21,22
Previously, we have shown that wortmannin, administered as a single agent or combined with sodium butyrate (NaBT), significantly inhibited colon cancer cell growth and increased apoptosis, suggesting that PI3K inhibition may be a useful therapeutic option in colon cancer treatment.17 In this current study, we have determined the distribution of PI3K pathway component expression in human colorectal adenocarcinomas, and addressed the function of type I PI3Ks in colon cancer cell growth using an RNAi approach and investigated whether specifically reducing the levels of the p85α regulatory or p110α catalytic subunits of PI3K protein expression in established colon cancer cell lines inhibits in vitro and in vivo proliferation. Here, we show that RNAi provides a useful methodology for evaluating the role of regulatory genes that control the proliferation of cancer cells. Finally, our in vivo studies suggest that RNAi directed against specific PI3K pathway components may suppress metastasis and provide a novel therapeutic strategy for certain colorectal cancers.
MATERIALS AND METHODS
Cell Lines, Reagents, and Antibodies
The human colon cancer cell line HT29 was purchased from American Type Culture Collection (Manassas, VA) and stably transfected with the pEGFPN1 vector. The human colon cancer cell line KM20 (derived from a Dukes’ D colon cancer) was obtained from Dr Isaiah Fidler (M. D. Anderson Cancer Center, Houston, TX). HT29 cells were grown in McCoy’s 5A medium supplemented with 10% fetal bovine serum. KM20 cells were grown in minimum Eagle medium supplemented with 10% fetal bovine serum, 1% sodium pyruvate and 1% nonessential amino acids, 2% MEM essential vitamin, and cultured at 37°C under an atmosphere containing 5% CO2. Tissue culture media and reagents were obtained from Life Technologies, Inc. (Grand Island, NY). SiSTABLE in vivo SMARTpool siRNA and regular SMARTpool reagents for p85α, p110α, and nontargeting control (NTC) siRNA duplexes were designed and synthesized by Customer SMARTpool siRNA Design from Dharmacon (Lafayette, CO). siSTABLE in vivo duplex is chemically modified to extend siRNA stability in vivo compared with unmodified siRNA. TransIT In Vivo Gene Delivery System was purchased from Mirus (Madison, WI). Lipofectamine 2000 transfection reagent was obtained from Invitrogen (Carlsbad, CA). Rabbit anti-Akt, phospho (Ser473) and anti-p110α were purchased from Cell Signaling (Beverly, MA). Mouse monoclonal anti-p85α antibody was purchased from Upstate (Charlottesville, VA). Mouse monoclonal anti-β-actin antibody was obtained from Sigma-Aldrich (St. Louis, MO). pEGFP vector was obtained from Clontech Laboratories (Mountain View, CA). Immobilon P membranes for Western blots were from Millipore Corp. (Bedford, MA), and x-ray film was purchased from Eastman Kodak (Rochester, NY). The enhanced chemiluminescence (ECL) system for Western immunoblot analysis was from Amersham Biosciences (Arlington Heights, IL). All other reagents were of molecular biology grade and purchased from Sigma-Aldrich.
Tissue Procurement
Primary colorectal adenocarcinomas and adjacent mucosa (approximately 5–10 cm from the cancer) were obtained from 40 patients undergoing elective surgical resection over a 4-year period from 2001 to 2005 at the University of Texas Medical Branch (UTMB), Galveston, TX. Tumor stage (TNM classification23) and differentiation grade were assessed. Immediately upon collection, samples were placed in liquid nitrogen and stored at −80°C until used for immunohistochemistry. Tissue acquisition and subsequent use were approved by the Institutional Review Board at UTMB.
Immunohistochemistry
Samples (paired colorectal cancers or polyps and adjacent normal mucosa) were removed from −80°C and placed into 10% neutral buffered formalin for 24 hours, then into 70% ethanol for 24 hours. Formalin-fixed tissues were embedded in paraffin, and sections (5 μm) were cut from the paraffin blocks. The sections were deparaffinized in xylene and rehydrated in descending ethanol series. Protein staining was performed using DAKO EnVision Kit (Dako Corp., Carpinteria, CA). Briefly, sections were incubated overnight at 4°C with monoclonal antibodies (diluted 1:100 in 0.05 mol/L Tris-HCL + 1% BSA) against p85α (Santa Cruz Biotechnology, Santa Cruz, CA), Akt1, Akt2, p110α, and PTEN (Cell Signaling). After 3 washes with TBST, the sections were incubated for 30 minutes with secondary antibody labeled with peroxidase, then washed 3 times with TBST. Lastly, peroxidase substrate DAB was added for staining. All sections were counterstained with hematoxylin and observed by light microscopy. For negative controls, primary antibody was omitted from the above protocol.
Transfection Techniques
siRNA directed against p85α and p110α, and nontargeting control (NTC) were introduced into HT29 and KM20 cells by electroporation (Gene Pulser, Bio-Rad). Exponentially growing cells (3 × 106) were resuspended in culture medium without FCS, supplemented with 20 mmol/L HEPES and electroporated with siRNA (100 nmol). The conditions of electroporation were: 400 V and 500 μF for HT29; 300 V and 600 μF for KM20. Lipofectamine 2000 transfection reagent was used to transfect HT29 cells with the GFP vector for assessment of in vivo metastasis. Briefly, Lipofectamine 2000 transfection reagent and GFP:DNA vector were incubated for 5 minutes in serum-free media and then mixed together for 20 minutes at room temperature. The DNA mixture was applied to HT29 cells for 4 hours at 37°C in serum-free media. After incubation, FBS was added to a final concentration of 10%. Cells were grown in 37°C and 5% CO2, and the culture media was changed daily. After 4 days, cells were selected using G418 antibiotic (500 μg/mL), and transfected clones were identified by FACS A219 cell sorter on the basis of GFP fluorescence. GFP expression was ∼35% as determined by fluorescence-activated cell sorting.
Protein Preparation and Western Immunoblot
Western immunoblot analyses were performed as described previously.17 Cells were lysed with TNN buffer at 4°C for 30 minutes. Lysates were clarified by centrifugation (10,000g for 30 minutes at 4°C) and protein concentrations determined using the method of Bradford.24 Briefly, total protein (60 μg) was resolved on a 10% polyacrylamide gel and transferred to Immobilon-P nylon membranes. Filters were incubated overnight at 4°C in blotting solution (Tris-buffered saline containing 5% nonfat dried milk and 0.1% Tween 20), followed by a 1-hour incubation with primary antibodies. Filters were washed 3 times in a blocking solution and incubated with horseradish peroxidase-conjugated secondary antibodies for 1 hour. After 3 additional washes, the immune complexes were visualized by ECL detection.
MTT Assay
Forty-eight hours after p85α, p110α, or NTC siRNA transfection, adherent cells were detached by rapid trypsinization, counted in a Hausser chamber, and replated into 96-well plates at a concentration of 1 × 103 cells/well. Assays were performed at 48, 72, 96, 120, and 144 hours posttransfection according to the manufacturer’s protocol and as we have previously described.25
Apoptosis Assays
Cells were replated in 96-well plates at 96 hours posttransfection; APOPercentage Dye uptake during the apoptotic process was measured the next day with APOPercentage APOPTOSIS Assay kit (Accurate Chemical & Scientific, Westbury, NY) according to manufacturer’s instructions. DNA fragmentation was quantitated by determination of cytoplasmic histone-associated DNA fragments (mono- and oligonucleosomes) using a Cell Death Detection ELISAPlus kit (Roche Molecular Biochemicals) according to the manufacturer’s instructions and as we have previously described.17
In Vivo Experiments
For in vivo studies, 4- to 6-week-old male nudenu/nu mice were obtained from Harlan Sprague Dawley (Indianapolis, IN) and housed in clean, pathogen-free rooms in an environment with controlled temperature (22°C), humidity, and a 12 hours light/dark cycle. The mice were fed standard chow (Formula Chow 5008; Purina Mills, St. Louis, MO) and tap water ad libitum and allowed to acclimate for 1 week. All studies were approved by the Institutional Animal Care and Use Committee of UTMB. Tumor cells were injected intrasplenically by methods previously described.26 Briefly, mice were anesthetized with halothane, a small left abdominal flank incision was created, and the spleen was exteriorized. Transfectants were harvested using only trypsin and resuspended as a single-cell suspension in Hanks Balanced Salt Solution, free of Mg2+ and Ca2+. Tumor cells (5 × 106 cells/400 μL) were injected into the spleen with a 27-gauge needle. The spleen was returned to the abdomen, and the wound was closed in one layer with wound clips. The mice were observed over the indicated time period using the Illumatool TLS (Lightools Research, Encinitas, CA). Animals were randomized into 3 experimental groups (5 animals per group) to receive p85α, p110α, or NTC siSTABLE siRNA (20 μg/mice, qod) by hydrodynamic tail vein injection27 24 hours after operation; mice were killed 35 days later. Experiments were performed in duplicate.
Statistical Analysis
DNA fragmentation for HT29 or KM20 and ADP analysis were analyzed using one-way classification analysis of variance. The group (NTC, p85α, and p110α) was assessed at the 0.05 level of significance. Fisher’s least significant difference procedure was used for multiple comparisons with Bonferroni adjustment for the number of comparisons. The effect of siRNA on cell growth was analyzed with the Kruskal-Wallis test. A P value of 0.05 was considered significant.
RESULTS
Expression of the PI3K p85α Regulatory Subunit, Akt2, and PTEN in Human Colorectal Polyps, Cancers, and Corresponding Normal Mucosa
Colorectal cancers and adjacent normal mucosa from 40 patients with either proximal (ie, cecal or ascending colon) or distal (ie, sigmoid or rectal) tumors were analyzed for expression of the PI3K/Akt pathway components p85α and p110α, Akt1, and Akt2, and the tumor suppressor PTEN, the natural PI3K inhibitor (Table 1) shows patient characteristics, tumor location, and TNM staging.28 In our current study, we have focused our comparison on proximal and distal cancers due to previous findings noting a differential expression pattern of PTEN with decreased expression in the distal colon.29,30 Representative staining patterns for p85α, Akt2, and PTEN are shown in Figure 1. Three patients had resections for tubulovillous adenomas; a representative patient specimen is shown in Figure 1A. PTEN, p85α, and Akt2 expression was highest in the surface epithelium of normal colon, with PTEN expression extending into the base of the crypts; PTEN expression was more pronounced than either p85α or Akt2. The glandular elements of the polyps expressed PTEN, p85α, and Akt2, with p85α expression stronger than either PTEN or Akt2; p85α and Akt2 expression in the stroma was limited to endothelium and inflammatory cells, with a predominantly cytoplasmic distribution, while there was a predominantly nuclear distribution of PTEN by fibroblasts and inflammatory cells in the stroma. Thus, in contrast to the adjacent normal mucosa, the polyps expressed higher levels of p85α and Akt2.
TABLE 1. Patient Demographics
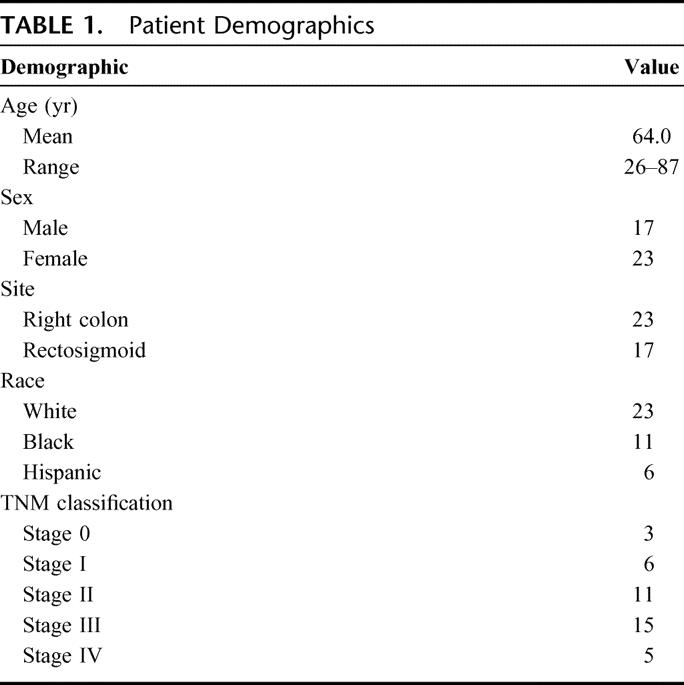
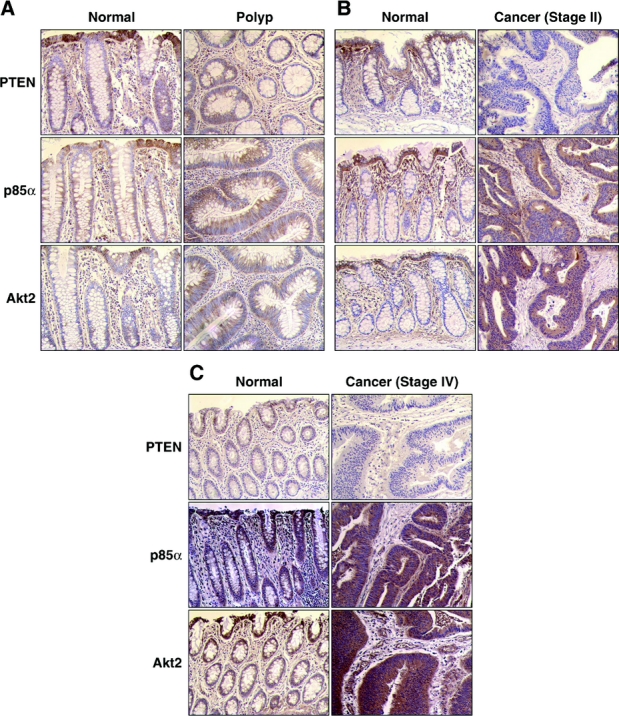
FIGURE 1. PI3K pathway components, including p85α and Akt2, are highly expressed in human colorectal colon cancer. The distribution and intensity of PTEN, p85α, and Akt2 expression were analyzed in patient-matched normal mucosa and tumor samples by immunohistochemistry. Representative expression patterns are shown in patients with tubulovillous adenomas (stage 0) (A), stage II cancer (B), and stage IV cancer (C).
We next analyzed stage I, stage II, and stage III colorectal cancers. Similar expression patterns were noted for these cancers (Fig. 1B shows representative samples from a patient with a Stage II colon cancer). Similar to the normal adjacent mucosa from patients with polyps, PTEN and p85α expression was highest in the surface epithelium with some expression noted in inflammatory cells in the superficial lamina propria; Akt2 expression was again limited to the surface epithelium. In contrast to the glandular elements of polyps, the glandular elements of the stage I, II, and III cancers expressed little to no PTEN, but strongly expressed p85α and Akt2 with a similar distribution and intensity. There was little to no PTEN expression in the stroma, with p85α and Akt2 expression again limited to stromal endothelium and inflammatory cells.
Five patients presented with liver metastasis (stage IV disease) (Fig. 1C shows representative sections from a patient with stage IV cancer). Compared with sections of normal mucosa from patients with polyps or stage I, II, or III cancers, which demonstrated predominant PTEN and p85α expression with little Akt2 expression, there was little to no PTEN expression in the surface epithelium of normal colon or lamina propria, with strong expression of both p85α and Akt2 in the surface epithelium descending into the base of the crypts. In the cancers, there was little to no PTEN expression in the glandular or stromal elements, but strong expression of both p85α and Akt2 in the glandular elements, and a similar cytoplasmic distribution of p85α and Akt2 in inflammatory cells and stromal endothelium. Akt2 expression was more pronounced in stage IV disease than in any other stage.
Overall, there were no apparent differences in the expression patterns of proximal or distal colorectal cancers. Consistent with previous studies,30,31 we noted an increase in PTEN expression in the normal proximal colonic mucosa compared with the normal distal (ie, rectosigmoid) mucosa. Expression of Akt1 was variable, with expression noted in some polyps and stage I and II cancers (data not shown). Little p110α expression was demonstrated in either the normal colonic mucosa or the cancers (data not shown). Collectively, our findings suggest increased expression of p85α and Akt2 in stage I, II, and III colorectal cancers compared with normal mucosa or benign polyps; this expression pattern appeared stronger in stage IV cancers where there also appeared to be increased p85α and Akt2 expression in the normal adjacent mucosa compared with normal mucosa of patients with stages I, II, and III cancers. PTEN expression was decreased in all cancers compared with polyps or normal mucosa.
p85α and p110α siRNA Decrease In Vitro Colon Cancer Cell Survival and Increase Apoptosis in Human Colon Cancer Cells KM20 and HT29
PI3K inhibition exhibits a potent antitumor effect in certain cancer cells including colon cancers;17,32 these effects appear to be due to inhibition of Akt/PKB phosphorylation.15 Therefore, we speculated that siRNA directed to PI3K pathway components would inhibit cell growth and induce apoptosis in human colon cancer cell lines. To determine the functional effects of RNAi treatment, we examined the effect of siRNA treatment on the viability of KM20 and HT29 cells by MTT assay (Fig. 2). Transfection with either p85α or p110α siRNA significantly suppressed cell viability in KM20 (Fig. 2A) and HT29 (Fig. 2B) cells at 120 and 144 hours after transfection compared with NTC. To confirm inhibition of expression by siRNA treatment, protein was extracted and analyzed by Western blot (Fig. 2C, 2D). Transfection with siRNA directed to either p85α or p110α into KM20 cells (Fig. 2C) or HT29 (Fig. 2D) reduced p85α and p110α protein levels, respectively, at 120 hours after transfection. Both p85α and p110α siRNA suppressed basal pAkt expression.

FIGURE 2. siRNA directed against p85α or p110α inhibits proliferation. The effect of siRNA directed to PI3K components on the viability of KM20 (A) or HT29 (B) cells was assessed. Cell viability was measured as described in Materials and Methods. Points represent means of triplicate determinations ± SD. *P < 0.05 for p85α, p110α siRNA compared with nontargeting control (NTC) siRNA. KM20 (C) or HT29 (D) cells transfected with p85α, p110α, or NTC. siRNA sequences were lysed and Western blots performed using anti-Akt, phospho (Ser473), anti-p85α, and anti-p110α; β-actin was used as a loading control (bottom row).
To determine whether this reduction in cell viability was a result of increase cell death, we analyzed apoptosis by 2 methods (Fig. 3). In the first, we measured APOPercentage Dye uptake after the various treatments with APOPercentage APOPTOSIS Assay kit. The APOPercentage Dye enters the cells following phosphatidylserine transmembrane movement; dye uptake continues until blebbing occurs. No further dye can then enter the cell, and dye that has accumulated within the cell is not released. An increase in APOPercentage Dye uptake was demonstrated in both KM20 and HT29 colon cancer cells treated with either p85α or p110α siRNA compared with NTC (Fig. 3A, B). In the second method, DNA fragmentation was measured by an ELISA assay (Fig. 3C, D). An increase in DNA fragmentation, which is characteristic of apoptosis, was demonstrated in both KM20 and HT29 colon cancer cells with either p85α or p110α siRNA compared with NTC. In HT29 cells, treatment with p110α siRNA achieves statistical significance. Even though siRNA to p85α and p110α increased apoptosis, the increase in cell death was not as dramatic as previously noted with other agents (eg, wortmannin, which irreversibly inhibits PI3K).33 Therefore, the effect of targeted treatment of PI3K components may be more directed to tumor cell suppression.
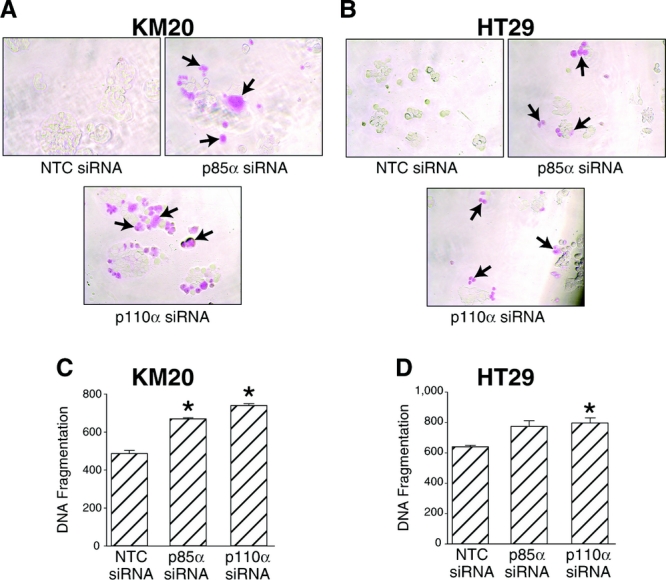
FIGURE 3. siRNA directed against p85α or p110α increases apoptosis. KM20 or HT29 cells were transfected with siRNA (100 nmol) directed against p85α, p110α, or nontargeting control (NTC) as described in Materials and Methods, and 72 hours posttransfection seeded in 96-well plates. Twenty-four hours later, APOPercentage Dye uptake was measured by APOPercentage Apoptosis Assay in KM20 (A) or HT29 (B) cells. Quantitative in vitro determination of cytoplasmic histone-associated DNA fragments was performed using a Cell Death Detection ELISAPLUS in KM20 (C) and HT29 (D) cells. Data represent mean ± SD. *P < 0.05 versus NTC. †P < 0.05 versus p85α siRNA or p110α siRNA alone.
Suppression of Metastatic Tumor Growth by p85α or p110α siRNA Treatment
The liver is a common site of systemic metastases from colorectal cancer.34 The involvement of the PI3K pathway has been linked to tumor cell migration and invasion in a number of cancers through multiple mechanisms,15,35 suggesting that this signaling pathway may contribute to invasion and metastasis in colorectal cancers. To begin to analyze the effects of siRNA treatment on colorectal cancer metastasis to the liver, we established a liver metastasis model using described techniques that involve injection of colorectal cancer cells into the spleen of athymic mice.26 Pilot studies were performed to establish the optimal conditions that would provide detectable liver metastases in all mice but not too numerous so that any treatment differences could be observed; HT29 cells (5 × 106) injected intrasplenically were noted to be optimal in our system. In addition, HT29 cells were transfected with a plasmid containing GFP, which allows for a real-time assessment of tumor metastasis using the Illumatool TLS (Fig. 4). Typically, metastases to the liver are detected 3 to 4 weeks after splenic injection (Fig. 4A, B). Using this model, we next determined whether intravenous siRNA injection directed against p85α or p110α could alter the metastasis of HT29 cells to the liver. Tumor cells were injected into the spleen by methods previously described.26 Animals were randomized into 3 experimental groups (5 animals per group) to receive p85α, p110α, or nontargeting siSTABLE siRNA (20 μg/mice, qday) by hydrodynamic tail vein injection36,37 starting 24 hours after intrasplenic tumor injection; mice were killed 35 days later. The development of liver metastasis was monitored in vivo by bioluminescent imaging. Mice treated with NTC siRNA demonstrated increased metastases compared with either the p85α or p110α siRNA-treated groups as noted by a qualitative assessment of GFP fluorescence; treatment with p85α siRNA appeared to be more effective than p110α siRNA (Fig. 4C). Results were further quantitated by measurement of fluorescence and values expressed as pixel numbers (Fig. 4D). Results demonstrate a significant decrease in tumor metastasis in the p85α and p110α siRNA-treated groups compared with NTC which correlates with our qualitative assessment. Therefore, systemic delivery of PI3K-specific siRNA could represent a unique strategy for the suppression of colorectal cancer metastasis.
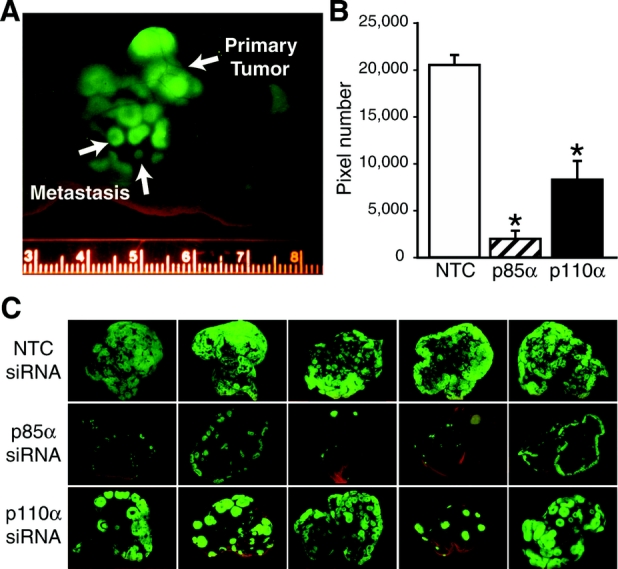
FIGURE 4. Suppression of metastatic tumor growth by p85α or p110α siRNA. A, HT29-GFP cells (5 × 106) were inoculated intrasplenically and mice were killed 5 weeks later. Animals were monitored individually for metastatic tumor growth using the Illumatool TLS. Representative images of nude mice at the end of the experiment on day 35 are shown. B, Animals were randomized into 3 experimental groups (5 animals per group) to receive p85α, p110α, or nontargeting siSTABLE siRNA (20 μg/mice, qod) by hydrodynamic tail vein injection 24 hours after intrasplenic injection; mice were killed 35 days later. Using Adobe Photoshop, the level of fluorescence was measured and expressed as a pixel number. All tests were assessed at the 0.05 level of significance. C, Representative images of nude mouse livers at the end of the experiment on day 35.
DISCUSSION
Activation of the PI3K/Akt pathway increases proliferation and cell survival of certain cancers, including colorectal cancers.11,15 Previously, we have demonstrated that inhibition of PI3K, using the chemical inhibitors wortmannin or LY294002, enhanced sodium butyrate (NaBT)-induced colorectal cancer apoptosis in vitro and suppressed growth of xenograft tumors.17 Moreover, we have recently shown that targeted RNAi to PI3K/Akt components, particularly p85α, can sensitize resistant colorectal cancers to TRAIL-mediated apoptosis.38 In our current study, we have extended these previous findings and demonstrate the following important points: 1) expression of p85α and Akt2 are increased in the glandular components of human colorectal cancers of all stages compared with polyps and normal mucosa; PTEN protein expression was decreased in these cancers, 2) treatment of colon cancers in vitro with siRNA directed to the p85α or p110α genes suppressed cell growth, and 3) using a murine model of colorectal cancer metastasis, we show that siRNA, directed to either p85α or p110α, significantly suppressed tumor metastases. Our study provides additional evidence that selective targeting of PI3K components, such as p85α, may provide a novel treatment strategy for colorectal cancers. This strategy may prove useful in the treatment of colorectal cancers either to increase tumor cell death or, more likely, to enhance the sensitivity of chemoresistant cancers to the effects of other chemotherapeutic agents.
Class I PI3Ks are heterodimers composed of a regulatory (p85) and catalytic (p110) subunit; the regulatory p85 subunit is essential for the stability of the p110 catalytic subunit and for its recruitment to activated growth factor receptors.39 Our previous studies have suggested a role for the p85α subunit in colorectal cancers;38 other investigators have shown that the p110 subunit likewise plays an important role in colorectal cancers.40 In our current study, we have performed immunohistochemical analyses of cancers or polyps and adjacent normal mucosa of 40 patients to assess expression of PI3K pathway components. We demonstrate increased p85α expression in the glandular elements of colorectal cancers at all stages. An increasing role for the surrounding stroma in tumorigenesis has been proposed.41 In this regard, we also noted p85α expression within the cytoplasm of endothelial cells and inflammatory cells present within the stroma. This is in marked contrast to the normal mucosa of patients with stage I, II, and III cancers, where p85α expression was limited to the surface epithelium, with little p85α expression in inflammatory cells present in the superficial lamina propria. Interestingly, increased p85α expression was noted in the normal mucosa of patients with stage IV cancers. These results may reflect a small sample size (5 patients with stage IV disease) and will require further analysis. Our results are in agreement with findings in breast cancers, where a majority of the cancers exhibit increased p85α expression,42 and in agreement with studies that have identified p85α/PI3K overactivity in approximately 86% of colorectal cancer specimens when compared with matched normal controls.43 We found little expression of p110α in cancers or normal mucosa which further supports a role for the increased expression of the p85α subunit in the growth or invasive properties of colorectal cancers.
We next assessed expression of the Akt1 and Akt2 isoforms, which are activated by upstream PI3K, and subsequently activate a number of proteins critical for cell growth or survival.13 Previous studies have identified increased expression of phosphorylated Akt1 and Akt2 in colorectal cancers and colon cancer cell lines.5,13 Because of the variable delay between the time of tumor resection and sample collection, we did not examine the expression of the labile phosphorylated Akt, concentrating instead on Akt1 and Akt2 distribution. A number of studies have focused on the role of Akt1 as the important isoform for PI3K-mediated cell proliferation;44 however, the demonstration of Akt1 expression was highly variable in our study, with minimal expression noted in many cancers. In contrast, our findings clearly showed increased Akt2 expression in all colorectal cancers, particularly in the stage IV cancers. Similar to the increased p85α expression in the adjacent normal mucosa of patients with stage IV cancers, Akt2 expression was also increased. Increasingly, studies are suggesting a more prominent role for Akt2 expression in cancer invasion. For example, Akt2 overexpression contributes to the up-regulation of β1 integrins and increased invasion and metastasis in human breast, ovarian, and colorectal cancers.13,45 Our findings of increased Akt2 in colorectal cancers may reflect an increase in PI3K activation, favoring enhanced cell survival and possibly invasion.
The PTEN protein plays an important role in the carcinogenesis of multiple human cancers including colorectal cancers.46 PTEN modulates cell growth and survival by negatively regulating PI3K/Akt, leading to cell cycle inhibition.47 PTEN expression was decreased in the colorectal cancers compared with expression in polyps and normal mucosa. This is consistent with evidence suggesting that PTEN expression is decreased in approximately 40% of colorectal cancers, often with associated PTEN mutation or deletion.48 In addition to colorectal cancers, decreased PTEN expression has been demonstrated in other cancers, most notably breast and prostate cancers.49 Finally, we have confirmed findings of decreased PTEN expression in the distal colon and rectum, which was previously noted in animal studies.31 It is interesting to speculate that decreased PTEN expression may contribute to the propensity for cancers in the more distal colon and rectum.
The PI3K pathway and downstream proteins are increasingly recognized as potential targets for anticancer therapies. For example, mammalian target of rapamycin (mTOR) kinases are among the downstream targets of Akt and are thought to link mitogenic stimulation to protein synthesis and cell cycle regulation.6,16 Several rapamycin analogs (eg, CCI-779 and RAD001) are currently being evaluated in clinical trials to test efficacy against certain cancers.50,51 Kinase inhibitors targeting Akt or PDK1 are being developed but, thus far, lack specificity.52 Another approach used in experimental models is to broadly inhibit PI3K using either LY294002 or wortmannin. LY294002 can inhibit the growth of certain cancers, including colorectal cancer53 in in vivo experimental models; however, usage is limited by the toxicity of the agent. In addition, we17 and others54 have shown that wortmannin treatment inhibits the in vivo growth of cancers through the irreversible inhibition of PI3K.55 However, the ubiquitous expression and function of PI3K will likely prevent the use of global inhibitors for chronic treatment, since these inhibitors (especially wortmannin) exert a significant nonspecific toxicity,52 precluding use as anticancer agents in patients. A more targeted approach to PI3K inhibition may allow for more effective and less toxic side effects.
Two potential targets include the p110 catalytic subunit, which promotes cell growth and invasion of human cancer cells,35 and the p85 regulatory subunit, which can activate class Ia PI3K by receptor tyrosine kinases.3 In our study, we show that siRNA directed to either p110α or p85α effectively suppressed colon cancer cell growth in vitro through mechanisms that include increased cell death. Consistent with the inhibition noted by p110α siRNA, Takeshita et al22 noted inhibition of prostate cancer proliferation in vitro and metastatic tumor growth after p110α siRNA treatment. Interestingly, the inhibition of p85α may prove to be a more selective form of therapy. Recent studies showed that p85α forms a complex with a protein network associated with oncogenic fusion tyrosine kinases (FTKs) (eg, BCR/ABL, TEL/ABL, TEL/JAK2, TEL/PDGFβR, and NPM/ALK) resulting in constitutive activation of the p110 catalytic subunit of PI3K. These results suggest that the BCR/ABL-p85 association may be a potential target for small molecules designed to disassemble/prevent this interaction and stop malignant growth.56 Together, these findings would support the selective targeting of p85α, or possibly p110α, in the treatment of certain cancers.
To further determine the potential efficacy of this RNAi approach in the treatment of colorectal cancer, we used a more clinically relevant in vivo model of liver metastasis injecting colon cancer cells into the spleen of athymic nude mice as has been described previously.26 We show that intravenous injection of either p85α siRNA or p110α siRNA significantly suppressed liver metastasis with p85α siRNA appearing to be more effective in this regard. These findings provide a proof of concept that therapies targeting the p85α or p110α subunits may be useful in the armamentarium of agents to suppress cancer growth and metastasis. These findings do not address other issues related to potential mechanisms of action. For example, the effects of p85α or p110α siRNA may be related to the direct suppression of cancer growth and metastasis or indirectly by effects on surrounding stromal cells. Further studies will address these possible effects as well as determining whether treatment with siRNA can suppress the growth of established liver metastases. Consistent with our current study, which suggests possible beneficial effects of selective RNAi in the treatment of colorectal cancer, recent studies, using other cancer models, have shown encouraging results. For example, siRNA targeting VEGF effectively inhibits growth of malignant melanoma and squamous cell carcinoma of head and neck cancers both in vitro and in vivo.57,58 Also, down-regulation of antiapoptotic gene expression (eg, survivin) by in vivo siRNA can decrease the radioresistance of breast cancer cells.59
RNAi has the potential to be more selective and, as a result, more effective and less toxic than traditional approaches. Two major obstacles must be overcome for this potential to be realized. First, drug delivery techniques must be refined to provide more specific uptake in cancer cells. Viral delivery methods are efficient but cause serious side effects.60 Cationic lipid complexes are effective siRNA delivery agents.61 A drawback of cationic lipid reagents is that, in some instances, they can be especially toxic and induce immune response in vivo.61 Second, the ability to modify RNA oligonucleotides so that they are more stable in vivo will be necessary before applying this technique for in vivo therapy. Proprietary chemical modifications have been developed that dramatically enhance both the stability and silencing longevity of siRNA while improving its potency and decreasing cellular toxicity.62 These modifications now enable studies that were previously not feasible due to instability of the siRNA duplex or short duration of siRNA-mediated silencing and may provide for agents that are more clinically applicable for treating disease states that require longer acting effects.
ACKNOWLEDGMENTS
The authors thank Drs. Kathleen O’Connor and Suimin Qiu for thoughtful comments, Eileen Figueroa and Karen Martin for manuscript preparation, Kim Chau for maintenance of the human tumor bank and technical assistance, and Tatsuo Uchida for statistical analysis.
Discussions
Dr. Benjamin D. L. Li (Shreveport, Louisiana): Dr. Evers and his group continue to expand our knowledge and appreciation for the importance of signal transduction in GI malignancy. In this latest contribution, they examined the distribution pattern of PI3K pathway components in human colorectal cancer and in the adjacent normal mucosa. They then determined whether the targeted inhibition of PI3K components using siRNA alters cancer cell line growth in vitro and the suppression of liver metastases in nude mice. This elegant piece of work is one in a long line representative of the productivity and excellence in their lab.
I have three questions: In vitro, siRNA targeted to PI3K components resulted in the inhibition of cell growth and increased apoptotic activity in the KM20 and HT29 cell lines. In vivo, though, siRNA stability is a little trickier. In your model, the siRNA was introduced by tail vein injection. Thus, can you discuss how you verified that the siRNA was actually taken up by the HT29 cells that were intrasplenically injected?
Second question. PI3K is relatively ubiquitous and, as such, may have significant systemic implications. Your siRNA targeted specific components of the pathway rather than the whole pathway. Thus, did your initial animal model demonstrate a significantly lower level of toxicity because of your target specificity?
Question number 3. Liver metastases are certainly a significant problem in recurrent colorectal cancer. However, nonhepatic systemic dissemination, such as peritoneal carcinomatosis and pulmonary metastases, is a significant source of morbidity and mortality. Has your group examined whether the use of siRNA lowered nonhepatic metastases as well?
Dr. B. Mark Evers (Galveston, Texas): With regard to the first question, we used a hydrodynamic tail vein injection in association with transfection reagent in order to obtain high level transfection. A majority of hepatocytes will take up the siRNA based upon this injection method. We have not looked specifically at the cancer cells. This is something obviously that we need to do. The mechanism for the effect of the siRNA is likely going to be a complex issue, which may involve both direct effects on the cancer cells and indirect effects on surrounding cells.
The second question addresses the lower toxicity. This is exactly why we are evaluating more specific targeting mechanisms such as siRNA. PI3K pathway chemical inhibitors decrease tumor growth but have significant problems with side effects. In our study, mice were treated every day with siRNA for over a month and they were healthy and showed no systemic sequelae.
The last question has to do with metastasis to other organs. This is not something that we have looked at yet. We wanted to first study the most clinically relevant site of metastasis for colon cancers, which is the liver. We have much more work to do in this current model, so we have not proceeded to evaluate other metastasis models as of yet.
Dr. David J. Cole (Charleston, South Carolina): This is the next in a series of studies which have significantly contributed to our understanding of the oncogenesis of colon cancer.
In the world of cancer therapy (molecular medicine), it is becoming increasingly apparent that it is going to have a significant impact on the diagnosis and treatment, and ultimately the prevention, of cancer. Furthermore, as we gain a more sophisticated understanding of the mechanisms of carcinogenesis, it is becoming apparent that targeted therapeutics are going to take center stage. It is for these reasons that these sorts of studies are very important.
I have four questions for the authors. Your initial immunohistochemical studies demonstrated little to no p100 alpha expression in normal and colon cancer specimens. I am curious, therefore, as to your rationale for using it as one of your targets for your siRNA, as opposed to possibly Akt-2, which is a potential downstream target. Also, given that, how would you explain at a mechanistic level your findings in terms p100 alpha inhibition?
Second, Akt-2 would appear to have promises of prognostic marker. I was curious if you had the opportunity to do an analysis of both primary and metastatic tumor tissues in terms of expression of this marker. Ultimately, what are your thoughts in terms of its applicability as a prognostic indicator?
Third, your data, although statistically significant in terms of MTT assay and in vivo therapy, has only a modest impact on the presence of tumor cells. So I was curious whether you think this is a delivery issue: ie, you have the perfect gene and if you can just knock it down with 100% efficacy, it will cure the animals, or whether this is a problem with a downstream or concomitant signaling pathway in which you are going to have to have multiple targets to have an effective therapy.
Finally, effectiveness and specificity of delivery are universal problems in cancer gene therapy today. And although hydrodynamic tail vein injection is a nice tool for demonstrating a point in animal models, it is not practically applicable in humans. So I am curious what your thoughts are in terms of the next step required to get these findings into a phase 1 patient trial.
Dr. B. Mark Evers (Galveston, Texas): We were surprised as well to find little to no p110 expression. We considered this a reasonable target since it is the catalytic subunit of PI3K. The problem with Akt2 is that there are no Akt isoforms. We worried that we would see less of an effect compared with targeting upstream components.
With regards of Akt2 as a prognostic indicator, we will need to evaluate more tumor samples before we can make these conclusions.
You asked about drug delivery and where to go with the next step. I think that we have established “proof of principle” using the hydrodynamic tail vein injection. We have several avenues that we are currently pursuing. We have a very active chemical biosynthesis group at UTMB, and we are working with them to develop a small molecule inhibitor to p85a. In addition, with chemical modification, more stable forms of siRNA may be feasible to use in vivo. Different companies are working on chemical modifications of siRNA that can be used for longer-term treatments. We are also working with a bioengineering group at UTMB to enhance drug delivery, which remains a huge obstacle for cancer treatment. So, in summary, there are multiple avenues that we are pursuing.
Dr. David R. Jones (Charlottesville, Virginia): What is the p16 and the p53 status of the HT29 and M20 cells? Have you looked at other colorectal cell lines such that you would believe that the cell lines used in your in vivo model truly representative of colorectal cancer?
Did you look at any of the other end organs histologically at the time of necropsy, such as small bowel mucosa, kidney, or lung, to determine whether the knockdown of Akt had any histologic effects on these organs? Did you look at the adjacent noncancerous hepatic tissue to see whether there was knockdown of Akt there, relative to the tumor cell?
I noticed that you continued your therapy for 4 weeks and then euthanized the animals. Did you or have you considered looking at stopping the therapy and then seeing what happens to the tumors after you have discontinued the knockdown therapy? One possibility is these tumors would then significantly grow, and that could be very problematic.
Finally, I noted that there was significantly less p110 in the normal or noncancerous tissue on your immunohistochemistry studies as you moved from a precancerous into a cancerous stage. Do you have any hypothesis why p110 levels fall in the noncancerous tissue that is adjacent the tumor? Is it some type of bystander effect?
Dr. B. Mark Evers (Galveston, Texas): I think that the decreased PTEN expression in adjacent mucosa is an interesting finding and certainly something that wasn’t expected. All 5 of our patients with stage IV cancers had little to no PTEN expression in the adjacent normal mucosa and expression of p85A and Akt2 was markedly elevated. We need to evaluate more patients with metastatic disease before we can say anything definitive regarding this expression pattern.
The reason that we looked at right-sided versus left-sided lesions is because we, as well as others, have noted a differential gradient of PTEN expression in the normal mucosa. PTEN is more abundantly expressed on the right side and progressively decreases in the distal colon and rectum. So it is interesting to speculate that this decreased expression may actually play a role in more left-sided cancers.
You asked a question about stopping the therapy to determine what happens with tumor growth. This is an experiment that we have planned for the future. We also plan to look at initiating therapy later in the course.
We have also looked at combination treatment with chemotherapeutic agents. We can show, for example, that resistant colon cancers are sensitive to TRAIL, an apoptotic protein in the TNF family, when pretreated with siRNA directed to p85a. Therefore, selective P13K pathway targeting may be useful to sensitive colorectal cancers to chemotherapeutic agents.
You asked about the expression of p13K components in normal tissues. We have not looked at this yet. Finally, you asked about PTEN and p53 status in the colon cancer cell lines. We have not specifically looked at p53. PTEN is intact in HT29 cells.
Footnotes
Supported by Grant Nos. RO1CA104748, RO1DK48498, PO1DK35608, and T32DK07639 from the National Institutes of Health.
Reprints: B. Mark Evers, MD, Department of Surgery, University of Texas Medical Branch, 301 University Boulevard, Galveston, TX 77555-0536. E-mail: mevers@utmb.edu.
REFERENCES
- 1.Fruman DA, Meyers RE, Cantley LC. Phosphoinositide kinases. Ann Rev Biochem. 1998;67:481–507. [DOI] [PubMed] [Google Scholar]
- 2.Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res. 1999;253:239–254. [DOI] [PubMed] [Google Scholar]
- 3.Jimenez C, Hernandez C, Pimentel B, et al. The p85 regulatory subunit controls sequential activation of phosphoinositide 3-kinase by Tyr kinases and Ras. J Biol Chem. 2002;277:41556–41562. [DOI] [PubMed] [Google Scholar]
- 4.Philp AJ, Campbell IG, Leet C, et al. The phosphatidylinositol 3′-kinase p85{alpha} gene is an oncogene in human ovarian and colon tumors. Cancer Res. 2001;61:7426–7429. [PubMed] [Google Scholar]
- 5.Khaleghpour K, Li Y, Banville D, et al. Involvement of the PI 3-kinase signaling pathway in progression of colon adenocarcinoma. Carcinogenesis. 2004;25:241–248. [DOI] [PubMed] [Google Scholar]
- 6.Shao J, Evers BM, Sheng H. Roles of phosphatidylinositol 3′-kinase and mammalian target of rapamycin/p70 ribosomal protein S6 kinase in K-Ras-mediated transformation of intestinal epithelial cells. Cancer Res. 2004;64:229–235. [DOI] [PubMed] [Google Scholar]
- 7.Campbell IG, Russell SE, Choong DY, et al. Mutation of the PIK3CA gene in ovarian and breast cancer. Cancer Res. 2004;64:7678–7681. [DOI] [PubMed] [Google Scholar]
- 8.Jimenez C, Jones DR, Rodriguez-Viciana P, et al. Identification and characterization of a new oncogene derived from the regulatory subunit of phosphoinositide 3-kinase. EMBO J. 1998;17:743–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. [DOI] [PubMed] [Google Scholar]
- 10.Semba S, Itoh N, Ito M, et al. Down-regulation of PIK3CG, a catalytic subunit of phosphatidylinositol 3-OH kinase, by CpG hypermethylation in human colorectal carcinoma. Clin Cancer Res. 2002;8:3824–3831. [PubMed] [Google Scholar]
- 11.Fresno Vara JA, Casado E, de Castro J, et al. PI3K/Akt signalling pathway and cancer. Cancer Treat Rev. 2004;30:193–204. [DOI] [PubMed] [Google Scholar]
- 12.Bellacosa A, Testa JR, Staal SP, et al. A retroviral oncogene, Akt, encoding a serine-threonine kinase containing an Sh2-like region. Science. 1991;254:274–277. [DOI] [PubMed] [Google Scholar]
- 13.Roy HK, Olusola BF, Clemens DL, et al. AKT proto-oncogene overexpression is an early event during sporadic colon carcinogenesis. Carcinogenesis. 2002;23:201–205. [DOI] [PubMed] [Google Scholar]
- 14.Asano T, Yao Y, Zhu J, et al. The PI 3-kinase/Akt signaling pathway is activated due to aberrant Pten expression and targets transcription factors NF-kappaB and c-Myc in pancreatic cancer cells. Oncogene. 2004;23:8571–8580. [DOI] [PubMed] [Google Scholar]
- 15.Itoh N, Semba S, Ito M, et al. Phosphorylation of Akt/PKB is required for suppression of cancer cell apoptosis and tumor progression in human colorectal carcinoma. Cancer. 2002;94:3127–3134. [DOI] [PubMed] [Google Scholar]
- 16.Liu X, Powlas J, Shi Y, et al. Rapamycin inhibits Akt-mediated oncogenic transformation and tumor growth. Anticancer Res. 2004;24:2697–2704. [PubMed] [Google Scholar]
- 17.Wang Q, Li N, Wang X, et al. Augmentation of sodium butyrate-induced apoptosis by phosphatidylinositol 3′-kinase inhibition in the KM20 human colon cancer cell cine. Clin Cancer Res. 2002;8:1940–1947. [PubMed] [Google Scholar]
- 18.Hortobagyi GN. Developments in chemotherapy of breast cancer. Cancer. 2000;88(suppl 12):3073–3079. [DOI] [PubMed] [Google Scholar]
- 19.Gunther R, Abbas HK, Mirocha CJ. Acute pathological effects on rats of orally administered wortmannin-containing preparations and purified wortmannin from Fusarium oxysporum. Food Chem Toxicol. 1989;27:173–179. [DOI] [PubMed] [Google Scholar]
- 20.Matzke MA, Birchler JA. RNAi-mediated pathways in the nucleus. Nat Rev Genet. 2005;6:24–35. [DOI] [PubMed] [Google Scholar]
- 21.Yin JQ, Gao J, Shao R, et al. siRNA agents inhibit oncogene expression and attenuate human tumor cell growth. J Exp Ther. 2003;3:194–204. [DOI] [PubMed] [Google Scholar]
- 22.Takeshita F, Minakuchi Y, Nagahara S, et al. Efficient delivery of small interfering RNA to bone-metastatic tumors by using atelocollagen in vivo. Proc Natl Acad Sci USA. 2005;102:12177–12782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macdonald PM, Struhl G. A molecular gradient in early Drosophila embryos and its role in specifying the body pattern. Nature. 1986;324:537–545. [DOI] [PubMed] [Google Scholar]
- 24.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. [DOI] [PubMed] [Google Scholar]
- 25.Iwase K, Evers BM, Hellmich MR, et al. Inhibition of neurotensin-induced pancreatic carcinoma growth by a nonpeptide neurotensin receptor antagonist, SR48692. Cancer. 1997;79:1787–1793. [DOI] [PubMed] [Google Scholar]
- 26.Bruns CJ, Harbison MT, Kuniyasu H, et al. In vivo selection and characterization of metastatic variants from human pancreatic adenocarcinoma by using orthotopic implantation in nude mice. Neoplasia. 1999;1:50–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watanabe H, Saito H, Rychahou PG, et al. Aging is associated with decreased pancreatic acinar cell regeneration and phosphatidylinositol 3-kinase/Akt activation. Gastroenterology. 2005;128:1391–1404. [DOI] [PubMed] [Google Scholar]
- 28.Hermanek P, Scheibe O, Spiessl B, et al. TNM classification of malignant tumors: the new 1987 edition. Rontgenblatter. 1987;40:200. [PubMed] [Google Scholar]
- 29.Kim R, Tanabe K, Uchida Y, et al. Current status of the molecular mechanisms of anticancer drug-induced apoptosis: the contribution of molecular-level analysis to cancer chemotherapy. Cancer Chemother Pharmacol. 2002;50:343–352. [DOI] [PubMed] [Google Scholar]
- 30.Di Cristofano A, Pesce B, Cordon-Cardo C, et al. Pten is essential for embryonic development and tumour suppression. Nat Genet. 1998;19:348–355. [DOI] [PubMed] [Google Scholar]
- 31.Kim S, Domon-Dell C, Wang Q, et al. PTEN and TNF-alpha regulation of the intestinal-specific Cdx-2 homeobox gene through a PI3K, PKB/Akt, and NF-kappaB-dependent pathway. Gastroenterology. 2002;123:1163–1178. [DOI] [PubMed] [Google Scholar]
- 32.Osaki M, Oshimura M, Ito H. PI3K-Akt pathway: its functions and alterations in human cancer. Apoptosis. 2004;9:667–676. [DOI] [PubMed] [Google Scholar]
- 33.Krystal GW, Sulanke G, Litz J. Inhibition of phosphatidylinositol 3-kinase-Akt signaling blocks growth, promotes apoptosis, and enhances sensitivity of small cell lung cancer cells to chemotherapy. Mol Cancer Ther. 2002;1:913–922. [PubMed] [Google Scholar]
- 34.Jemal A, Murray T, Samuels A, et al. Cancer statistics, 2003. CA Cancer J Clin. 2003;53:5–26. [DOI] [PubMed] [Google Scholar]
- 35.Samuels Y, Diaz LA Jr, Schmidt-Kittler O, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7:561–573. [DOI] [PubMed] [Google Scholar]
- 36.Song E, Lee SK, Wang J, et al. RNA interference targeting Fas protects mice from fulminant hepatitis. Nat Med. 2003;9:347–351. [DOI] [PubMed] [Google Scholar]
- 37.Zhang G, Budker V, Wolff JA. High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum Gene Ther. 1999;10:1735–1737. [DOI] [PubMed] [Google Scholar]
- 38.Rychahou PG, Murillo CA, Evers BM. Targeted RNA interference of PI3K pathway components sensitizes colon cancer cells to TNF-related apoptosis-inducing ligand (TRAIL). Surgery. 2005;138:391–397. [DOI] [PubMed] [Google Scholar]
- 39.Yu J, Zhang Y, McIlroy J, et al. Regulation of the p85/p110 phosphatidylinositol 3′-kinase: stabilization and inhibition of the p110alpha catalytic subunit by the p85 regulatory subunit. Mol Cell Biol. 1998;18:1379–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ikenoue T, Kanai F, Hikiba Y, et al. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005;65:4562–4567. [DOI] [PubMed] [Google Scholar]
- 41.Micke P, Ostman A. Exploring the tumour environment: cancer-associated fibroblasts as targets in cancer therapy. Exp Opin Ther Targets. 2005;9:1217–1233. [DOI] [PubMed] [Google Scholar]
- 42.Gershtein ES, Shatskaya VA, Ermilova VD, et al. Phosphatidylinositol 3-kinase expression in human breast cancer. Clin Chim Acta. 1999;287:59–67. [DOI] [PubMed] [Google Scholar]
- 43.Phillips WA, St. Clair F, Munday AD, et al. Increased levels of phosphatidylinositol 3-kinase activity in colorectal tumors. Cancer. 1998;83:41–47. [DOI] [PubMed] [Google Scholar]
- 44.Hutchinson JN, Jin J, Cardiff RD, et al. Activation of Akt-1 (PKB-alpha) can accelerate ErbB-2-mediated mammary tumorigenesis but suppresses tumor invasion. Cancer Res. 2004;64:3171–3178. [DOI] [PubMed] [Google Scholar]
- 45.Arboleda MJ, Lyons JF, Kabbinavar FF, et al. Overexpression of AKT2/protein kinase Bbeta leads to up-regulation of beta1 integrins, increased invasion, and metastasis of human breast and ovarian cancer cells. Cancer Res. 2003;63:196–206. [PubMed] [Google Scholar]
- 46.Guanti G, Resta N, Simone C, et al. Involvement of PTEN mutations in the genetic pathways of colorectal cancerogenesis. Hum Mol Genet. 2000;9:283–7. [DOI] [PubMed] [Google Scholar]
- 47.Cantley LC, Neel BG. New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway. Proc Natl Acad Sci USA. 1999;96:4240–4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Goel A, Arnold CN, Niedzwiecki D, et al. Frequent inactivation of PTEN by promoter hypermethylation in microsatellite instability-high sporadic colorectal cancers. Cancer Res. 2004;64:3014–3021. [DOI] [PubMed] [Google Scholar]
- 49.Li J, Yen C, Liaw D, et al. PTEN, a putative protein tyrosine phosphatase gene mutated in human brain, breast, and prostate cancer. Science. 1997;275:1943–1947. [DOI] [PubMed] [Google Scholar]
- 50.Chan S, Scheulen ME, Johnston S, et al. Phase II Study of Temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol. 2005;23:5314–5322. [DOI] [PubMed] [Google Scholar]
- 51.Raymond E, Alexandre J, Faivre S, et al. Safety and pharmacokinetics of escalated doses of weekly intravenous infusion of CCI-779, a novel mTOR inhibitor, in patients with cancer. J Clin Oncol. 2004;22:2336–2347. [DOI] [PubMed] [Google Scholar]
- 52.Davies SP, Reddy H, Caivano M, et al. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351(Pt 1):95–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Semba S, Itoh N, Ito M, et al. The in vitro and in vivo effects of 2-(4-morpholinyl)-8-phenyl-chromone (LY294002), a specific inhibitor of phosphatidylinositol 3′-kinase, in human colon cancer cells. Clin Cancer Res. 2002;8:1957–1963. [PubMed] [Google Scholar]
- 54.Ng SSW, Tsao M-S, Nicklee T, et al. Wortmannin inhibits PKB/Akt phosphorylation and promotes gemcitabine antitumor activity in orthotopic human pancreatic cancer xenografts in immunodeficient mice. Clin Cancer Res. 2001;7:3269–3275. [PubMed] [Google Scholar]
- 55.Powis G, Bonjouklian R, Berggren MM, et al. Wortmannin, a potent and selective inhibitor of phosphatidylinositol-3-kinase. Cancer Res. 1994;54:2419–2423. [PubMed] [Google Scholar]
- 56.Ren S-y, Bolton E, Mohi MG, et al. Phosphatidylinositol 3-kinase p85{alpha} subunit-dependent interaction with BCR/ABL-related fusion tyrosine kinases: molecular mechanisms and biological consequences. Mol Cell Biol. 2005;25:8001–8008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Tao J, Tu YT, Huang CZ, et al. Inhibiting the growth of malignant melanoma by blocking the expression of vascular endothelial growth factor using an RNA interference approach. Br J Dermatol. 2005;153:715–724. [DOI] [PubMed] [Google Scholar]
- 58.Zhang X, Chen Z, Choe MS, et al. Tumor growth inhibition by simultaneously blocking epidermal growth factor receptor and cyclooxygenase-2 in a xenograft model. Clin Cancer Res. 2005;11:6261–6269. [DOI] [PubMed] [Google Scholar]
- 59.Uchida H, Tanaka T, Sasaki K, et al. Adenovirus-mediated transfer of siRNA against survivin induced apoptosis and attenuated tumor cell growth in vitro and in vivo. Molecular Therapy. 2004;10:162. [DOI] [PubMed] [Google Scholar]
- 60.Lundstrom K, Boulikas T. Viral and non-viral vectors in gene therapy: technology development and clinical trials. Technol Cancer Res Treat. 2003;2:471–486. [DOI] [PubMed] [Google Scholar]
- 61.Sioud M, Sorensen DR. Cationic liposome-mediated delivery of siRNAs in adult mice. Biochem Biophys Res Commun. 2003;312:1220–1225. [DOI] [PubMed] [Google Scholar]
- 62.Chiu Y-L, Rana TM. siRNA function in RNAi: a chemical modification analysis. RNA. 2003;9:1034–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]