

Abstract
Voltage-gated Na channels, which are potential targets for general anaesthetics, are substrates for PKC, which phosphorylates a conserved site in the channel inactivation gate. We investigated the idea that PKC modulates the effect of volatile anaesthetics on Na channels via phosphorylation of this inactivation gate site.
Na currents through rat skeletal muscle Na channel α-subunits expressed in Xenopus oocytes were measured by two-microelectrode voltage clamp in the presence of the volatile anaesthetic agent halothane (2-bromo-2-chloro-1,1,1-trifluroethane). PKC activity was modulated by co-expression of a constitutively active PKC α-isozyme.
Halothane (0.4 mM) had no effect on Na currents. With co-expression of PKC, however, halothane dose-dependently enhanced the rate of Na current decay and caused a small, but statistically significant reduction in Na current amplitude.
The enhancement of Na current decay was absent in a Na channel mutant in which the inactivation gate phosphorylation site was disabled. Effects of halothane on amplitude were independent of this mutation.
Co-expression of a PKC α-isozyme permits an effect of halothane to hasten current decay and reduce current amplitude, at least in part through interaction with the inactivation gate phosphorylation site. We speculate that the interaction between halothane and Na channels is direct, and facilitated by PKC activity and by phosphorylation of a site in the channel inactivation gate.
Keywords: Na channels, halothane, protein kinase C
Introduction
Two lines of evidence link anaesthetic agents with voltage gated Na channels. First, Na channel blockade by local anaesthetics reduces the MAC of general anaesthetics (Himes et al., 1977; 1979; DiFazio et al., 1976). Second, suppression of Na channel activity by volatile general anaesthetics has been reported in a variety of tissues (Bean et al., 1981; Weigt et al., 1997b; Ruppersberg & Rudel, 1988; Eskinder et al., 1993; Rehberg et al., 1996).
Na channels are modulated by phosphorylation. In particular, protein kinase C (PKC) phosphorylates the channel inactivation gate (Numann et al., 1991; West et al., 1991; 1992; Bendahhou et al., 1995), and volatile anaesthetics modulate PKC activity (Lester & Baumann, 1991; Hemmings & Adamo, 1994; 1996; Tsuchia et al., 1988; Tas & Koschel, 1997). A unifying explanation may be that the effect of volatile anaesthetics on Na channels is dependent on PKC activity.
We studied the mechanism of the interaction of muscle Na channels and the volatile anaesthetic agent halothane. We explored the possibility that the interaction with halothane may be modulated by PKC by co-expressing Na channels with a constitutively active PKC isozyme in Xenopus oocytes (West et al., 1991; 1992; Bendahhou et al., 1995). To our surprise we found that halothane altered muscle Na currents in RNA-injected oocytes only if PKC is co-expressed. These new findings add to our understanding of the mechanism of anaesthetic effects on Na channels.
Methods
Oocyte preparation and electrophysiology
Our methods for preparation and two-microelectrode voltage clamping of Xenopus laevis oocytes have been described (Durieux et al., 1992; Moorman et al., 1990; Mounsey et al., 1995). Frogs were anaesthetized by immersion in ice until immobile, and oocytes were harvested under sterile conditions by laparotomy. Two to three ovarian lobules, each containing 200–300 oocytes, were excised. Frogs were utilized for oocyte harvesting up to four times, at intervals of about 1 month after which the frog was sacrificed by decapitation under anaesthesia. Oocytes were isolated manually in modified Barth's solution (which contained (mM); NaCl 88, KCl 1, NaHCO3 2.4, HEPES 15, CaNO3·4H2O 0.3, CaCl2·6H2O 0.41, MgSO4·7H2O 0.82, sodium penicillin (10 μg ml−1), streptomycin sulphate (10 μg ml−1), pH 7.6 (NaOH)), and were not exposed to collagenase. Injection of RNA (20–50 nl of 0.2–1.5 μg μl−1 in 100 mM KCl) was accomplished using a graduated, mechanical 10 μl micropipettor (Drummond). Injected oocytes were incubated in plastic Petri dishes and continuously rotated in an incubator at 19°C. The oocytes were defolliculated manually the day of the electrophysiologic experiments, 2–5 days after injection. Large-tipped current and voltage electrodes (0.5 MΩ) were filled with 3 M KCl and 10 mM HEPES; pH was adjusted to 7.35 (KOH). The bath solution contained (mM) NaCl 150, KCl 5, CaCl2 2, MgCl2 1.0, glucose 10, HEPES 10, pH was adjusted to 7.4 (NaOH); the flow rate was 3–4 ml min−1 and the volume of the experimental chamber was 3 ml. Defolliculated oocytes were clamped from a holding potential of −100 mV (Oocyte clamp, Warner Instruments, New Haven, CT, U.S.A.). Voltage protocols were delivered, and currents were digitized, using pCLAMP (Axon Instruments) hardware and software. We used scaled versions of currents during voltage steps that did not open channels to correct for passive components of leak and capacitance. Data were analysed only when peak Na current was ⩽10 μA and time to peak Na current was ⩽3 msec. All experiments were performed at room temperature. We tested uninjected oocytes from each batch, and none had the very rare occurrence of endogenous fast Na channels (Bourinet et al., 1992; Parker & Miledi, 1987).
For the data shown in Figures 5 and 6, the oocytes were held at −100 mV and clamped from −50 mV to 50 mV in 10 mV increments; the peak current was recorded for each oocyte. Oocytes were injected with identical quantities of wild-type or phos(−) Na channel RNA; half of each batch was co-injected with PKC mRNA.
Figure 5.

Box plots of the effect of PKC co-expression on the amplitude of currents through wild-type and phos(−) Na channels. Currents were normalized so that the mean current amplitude in oocytes expressing wild-type or phos(−) channels alone in each batch was 1.0. In the box plot symbol, the line represents the median, the boxes enclose 50% of the data and the hatches enclose 80% of the data. Data were derived from 30–64 oocytes, four to six frogs. *P<0.001 (rank sum test).
Figure 6.
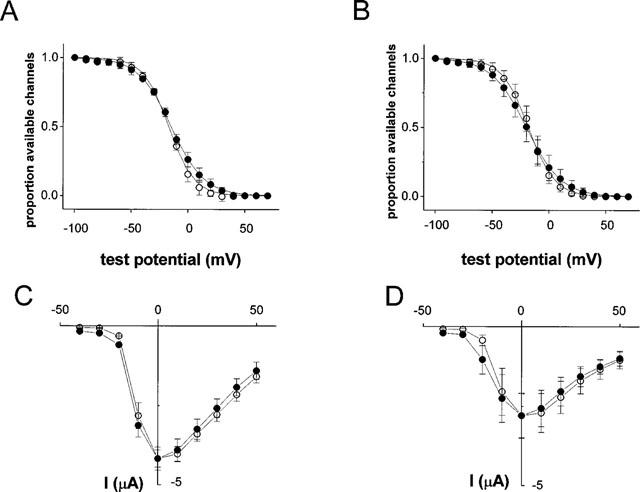
(A and B) The effect of halothane (0.4 mM, 10 min) on the proportion of Na channels available in oocytes expressing Na channels alone (A), and in oocytes co-expressing PKC (B). The lines are least squares fits of the data to a Boltzmann function. The slope factor and midpoints were −11.0 and −17 mV in oocytes expressing Na channels alone, and −11.4 and −18 mV in oocytes expressing PKC. Halothane had no significant effect on either parameter (t-test). The data points are mean (s.d.) from five oocytes, two frogs. (C and D) The effect of halothane (0.4 mM, 10 min) on the current-voltage relationship in oocytes expressing Na channels alone (C), and in oocytes co-expressing PKC (D). The data points are mean (s.d.) from five oocytes, two frogs. The current amplitudes were normalized to the pre-halothane value to emphasize effects on the potential dependence of amplitude. Open circles represent control data; filled circles were obtained after exposure to halothane.
Steady-state inactivation was determined using a standard two-pulse protocol, with a 100 msec step from the holding potential of −100 mV to the test potential. Currents were measured at a test potential of 0 mV. The curves were obtained from a least-squares fit of the data to:
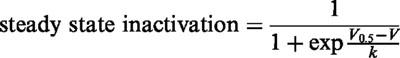 |
where V0.5 is the voltage at which half the channels are inactivated at equilibrium, and k is a slope factor.
RNA preparation
RNA encoding the α-subunit of the μ1 rat skeletal muscle channel was prepared from the Na channel-pALTER-T construct as previously described (Mounsey et al., 1995). We have previously detailed the fabrication of the S1321A (phosphorylation(−)) Na channel mutant (Mounsey et al., 1995). Wild-type and mutant cDNAs were linearized with SalI, and capped RNA was transcribed using the SP6 promoter (Ambion Megascript). RNA encoding a protein kinase C (PKC) α-isozyme was generously supplied by Drs K.P. Lu and E.N. Olson. The expressed PKC lacks the coding region for the regulatory domain, and is thus constitutively active. The biochemical evidence for enzymatic activity has been described (Li et al., 1992; James & Olson, 1992).
Halothane solution preparation
Halothane- (2-bromo 2-chloro 1,1,1-trifluoroethane, Halocarbon Laboratories, River Edge, NJ, U.S.A.) containing solutions were prepared by bubbling halothane vapour at room temperature through the solution used for superfusion of oocytes. Halothane was vaporized in a conventional clinical general anaesthesia vaporizer (OHIO Medical Products, Madison, WI, U.S.A.) using compressed air at 5 l min−1. Solutions were equilibrated with halothane for at least 30 min and supplied to the experimental chamber through Teflon tubing. The concentration of halothane present at the cell was determined from samples of flowing control solution equilibrated with halothane.
Halothane standards were prepared from a saturated solution of halothane (17.4 mM determined by the solubility of halothane in water: 0.345%, Merck Index, 11th edition) and used to construct a calibration curve. Solutions were equilibrated with halothane by being bubbled for ⩾30 min at each setting on the halothane vaporizer. The recording chamber was then perfused with the experimental solution, and samples at the experimental site were taken. The concentration of the anaesthetic in the effluent was determined using a gas chromatograph (Hewlett Packard 5890A series II using a 30 m HP-1 capillary column). Injections into the gas chromatograph were performed manually using a 1 μl syringe. Samples were dissolved in a methanol solvent in order to determine the injection volume. All experimental results were compared to standard solutions made and analysed at the time of the unknown sample analysis.
Data analysis
We determined peak current amplitude and current decay kinetics from traces corrected for leak and capacitance (pCLAMP, Axon). The linear drift of Na current decay τ and amplitude was corrected for each experiment (Excel, Microsoft) as detailed below. Corrected current amplitudes were then normalized to the current amplitude before application of halothane to facilitate comparison between oocytes with different initial levels of Na current amplitude.
To determine significance of differences between groups we used t-tests for data with a normal distribution as determined by the Kolmogorov-Smirnov test, or the Rank Sum test for non-normal data (SigmaStat, Jandel). Data are expressed as mean±s.e.mean unless otherwise stated, and P<0.05 was taken to be a statistically significant result.
Results
Changes in Na current decay and amplitude over time
The effects of halothane and PKC co-expression on Xenopus oocyte-expressed rat skeletal muscle Na channel α-subunits were assessed by studying the Na currents during repetitive depolarizing clamp pulses from a holding potential of −100 mV to a test potential of 0 mV, applied 30 s apart. Figure 1A shows Na currents recorded in the absence of halothane and demonstrates that both the decay time and amplitude were reduced over the 20-min course of a typical experiment. Na current decay was fitted to a single exponential decay function, and the change in the time constant, τ, over time was linear (Figure 1B). Similarly, the decay of peak current amplitude was linear (Figure 1C). In five oocytes from two frogs, the rate of decay of τ was 0.09±0.03 msec min−1 and the rate of decay of normalized amplitude was 0.2±0.06% min−1. These linear drifts of τ and amplitude were corrected in individual data sets by subtracting a regression line drawn through data points recorded in the first 2 min, before halothane was applied, and last 2 min, after 8 min wash, of each experiment, as shown in Figure 1B,C. The drifts of amplitude and τ were the same for wild-type and mutant Na channels, and were independent of PKC co-expression and the halothane dose (legends to Figures 4 and 7). One possible explanation for the slow decline in peak Na current amplitude is slow Na channel inactivation gating (Simoncini & Stuhmer, 1987; Ruff et al., 1988) and the change in decay kinetics might have been the result of changes in modal gating of expressed Na channels (Zhou et al., 1991; Moorman et al., 1990).
Figure 1.

Effect of repeated depolarizations, 30 s apart, on currents through oocyte-expressed rat skeletal muscle Na channels. The original current records (A) reveal a steady decrement in amplitude and decay kinetics. The individual current traces were fitted to a single exponential, and the decay time constant (τ) was a linear function of time (B, squares). A regression line had a slope of −0.09 msec min−1, (r=0.980, P<0.001). This linear decay was subtracted from the raw data to yield an unchanging corrected τ over the 22 min course of the experiment (circles). The drift of amplitude was also linear (C), and a regression line had a slope of 0.1 μA min−1 (r=0.998, P<0.001). When this was subtracted from the raw data, corrected amplitude was unchanging.
Figure 4.
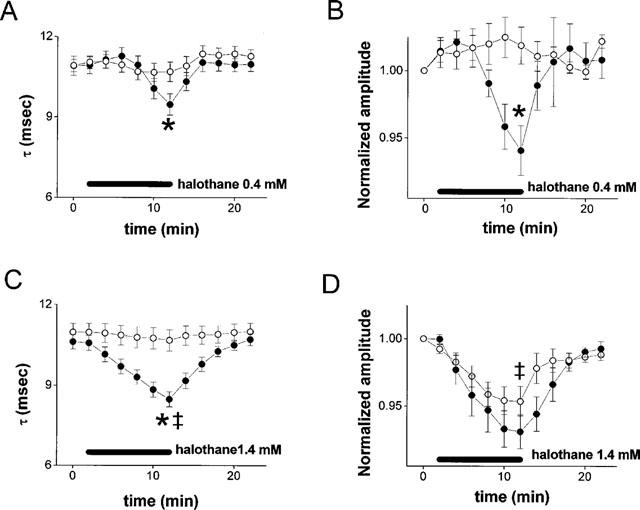
Summary data for the effects of 0.4 and 1.4 mM halothane on Na current decay τ and amplitude. Open circles represent oocytes expressing Na channels alone; filled circles represent oocytes co-expressing PKC. Data have been corrected for linear drift. In oocytes expressing Na channels alone, the mean drift in decay τ was 0.07 msec min−1 in 0.4 mM halothane and 0.06 msec min−1 in 1.4 mM halothane. The mean drift in amplitude was 1.8% min−1 in 0.4 mM halothane compared with 2.3% min−1 in 1.4 mM halothane (P=NS, t-test). In oocytes co-expressing PKC, the mean drifts in decay τ were 0.06 and 0.09 msec min−1, and the mean drifts in amplitude were 1.5% and 1.8% min−1 in 0.4 and 1.4 mM halothane (P=NS, t-test). *P<0.05 (t-test) for the comparison between oocytes expressing Na channels alone and oocytes co-expressing PKC. ‡P<0.05 (t-test) for the comparison between halothane 0.4 and 1.4 mM. Data were derived from 10–21 oocytes, three to six frogs.
Figure 7.

Summary data for the effects of halothane on Na current decay rate and amplitude in phos(−) Na channels. Data were derived from 10–15 oocytes, two to four frogs. Experimental details and symbols as in Figure 4. In oocytes expressing Na channels alone, the mean drift in decay τ was 0.07 and 0.09±0.01 msec min−1 in 0.4 and 1.4 mM halothane. The mean drifts in amplitude were 1.2% min−1 compared with 1.1% min−1 (P=NS, t-test). In oocytes co-expressing PKC the mean drifts in decay τ were 0.05 and 0.1 msec min−1, and the mean drifts in amplitude were 1.8 and 2.5% min−1 in 0.4 and 1.4 mM halothane (P=NS, t-test).
PKC co-expression modulates halothane effects on skeletal muscle Na channels
Figure 2 shows examples of the effects of halothane on Na currents. Halothane (0.4 mM for 10 min) had no effect on the decay kinetics or the amplitude of Na currents in an oocyte expressing wild-type Na channels alone, over and above the linear drifts in the preparation (Figure 2A, see also Figure 3A,C). In an oocyte from the same batch co-injected with RNA encoding a PKC α-isozyme, the current decay rate was similar at baseline (Figure 2B, trace (i)). Exposure to halothane, however, induced a reduction in Na current amplitude and enhanced the speed of Na current decay (Figure 2B trace (ii)), both of which reversed to control during the washout period (Figure 2B trace (iii)). These changes were also distinct from linear drifts, as emphasised in Figure 3B,D.
Figure 2.

The effect of halothane on oocytes expressing rat skeletal muscle Na channels alone (left panels) and with PKC co-expression (right panels). (A and B) show currents recorded from oocytes expressing Na channels α-subunits alone; (C and D) show data from oocytes co-expressing the β-subunit. Sample recordings taken under control conditions (i), after superfusion with halothane for 10 min (ii), and after 10 min wash (in oocytes expressing Na channels alone) (iii), are shown superimposed. Note that currents recorded from the oocyte co-expressing the β-subunit were larger and decayed much more quickly than the currents recorded from the oocyte expressing Na channels alone.
Figure 3.
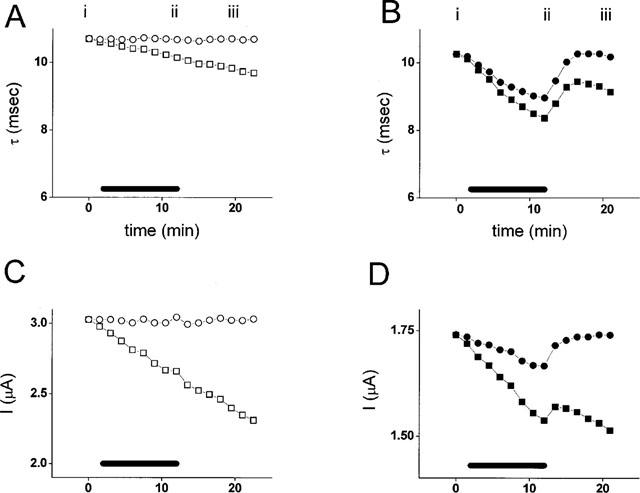
Subtraction of the linear drift in decay τ (A and B) and amplitude (C and D) for the examples shown in Figure 2A and B. (A and C) show data from the oocyte expressing Na channels alone; (B and D) show data from the oocyte co-expressing PKC. Halothane was applied between minutes 2 and 12 as shown by the bars. Raw data are displayed as squares; corrected data as circles. The drift of τ and amplitude in this example were 0.05 msec min−1 and 0.3 μA min−1 in oocytes expressing Na channels alone, and 0.05 msec min−1 and 0.1 μA min−1 in oocytes co-expressing PKC.
Figure 2C,D confirm that the effects of halothane were similar in oocytes co-expressing Na channels with the β-subunit. In an oocyte expressing Na channels alone (Figure 2C), halothane induced a small reduction in amplitude, but was without effect on current decay. In an oocyte co-expressing PKC, however, there was a larger reduction in Na current amplitude, and enhancement of Na current decay (Figure 2D). Similar effects were observed in four other oocytes. Note that currents in oocytes co-expressing the β-subunit were larger and decayed much more quickly than currents in oocytes expressing Na channels alone. The effects of halothane were qualitatively similar to those seen in oocytes not expressing the β-subunit (compare Figure 2A,B). We did not pursue these experiments further because of concerns about adequacy of voltage clamp control for the large, rapidly inactivating currents observed in oocytes co-expressing the β-subunit.
Figure 4 shows summary results from oocytes expressing Na channel α-subunits. Figure 4A shows that co-expression of PKC did not affect Na current decay τ. The decay τ before addition of halothane was 10.9±0.2 msec in oocytes expressing Na channels alone (open circles) and 10.8±0.2 in oocytes co-expressing PKC (filled circles; P=NS). After exposure to halothane, τ was 10.6±0.3 msec in oocytes expressing Na channels alone, compared with 9.5±0.4 msec in oocytes co-expressing PKC (P<0.05).
Figure 2A,B suggested that co-expression of PKC resulted in a reduction of current amplitude. Figure 5 (left two boxes) shows summary results. Co-expression of PKC led to a 60% reduction Na current amplitude in the absence of halothane (P<0.001). To facilitate comparison of the effects of halothane in oocytes expressing Na channels and in oocytes co-expressing PKC, the initial current in each group of oocytes was set to 1.0. As shown in Figure 4B, halothane had no effect on current amplitude in oocytes expressing Na channels alone. In oocytes co-expressing PKC, however, exposure to halothane resulted in a decrease in amplitude to 94±2% of control (P<0.05).
Figure 4C,D show that the effects of halothane on Na current decay kinetics and amplitude were dose-dependent. At 1.4 mM, halothane had no effect on decay τ in oocytes expressing Na channels alone (Figure 4C, P=NS), but in oocytes co-expressing PKC, halothane reduced decay τ to 8.5±0.3 msec, significantly less than the reduction of decay τ to 9.5±0.4 seen with 0.4 mM halothane (P<0.05).
Figure 4D shows that this high dose of halothane induced a reduction of current amplitude to 95±1% of control in oocytes expressing Na channels alone. This reduction was not significantly enhanced in oocytes co-expressing PKC, where amplitude was reduced to 93±1% (P=NS).
Steady state properties of Na channels were not affected by PKC or halothane
The changes in Na current decay τ and amplitude induced by halothane were not the result of alterations in the availability of Na channels. Figure 6A,B show plots of steady-state inactivation obtained by a standard two-pulse protocol before and after 10 min exposure to halothane. Figure 6A shows data obtained from oocytes expressing Na channels alone and Figure 6B shows data from oocytes co-expressing PKC. Co-expression of PKC did not affect channel availability, and for both groups of oocytes the curves were superimposable before and after exposure to halothane.
The changes in Na current kinetics and amplitude induced by halothane were not the result of a large change in the voltage-dependence of channel opening. Figure 6C,D show current-voltage relationships obtained before and 10 min after exposure to halothane in oocytes expressing Na channels alone and in oocytes co-expressing PKC. The amplitudes have been normalized to the peak current in the absence of halothane in each case to emphasize the absence of a shift in the voltage dependence of activation in either group of oocytes.
PKC effects were blocked by mutation of a phosphorylation site in the channel inactivation gate
To investigate whether the effects of PKC co-expression were the result of Na channel phosphorylation, or were caused by a non-specific effect of PKC on oocyte physiology, we re-tested the effects of PKC co-expression and halothane in oocytes expressing a Na channel mutant that lacked a PKC phosphorylation site. The rationale for these experiments was that any PKC or halothane effects that were the result of phosphorylation at any site in the channel molecule should be reduced in a mutant channel molecule which lacked that site. A very highly conserved PKC phosphorylation site is in the inactivation gate of the Na channel molecule (West et al., 1991; 1992; Numann et al., 1991; Bendahhou et al., 1995), at Serine 1321. This site was disabled by Serine to Alanine mutation (S1321A, phos(−) Na channels) and the effects of halothane and PKC co-expression were re-tested.
Figure 7A,C show that, as in wild-type channels, co-expression of PKC with these phos(−) channels had no effect on Na current decay rates (P=NS). In contrast to wild-type channels, however, exposure to halothane had no effect on the Na current decay τ in oocytes expressing phos(−) mutant channels alone or in oocytes co-expressing PKC (Figure 7A). Moreover, when the dose of halothane was increased to 1.4 mM, phos(−) Na channels were similarly unaffected by exposure to halothane, even with co-expression of PKC (Figure 7C). This finding suggests that phosphorylation of S1321 was a necessary step in the interaction between muscle Na channels and halothane.
Co-expression of PKC resulted in a reduction of current amplitude in wild-type channels. If this reduction resulted solely from phosphorylation of the inactivation gate site (S1321), it would be anticipated that the effect would be lost in phos(−) channels. Figure 5, right two boxes, shows this not to be the case. Co-expression of PKC with phos(−) Na channels resulted in a significant reduction in current amplitude, of 46%, which was not different from the reduction observed in wild-type channels (P=NS).
Figure 7B shows that, as for wild-type Na channels, halothane (0.4 mM) had no effect on the amplitude of currents through phos(−) channels expressed alone, whereas in oocytes co-expressing PKC there was a significant reduction in current amplitude to 97±0.7% of control. Figure 7D shows that the higher dose of halothane (1.4 mM) induced a reduction in amplitude to 97±1% which was independent of PKC co-expression.
Discussion
We studied the effects of halothane on Xenopus oocyte-expressed skeletal muscle Na channels, and the effects of co-expression of an active PKC α-isozyme. Our major findings were:
Halothane had no effect on Na currents at a dose of 0.4 mM.
After co-expression of PKC, halothane led to a reversible enhancement in the decay τ of Na currents and a reversible reduction in their amplitude.
This effect on decay τ was absent in a Na channel mutant lacking a PKC phosphorylation site in the inactivation gate, but the effect on amplitude was independent of mutation of this site.
The effect on decay τ was dose-dependent. Na current decay τ was significantly shorter at 1.4 mM halothane than at 0.4 mM in oocytes co-expressing PKC.
The effect on amplitude was dose-dependent. At 0.4 mM halothane, amplitude was reduced only when PKC was co-expressed. At 1.4 mM, amplitude was reduced regardless of PKC co-expression.
Thus halothane has dual effects on muscle Na channels – to suppress Na current amplitude and to hasten Na current decay. The findings were the same whether or not the β-subunit was co-expressed. Both of these actions would have the effect of reducing Na influx during the action potential, and hence to suppress excitability. The lower dose of halothane tested (0.4 mM) is possibly relevant clinically at 1–2 MAC (Franks & Lieb, 1994) and is in keeping with other studies of the effect of halothane on ion channels (Buljubasic et al., 1992; Weigt et al., 1997b; Rehberg et al., 1996). Our data suggest that phosphorylation of the Na channel inactivation gate is an essential step in the action of halothane to speed Na current decay. Suppression of Na current amplitude is also likely to be dependent on channel phosphorylation, although at a site distant from the inactivation gate, in that halothane suppressed current amplitude at a lower dose after PKC co-expression in both wild-type channels, and in channels lacking the inactivation gate phosphorylation site. At higher doses the reduction of amplitude was greater, and also independent of PKC co-expression, suggesting that PKC sensitizes current amplitude to halothane, but is not a necessary mediator in the effect on amplitude.
Na channels are at the basis of cellular excitability and effects of volatile anaesthetics on a variety of Na channel isoforms have been studied. In general suppression of current amplitude and enhancement of current decay have been reported. Bean and co-workers demonstrated that halothane and ether reduced Na current amplitude and hastened current decay in crayfish giant axons (Bean et al., 1981). Rehberg and co-workers reported that volatile anaesthetics decreased current amplitude through mammalian neuronal Na channels (Rehberg, et al., 1996), but current decay kinetics were not assessed. In canine cardiac Purkinje fibres, halothane and isoflurane reduced slow Na current amplitude, which had the effect of enhancing Na current decay kinetics with little appreciable effect of overall Na current amplitude (Eskinder et al., 1993). Halothane and isoflurane reduce Na current amplitude and enhance Na current decay of Na currents through human cardiac Na channels expressed in a mammalian cell line (Weigt et al., 1997a). Most relevant, in human skeletal muscle, halothane suppressed Na current amplitude and enhanced the time constant of current inactivation, albeit at high doses (Ruppersberg & Rudel, 1988).
We wished to test hypotheses about phosphorylation of the inactivation gate, and therefore chose to study muscle Na channels because they are structurally simpler than brain channels. Specifically, they lack a 200 amino acid portion of the intracellular region linking domains I and II that contains several other consensus phosphorylation sites. PKC phosphorylation of sites in the I-II linker has effects on Na currents, and prior phosphorylation of the site in the inactivation gate by PKC is required before phosphorylation of the I-II linker sites by PKA has functional effects (Li et al., 1993). Because there is less opportunity for convergent regulation by multiple kinases, skeletal muscle channels provide a simpler experimental system to test for possible interactions between PKC, the inactivation gate and halothane in regulation of Na channels.
Na channels are substrates for phosphorylation by PKC, and PKC activation has profound effects on Na currents. Our observation that co-expression of PKC with wild-type Na channels resulted in a reduction of Na current amplitude, is consistent with previous reports for both brain, cardiac and skeletal muscle channels in which activation of PKC reduced Na current amplitude (Numann et al., 1991; 1994; West et al., 1991; 1992; Sigel & Baur, 1988; Lotan et al., 1990; Dascal & Lotan, 1991; Bendahhou et al., 1995). The observation that PKC co-expression has no effect on current decay, however, is not consistent with published reports. In brain channels PKC activation results in slowing of Na current decay (Numann et al., 1991), whereas in skeletal muscle channels current decay is enhanced in some preparations and slowed in others (Numann et al., 1994; Bendahhou et al., 1995). In heart channels decay is unaffected (Murray et al., 1997). Our observed absence of an effect of PKC co-expression on steady state inactivation is also inconsistent with previous reports. In skeletal muscle channels a negative shift has been reported (Bendahhou et al., 1995), although in cardiac channels there is no change (Murray et al., 1997). These discrepancies may be related to experimental techniques. We co-expressed a constitutively active PKC α-isozyme whereas in previous reports PKC was activated pharmacologically. Pharmacologic activation would result in activation of multiple PKC isozymes, which may interact with multiple sites on the channel, possibly causing different effects on channel behaviour.
Phosphorylation of Na channels in the inactivation gate and probably at other sites modulates channel function and is likely to be an important mechanism of modulation of muscle and nerve excitability. In brain Na channels, mutation of the inactivation gate phosphorylation site abolishes the effects of PKC (West et al., 1992). In contrast, PKC effects on cardiac and skeletal muscle Na channels are independent of mutation of this site (Bendahhou et al., 1995; Murray et al., 1997), suggesting that PKC may interact also with other sites in the channel. This idea is supported by our observation that the effects of halothane on current decay were dependent on the presence of the inactivation gate phosphorylation site whilst the effects on amplitude were not. Nevertheless, the effects of PKC on muscle channels are likely to result from phosphorylation of the channel, since they are blocked by PKC inhibitors and enhanced by phosphatase inhibitors (Bendahhou et al., 1995; Murray et al., 1997).
Volatile anaesthetics have little effect on channel activation in cardiac, skeletal muscle and brain Na channels, but hyperpolarizing shifts in steady state inactivation have been reported (Ruppersberg & Rudel, 1988; Weigt et al., 1997a; Rehberg et al., 1996; Stadnika et al., 1998). We did not reproduce the effect on steady state inactivation in our preparation either in the presence or absence of PKC co-expression. This discrepancy is difficult to explain. Co-expression of the β-subunit with skeletal muscle Na channels in Xenopus oocytes in itself shifts steady state inactivation to more negative potentials (Nuss et al., 1995), and it is possible that the absence of the β-subunit in our measurements of steady state inactivation may modulate effects of anaesthetics on channel availability. These measurements require accurate assessment of peak currents, which are better resolved in our experiments using α-subunits expressed alone.
By what mechanism do halothane and PKC co-expression interact to suppress muscle Na channels? Halothane could, in principle, exert its effects via activation of PKC, indirectly through an alteration in lipid behaviour, or through preferential interaction of the drug with phosphorylated channels. Modulation of PKC activity by volatile anaesthetics has been reported (Lester & Baumann, 1991; Slater et al., 1993), but this seems an unlikely mechanism to explain the current data because our experiments were performed using a constitutively active PKC isozyme. Unless it was modulated by oocyte PKC regulatory subunit structures, the expressed PKC should be fully active. Alteration of the lipid behaviour of the cell membrane is also a credible explanation of our data. Volatile general anaesthetics insert preferentially into the hydrocarbon regions of phospholipid lipid bilayers rather than the polar head groups (Baber et al., 1995), and halocarbons which lack anaesthetic potency do not localize within the membrane interface (North & Cafiso, 1997). These effects might interface with the interaction between PKC and the cell membrane, but do not explain the difference we observed between wild-type and phos(−) Na channels, which differed by only a point mutation but were studied in an identical lipid milieu. The simplest interpretation is that halothane interacts directly on the channel molecule itself. Prior phosphorylation of the channel inactivation gate would be required to sensitize the channel to halothane, resulting in enhanced current decay. Current amplitude is diminished by high dose halothane, but prior phosphorylation of as yet unidentified site(s) permits halothane to exert an effect at lower doses.
In conclusion, in muscle Na channels expressed in Xenopus oocytes, halothane causes a reduction in Na current amplitude and enhancement of Na current decay. The effect of halothane on current decay requires co-expression of constitutively active PKC, and phosphorylation of a site in the Na channel inactivation gate. Co-expression of PKC enhances halothane's effect on Na current amplitude, but phosphorylation of the inactivation gate is not required for the effect and, at high dose, the effect is independent of PKC co-expression.
Acknowledgments
We thank Dr Carl Lynch III for valuable and stimulating discussion of these data. We thank also Drs K.P. Lu and E.N. Olson for the PKC clone. The financial support of the British Heart Foundation is gratefully acknowledged (J.P. Mounsey & M.K. Patel).
Abbreviations
- Decay τ
exponential decay time constant
- MAC
minimum alveolar concentration
- (m)RNA
(messenger) ribonucleic acid
- PKC
protein kinase C
References
- BABER J., ELLENA J.F., CAFISO D.S. Distribution of general anesthetics in phospholipid bilayers determined using 2H NMR and 1H-1H NOE spectroscopy. Biochemistry. 1995;34:6533–6539. doi: 10.1021/bi00019a035. [DOI] [PubMed] [Google Scholar]
- BEAN B.P., SHRAGER P., GOLDSTEIN D.A. Modification of sodium and potassium channel gating kinetics by ether and halothane. J. Gen. Physiol. 1981;77:233–253. doi: 10.1085/jgp.77.3.233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENDAHHOU S., CUMMINS T.R., POTTS J.F., TONG J., AGNEW W.S. Serine-1321-independent regulation of the mu 1 adult skeletal muscle Na+ channel by protein kinase C. Proc. Natl. Acad. Sci. (U.S.A.) 1995;92:12003–12007. doi: 10.1073/pnas.92.26.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOURINET E., NARGEOT J., CHARNET P. Electrophysiological characterization of a TTX-sensitive sodium current in native Xenopus oocytes. Proceedings of the Royal Society of London-Series B: Biological Sciences. 1992;250:127–132. doi: 10.1098/rspb.1992.0140. [DOI] [PubMed] [Google Scholar]
- BULJUBASIC N., RUSCH N.J., MARIJIC J., KAMPINE J.P., BOSNJAK Z.J. Effects of halothane and isoflurane on calcium and potassium channel currents in canine coronary arterial cells. Anesthesiology. 1992;76:990–998. doi: 10.1097/00000542-199206000-00020. [DOI] [PubMed] [Google Scholar]
- DASCAL N., LOTAN H. Activation of protein kinase C alters voltage dependence of a Na+ channel. Neuron. 1991;6:165–175. doi: 10.1016/0896-6273(91)90131-i. [DOI] [PubMed] [Google Scholar]
- DIFAZIO C.A., NEIDERLEHNER J.R., BURNEY R.G. The anesthetic potency of lidocaine in the rat. Anesth. Analg. 1976;55:818–821. doi: 10.1213/00000539-197611000-00016. [DOI] [PubMed] [Google Scholar]
- DURIEUX M.E., SALAFRANCA M.N., LYNCH K.R., MOORMAN J.R. Lysophosphatidic acid induces a pertussis toxin-sensitive Ca2+-activated Cl− current in Xenopus laevis oocytes. Am. J. Physiol. 1992;263:C896–C900. doi: 10.1152/ajpcell.1992.263.4.C896. [DOI] [PubMed] [Google Scholar]
- ESKINDER H., SUPAN H.D., TURNER L.A., KAMPINE J.P., BOSNJAK Z.J. The effects of halothane and isoflurane on slowly inactivating sodium currents in canine cardiac purkinje cells. Anesth. Analg. 1993;77:32–37. doi: 10.1213/00000539-199307000-00007. [DOI] [PubMed] [Google Scholar]
- FRANKS N.P., LIEB W.R. Molecular and cellular mechanisms of general anaesthesia. Nature. 1994;367:607–614. doi: 10.1038/367607a0. [DOI] [PubMed] [Google Scholar]
- HEMMINGS H.C., JR, ADAMO A.I.B. Effects of halothane and propofol on protein kinase C activation. Anesthesiology. 1994;81:147–155. doi: 10.1097/00000542-199407000-00021. [DOI] [PubMed] [Google Scholar]
- HEMMINGS H.C., JR, ADAMO A.I.B. Activation of endogenous protein kinase C by halothane in synaptosomes. Anesthesiology. 1996;84:652–656. doi: 10.1097/00000542-199603000-00021. [DOI] [PubMed] [Google Scholar]
- HIMES R.S., DIFAZIO C.A., BURNEY R.G. Effects of lidocaine on the anesthetic requirements for nitrous oxide. Anesthesiology. 1977;47:437–440. doi: 10.1097/00000542-197711000-00010. [DOI] [PubMed] [Google Scholar]
- HIMES R.S., MUNSON E.S., EMBRO W.J. Enflurane requirement and ventilatory response to carbon dioxide during lidocaine infusion in dogs. Anesthesiology. 1979;51:131–134. doi: 10.1097/00000542-197908000-00008. [DOI] [PubMed] [Google Scholar]
- JAMES G., OLSON E. Deletion of the regulatory domain of protein kinase C alpha exposes regions in the hinge and catalytic domains that mediate nuclear targeting. J. Cell. Biol. 1992;116:863–874. doi: 10.1083/jcb.116.4.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LESTER D.S., BAUMANN D. Action of organic solvents on protein kinase C. Eur. J. Pharmacol. 1991;206:301–308. doi: 10.1016/0922-4106(91)90114-w. [DOI] [PubMed] [Google Scholar]
- LI M., WEST J.W., NUMANN R., MURPHY B.J., SCHEUER T., CATTERALL W.A. Convergent regulation of sodium channels by protein kinase C and cAMP-dependent protein kinase. Science. 1993;261:1439–1442. doi: 10.1126/science.8396273. [DOI] [PubMed] [Google Scholar]
- LI L., ZHOU J., JAMES G., HELLER-HARRISON R., CZECH M.P., OLSON E.N. FGF inactivates myogenic helix-loop-helix proteins through phosphorylation of a conserved protein kinase C site in their DNA-binding domains. Cell. 1992;71:1181–1194. doi: 10.1016/s0092-8674(05)80066-2. [DOI] [PubMed] [Google Scholar]
- LOTAN I., DASCAL N., NAOR Z., BOTON R. Modulation of vertebrate brain Na+ and K+ channels by subtypes of protein kinase C. FEBS Lett. 1990;267:25–28. doi: 10.1016/0014-5793(90)80279-r. [DOI] [PubMed] [Google Scholar]
- MOORMAN J.R., KIRSCH G.E., VANDONGEN A.M.J., JOHO R.H., BROWN A.M. Fast and slow gating of sodium channels encoded by a single mRNA. Neuron. 1990;4:243–252. doi: 10.1016/0896-6273(90)90099-2. [DOI] [PubMed] [Google Scholar]
- MOUNSEY J.P., XU P., JOHN J.E., HORNE L.T., GILBERT J., ROSES A.D., MOORMAN J.R. Modulation of skeletal muscle sodium channels by human myotonin protein kinase. J. Clin. Invest. 1995;95:2379–2384. doi: 10.1172/JCI117931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MURRAY K.T., HU N.N., DAW J.R., SHIN H.G., WATSON M.T., MASHBURN A.B., GEORGE A.L.J. Functional effects of protein kinase C activation on the human cardiac Na+ channel. Circ. Res. 1997;80:370–376. doi: 10.1161/01.res.80.3.370. [DOI] [PubMed] [Google Scholar]
- NORTH C., CAFISO D.S. Contrasting membrane localization and behavior of halogenated cyclobutanes that follow or violate the Meyer-Overton hypothesis of general anesthetic potency. Biophys. J. 1997;72:1754–1761. doi: 10.1016/S0006-3495(97)78821-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NUMANN R., CATTERALL W.A., SCHEUER T. Functional modulation of brain sodium channels by protein kinase C phosphorylation. Science. 1991;254:115–118. doi: 10.1126/science.1656525. [DOI] [PubMed] [Google Scholar]
- NUMANN R., HAUSCHKA S.D., CATTERALL W.A., SCHEUER T. Modulation of skeletal muscle sodium channels in a satellite cell line by protein kinase C. J. Neurosci. 1994;14:4226–4236. doi: 10.1523/JNEUROSCI.14-07-04226.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NUSS H.B., CHIAMVIMONVAT N., PEREZ-GARCIA M.T., TOMASELLI G.F., MARBAN E. Functional association of the beta 1 subunit with human cardiac (hH1) and rat skeletal muscle (mu 1) sodium channel alpha subunits expressed in Xenopus oocytes. J. Gen. Physiol. 1995;106:1171–1191. doi: 10.1085/jgp.106.6.1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARKER I., MILEDI R. Tetrodotoxin-sensitive sodium current in native Xenopus oocytes. Proceedings of the Royal Society of London - Series B: Biological Sciences. 1987;232:289–296. doi: 10.1098/rspb.1987.0075. [DOI] [PubMed] [Google Scholar]
- REHBERG B., XIAO Y.-H., DUCH D.S. Central nervous system sodium channels are significantly suppressed at clinical concentrations of volatile anesthetics. Anesthesiology. 1996;84:1223–1233. doi: 10.1097/00000542-199605000-00025. [DOI] [PubMed] [Google Scholar]
- RUFF R.L., SIMONCINI L., STÜHMER W. Slow sodium channel inactivation in mammalian muscle: a possible role in regulating excitability. Muscle & Nerve. 1988;11:502–510. doi: 10.1002/mus.880110514. [DOI] [PubMed] [Google Scholar]
- RUPPERSBERG J.P., RUDEL R. Differential effects of halothane on adult and juvenile sodium channels in human muscle. Pflugers Archiv - European Journal of Physiology. 1988;412:17–21. doi: 10.1007/BF00583726. [DOI] [PubMed] [Google Scholar]
- SIGEL E., BAUR R. Activation of protein kinase C differentially modulates neuronal Na+, Ca2+, and gamma-aminobutyrate type A channels. Proc. Natl. Acad. Sci. (U.S.A.) 1988;85:6192–6196. doi: 10.1073/pnas.85.16.6192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMONCINI L., STÜHMER W. Slow sodium channel inactivation in rat fast-twitch muscle. J. Physiol. 1987;383:327–337. doi: 10.1113/jphysiol.1987.sp016411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SLATER S.J., COX K.J.A., LOMBARDI J.V., HO C., KELLY M.B., RUBIN E., STUBBS C.D. Inhibition of protein kinase C by alcohols and anaesthetics. Nature. 1993;364:82–84. doi: 10.1038/364082a0. [DOI] [PubMed] [Google Scholar]
- STADNIKA A., KWOK H.A., HARTMANN H.A., BOSNJAK Z.J.Fast and slow inactivation of the human cardiac sodium channel alpha subunit is differentially altered by volatile anesthetics Biophys. J. 1998742A399(Abstract) [Google Scholar]
- TAS P.W.L., KOSCHEL K. Volatile anesthetics stimulate the phorbol ester evoked neurotransmitter release from PC12 cells through an increase of the cytosolic Ca ion concentration. Biochim. Biophys. Acta. 1997;1991:401–404. doi: 10.1016/0167-4889(91)90206-d. [DOI] [PubMed] [Google Scholar]
- TSUCHIA M., OKIMASU E., UEDA W., HIRAKAWA M., UTSUMI K. Halothane, an inhalational anesthetic, activates PKC and superoxide generation by neutrophils. FEBS Lett. 1988;242:101–105. doi: 10.1016/0014-5793(88)80994-3. [DOI] [PubMed] [Google Scholar]
- WEIGT H.U., KWOK W.M., REHMERT G.C., TURNER L.A., BOSNJAK Z.J. Voltage-dependent effects of volatile anesthetics on cardiac sodium current. Anesth. Analg. 1997a;84:285–293. doi: 10.1097/00000539-199702000-00009. [DOI] [PubMed] [Google Scholar]
- WEIGT H.U., REHMERT G.C., BOSNJAK Z.J., KWOK W.M. Conformational state-dependent effects of halothane on cardiac Na+ current. Anesthesiology. 1997b;87:1494–1506. doi: 10.1097/00000542-199712000-00029. [DOI] [PubMed] [Google Scholar]
- WEST J.W., NUMANN R., MURPHY B.J., SCHEUER T., CATTERALL W.A. A phosphorylation site in the Na+ channel required for modulation by protein kinase C. Science. 1991;254:866–868. doi: 10.1126/science.1658937. [DOI] [PubMed] [Google Scholar]
- WEST J.W., NUMANN R., MURPHY B.J., SCHEUER T., CATTERALL W.A. Phosphorylation of a conserved protein kinase C site is required for modulation of Na+ currents in transfected Chinese hamster ovary cells. Biophys. J. 1992;62:31–33. doi: 10.1016/S0006-3495(92)81769-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU J., POTTS J.F., TRIMMER J.S., AGNEW W.S., SIGWORTH F.J. Multiple gating modes and the effect of modulating factors on the μI sodium channel. Neuron. 1991;7:775–785. doi: 10.1016/0896-6273(91)90280-d. [DOI] [PubMed] [Google Scholar]