

Abstract
Tumour necrosis factor-α (TNF-α)- and lipopolysaccharide (LPS)-induced apoptosis of bovine glomerular endothelial cells is now recognized as an important part in the pathogenesis of glomerulonephritis characterized by early mitochondrial cytochrome c release, mitochondrial permeability transition, Bak protein upregulation, Bcl-XL protein downregulation and caspase-3 activation.
Co-treatment of cells with 10 nM dexamethasone and TNF-α or LPS blocked roughly 90% of apoptotic cell death in glomerular endothelial cells. The action of glucocorticoids could be documented in that they prevented all apoptotic markers such as DNA laddering, DNA fragmentation measured by the diphenylamine assay as well as morphological alterations.
To mechanistically elucidate the action of glucocorticoids we evaluated whether glucocorticoids elicit a time-dependent effect. For dexamethasone, to maximally inhibit DNA fragmentation a preincubation period was not required. Even if dexamethasone was supplemented 6 h following TNF-α or LPS we observed a maximal inhibitory effect.
Concerning its influence on TNF-α and LPS signal transduction, we found that dexamethasone only partially prevented cytochrome-c-release as a first sign of apoptotic cell death but efficiently blocked mitochondrial permeability transition. Moreover, TNF-α- and LPS-induced Bak upregulation, Bcl-XL-downregulation, and the activation of caspase-3-like proteases, measured fluorometrically using DEVD-AMC and PARP cleavage, were efficiently blocked by dexamethasone.
We postulate that glucocorticoids exert their inhibitory action upstream of the terminal death pathways but downstream of primary receptor mediated signals by blocking pro-apoptotic signals pre- and/or post cytochrome c release and mitochondrial signalling.
Keywords: Glucocorticoids, apoptosis, TNF-α, LPS, Bak, Bcl-XL, caspases
Introduction
The adrenal glucocorticoids, cortisol and corticosterone, have important physiological actions on growth, development, the immune system, and inflammatory processes (Cole et al., 1995). Dexamethasone and other powerful synthetic glucocorticoids are in widespread clinical use in the treatment of rheumatoid arthritis, in a variety of autoimmune disorders, and in many other disease states (Da Silva, 1995). Glucocorticoids are the most potent and effective agents in controlling chronic inflammatory diseases. Inhaled steroids have become a first-line therapy for chronic asthma, one of the most prevalent inflammatory diseases in industrialized countries (Lipworth, 1999). Glucocorticoids exert their effects by binding to their cytoplasmatic, specific glucocorticoid receptor which belongs to a family of transcriptional regulatory proteins that interact with specific target sequences upon association with their cognate ligand. The interaction between the glucocorticoid receptor and other transcription factors such as activator protein 1 (AP-1) and nuclear factor κB (NF-κB) has also been shown to play a role in altering glucocorticoid action, suggesting extensive transcriptional cross-talk with other signalling pathways capable of modulating the effect of glucocorticoids (Wissink et al., 1998; Auphan et al.; 1995, Xie et al., 1997, Pfeilschifter & Mühl, 1999).
As many other chemotherapeutic agents, glucocorticoids are well known to inhibit cell growth and to induce apoptosis in lymphocytes and in several lymphoid cell lines (Osborne et al., 1996). The physiological role of apoptosis in immature T cell development is now relatively well understood. After entering the thymus, immature thymocytes undergo a complex series of differentiation steps involving proliferation, expression of accessory molecules, and T cell receptor (TCR) gene rearrangements. Immature CD4+CD8+ thymocytes expressing potentially self-reactive TCRs are subjected to negative selection, by the induction of programmed cell death (Anderson et al., 1996). Induction of thymocyte apoptosis by glucocorticoids also occurs under physiological conditions, i.e. by stimulation of endogenous glucocorticoid release via immunization with exogenous antigens (Gruber et al., 1994). Apart from its well-known apoptosis inducing effect in thymocytes and eosinophils (Nittoh et al., 1998; Liu et al., 1999), non-lymphoid cells are resistant to glucocorticoid-induced apoptosis and moreover, dexamethasone has been shown to prevent apoptosis in drug-induced glioma cells (Naumann et al., 1998) and in bovine glomerular endothelial cells (Meßmer et al., 1999b).
Glomerular inflammatory diseases and their progression to renal scaring (glomerulosclerosis and interstitial fibrosis) are leading causes of end stage renal failure (Sterzel & Lovett, 1988; Edelstein et al., 1997). Apoptosis of glomerular endothelial cells may contribute to the development of glomerulosclerosis during severe forms of glomerulonephritis (Kitamura et al., 1998). The contribution of proinflammatory agents such as the cytokine TNF-α and LPS, the cell wall component of gram negative bacteria to apoptosis induction in glomerular endothelial cells was concluded from in vivo experiments (Shimizu et al., 1997; 1998) and recently described in cultured bovine glomerular endothelial cells (Meßmer et al., 1999a).
TNF-α is one of the most intensively investigated pro-inflammatory cytokines which induces apoptosis via the TNF receptor (TNF-R) subtype I in several tumour cell lines and other cell types (Baker & Reddy, 1998). Following the TNF/TNF-RI-trimerization the intracellular signal transduction cascade starts by the interaction of the intracellular receptor ‘death domain' with other ‘death domain' containing proteins such as TNF receptor-associated death domain protein (TRADD), Fas-associated death domain protein (FADD), and receptor-interacting protein (RIP) (Darnay & Aggarwal, 1997). Involving several other pathways such as apoptosis signal-regulating kinase 1 (ASK1)/c-Jun N-terminal kinase (JNK)/NF-κB-inducing kinase (NIK) the pro-apoptotic signal is passed to the caspase protease family which acts as a terminal cascade of cysteine proteases causing the coordinated morphological changes of the cell structure. Modulatory effects on these pro-apoptotic signalling pathways are achieved by several other proteins such as members of the Bcl-2 protein family (like Bcl-2, Bcl-xL, Mcl-1, A1, and Bcl-w) (Reed, 1998) and the IAP protein family, the latter showing anti-apoptotic activities (Deveraux & Reed, 1999). In our previous works we characterized the interaction of TNF-α with bovine glomerular endothelial cells and compared its cytotoxic effects with those of LPS (Meßmer et al., 1999a). Glomerular endothelial cells die by apoptosis within 24 h following exposure to either TNF-α or bacterial LPS. Signalling pathways comprise a sequence of cytochrome c efflux from mitochondria into the cytosol, mitochondrial permeability transition, upregulation of the pro-apoptotic Bak protein and concomitant downregulation of the anti-apoptotic Bcl-xL molecule as well as caspase-3 activation (Meßmer et al., 1999a). Further on, we described that TNF-α and LPS-induced apoptosis was prevented by glucocorticoids (Meßmer et al., 1999b). We reported on the anti-apoptotic potency of naturally and synthetic glucocorticoids which could be ordered in the sequence fluocinolone>dexamethasone>prednisolone>hydrocortisone>corticosterone and which correlates well with their anti-inflammatory activities. Although we identified inhibitory effects upstream of terminal death pathways, the mechanism of the survival effect of glucocorticoids on glomerular endothelial cells (as well as in other cell types) remains to be clarified (Meßmer et al., 1999b). Therefore, in our current work we clearly characterized the effect of glucocorticoids on glomerular endothelial cell death with respect to a detailed time kinetic context and distinct apoptotic pathways. Glucocorticoids were found to block the lethal signal delivered by TNF-α and LPS signal even if they administered several hours after the apoptotic insult and they block apoptotic signal transduction upstream of Bak upregulation, Bcl-xL downregulation and caspase-3 activation.
Methods
Materials
Diphenylamine, lipopolysaccharide (LPS, E. coli serotype 0127:B8), heparin sodium, cycloheximide, dexamethasone, fluocinulone acetonide (fluocinulone), prednisolone, 17-hydroxycorticosterone (hydrocortisone), corticosterone-21-acetate (corticosterone), 4-androsterone-17β-ol-3-one (testosterone), and 17β-estradiol (estradiol) were purchased from Sigma (Deisenhofen, Germany). N-acetyl-aspartyl-glutamyl-valinyl-aspartyl-7-amino-4-coumarin (DEVD-AMC) was delivered by Bachem (Heidelberg, Germany). RU-486 was from Roussel-UCLAF. Bovine acidic fibroblast growth factor (aFGF) and human basic fibroblast growth factor (bFGF) were purchased from R&D Systems (Wiesbaden, Germany), and recombinant human tumour necrosis factor-α (specific activity: 6.6×106 units/mg) was a generous gift from Knoll AG, Germany. RPMI 1640, cell culture supplements and foetal calf serum were from Gibco (Eggenstein, Germany). All other chemicals were of the highest grade of purity commercially available.
Cell culture and cell treatment
Bovine glomerular endothelial cells were cultivated as described previously (Briner & Kern, 1994). In brief, approximately 10 g of renal cortex tissue were minced, passed through a sterile 240 μm stainless steel sieve, and suspended in HBSS. This suspension was then poured through a 180 μm stainless sieve followed by a 100 μm mesh. The glomeruli retained by the 100 μm sieve were washed three times in HBSS and were then incubated for 10 to 15 min at 37°C in HBSS containing 1 mg ml−1 collagenase (type V, Sigma, Deisenhofen, Germany). After digestion, glomerular remnants were sedimented at 500 g for 5 min. The supernatant was centrifuged at 1000×g for 5 min, and the pellet was suspended in RPMI 1640 medium containing 20% FCS, 100 u ml−1 penicillin, 100 μg ml−1 streptomycin, 50 μg ml−1 heparin sodium, and 5 ng ml−1 of acidic fibroblast growth factor. Cells were plated on 0.2% gelatin-coated tissue culture plates. Primary cultures of endothelial cell clones were isolated with cloning cylinders, detached with trypsin-EDTA, and passaged at cloning density onto gelatin-coated 35-mm diameter plates. Individual clones of endothelial cells were characterized by positive staining for Factor VIII-related antigen and uniform uptake of fluorescent acetylated low-density lipoproteins (Ballermann, 1989). Negative staining for smooth muscle actin and cytokeratin excluded mesangial cell and epithelial cell contaminations, respectively. For the experiments, passages 9 to 19 of endothelial cells were used.
For experiments, endothelial cells were grown to confluency in 60-mm or 100-mm petri-dishes with RPMI 1640 medium containing 15% FCS, 100 u ml−1 penicillin, 100 μg ml−1 streptomycin, 50 μg ml−1 heparin sodium, and 5 ng ml−1 of acidic fibroblast growth factor and incubated in RPMI 1640 containing 2% FCS, 100 u ml−1 penicillin, and 100 μg ml−1 streptomycin.
Quantitation of DNA fragmentation
DNA fragmentation was essentially assayed as reported previously (Meßmer et al., 1998). Briefly, after incubation cells were scraped off the culture plates, resuspended in 250 μl 10 mM Tris, 1 mM EDTA, pH 8.0 (TE-buffer), and incubated with an additional volume lysis-buffer (5 mM Tris, 20 mM EDTA, pH 8.0, 0.5% Triton X-100) for 30 min at 4°C. After lysis, the intact chromatin (pellet) was separated from DNA fragments (supernatant) by centrifugation for 15 min at 13,000×g. Pellets were resuspended in 500 μl TE-buffer and samples were precipitated by adding 500 μl 10% trichloroacetic acid at 4°C. Samples were pelleted at 4000 r.p.m. for 10 min and the supernatant was removed. After addition of 300 μl 5% trichloroacetic acid, samples were boiled for 15 min. DNA contents were quantitated using the diphenylamine reagent (Burton, 1956). The percentage of DNA fragmented was calculated as the ratio of the DNA content in the supernatant to the amount in the pellet.
Morphological investigations
Glomerular endothelial cells were grown in 60-mm culture plates to nearly confluency. Cells were stimulated, followed by fixation with 3% paraformaldehyde for 5 min onto glass slides. Samples were washed with phosphate-buffered saline, stained with Hoechst dye H33258 (8 μg ml−1) for 5 min, washed with distilled water, and mounted in KAISER's glycerol gelatin. Nuclei were visualized using a Zeiss Axiovert fluorescence microscope. For each preparation around 500 cells were counted by two different investigators which were blinded toward the treatment. Each evaluation was repeated three times by three independent experiments.
Western blot analysis
Cells were cultured and incubated as described. In general, cell lysis was achieved with lysis buffer (50 mM Tris, 5 mM EDTA, 150 mM NaCl, 0.5% Nonidet-40, 1 mM PMSF, pH 8.0) and sonication (Branson sonifier; 10 s, duty cycle 100%, output control 10%), followed by centrifugation (4000×g, 5 min), and Bradford protein determination (Bradford, 1976). For the analysis of mitochondrial cytochrome c efflux glomerular endothelial cells were harvested by trypsinization, pelleted by centrifugation, resuspended in 300 μl of homogenization buffer (in mM: HEPES 20, pH 7.5, KCl 10, MgCl2 1.5, EDTA 1, EGTA 1, DTT 1, Pefabloc 4, 5 μg ml−1 aprotinin, 10 μg ml−1 leupeptin, 250 mM sucrose) and incubated for 10 min on ice. Cells were broken by 2×15 passages through a syringe fitted with a 25-gauge needle. The lysate was centrifuged at 750×g for 10 min at 4°C to pellet nuclei. The remaining supernatant was centrifuged for 15 min at 10,000×g, the pellet was used as mitochondrial fraction and the supernatant as cytosolic fraction.
Proteins were normalized to 100 μg lane−1 (PARP), 50 μg lane−1 (cytochrome c) or to 40 μg lane−1 (Bcl-2 proteins and TNF-RI), resolved on 7.5% (PARP), 10% (TNF-RI), 12.5% (Bcl-2 proteins) or 14% (cytochrome c) polyacrylamide gels and blotted onto PVDF sheets. Sheets were washed twice with TBS (NaCl 140 mM, Tris 50 mM, pH 7.2) containing 0.1% Tween-20 before blocking unspecific binding with TBS/5% skim milk. Filters were incubated either with the mouse anti-cytochrome c antibody (clone 7H8.2C12, PharMingen, 1 μg ml−1 in TBS/2% skim milk/0.7% FCS), goat anti-TNF-RI antibody (0.2 μg ml−1 in TBS/0.5% skim milk), mouse anti-PARP antibody (Biomol, clone C-II-10, 1 μg ml−1, in TBS+0.5% skim milk), mouse anti-Bcl-2 antibody (Immunotech, clone 83-8B, 1 μg ml−1), rabbit anti-Bcl-x antibody (Transduction Laboratories, 1 : 1000 in TBS+0.5% skim milk), mouse anti-Bad antibody (Transduction Laboratories, 1 : 500 in TBS+0.5% skim milk), rabbit anti-Bax antibody raised against a peptide MDGSGEQPRGGGPTSSEQIMK coupled to KLH by the MBS method (1 : 2000 in TBS+0.5% skim milk), and rabbit anti-Bak antibody raised against a peptide WIARGGWVAALNLG coupled to KLH by the MBS method (1 : 1500 in TBS+0.5% skim milk) overnight at 4°C. Sheets were washed five times and unspecific binding was blocked as described. Detection was by horse-radish peroxidase-conjugated goat anti-mouse monoclonal antibodies (1 : 5000) or goat anti-rabbit monoclonal antibodies (1 : 5000) for 1.5 h at room temperature using the ECL method (Amersham). The primary bak and bax antibodies were tested by comparing with antibodies commercially available (Santa Cruz clone P-19 anti-bax; Calbiochem Ab-2 anti-bak) using mouse and human cell preparations (RAW 264.7 and U937). The antibodies exhibited no cross-reactivity with other Bcl-2 family members.
FACS analysis
For the determination of the mitochondrial membrane potential (ΔΨm) 3,3′-dihexyloxacarbocyanide iodide (DiOC6(3); final concentration 10 nM) was used (Petit et al., 1990). For these experiments glomerular endothelial cells were cultured in 60-mm culture dishes, incubated with the different apoptotic stimuli and for the last 15 min 10 nM DiOC6(3) was added. Afterwards, cells were harvested by trypsinization, and resuspended in 500 μl PBS containing 10 μg ml−1 propidium iodide. Within 30 min cells were analysed using a FACS calibur (Becton Dickinson, Heidelberg). Cells exhibiting a normal forward/side scatter ratio were selected followed by the determination of the DiOC6(3)/propidium iodide staining properties.
For cell cycle analysis cells were cultured in 60-mm culture dishes, incubated, harvested by trypsinization, resuspended in 900 μl PBS, supplemented with 2.1 ml ethanol, and fixed for at least 2 h at −20°C. Afterwards, pelleted cells were resuspended in 500 μl PBS containing 100 μg ml−1 RNase A and 20 μg ml−1 propidium iodide and incubated for 30 min at room temperature. Cells exhibiting a normal forward/side scatter ratio were selected followed by the determination of the propidium iodide staining properties.
Caspase-3 enzyme activity
For detection of caspase-3 activity, glomerular endothelial cells were incubated as indicated and lysed in lysis buffer (Tris/HCl 10 mM, sucrose 0.32 M, EDTA 5 mM, 1% Triton X-100, phenylmethanesulfonyl fluoride 1 mM, 1 μg ml−1 aprotinin, 10 μg ml−1 leupeptin, DTT 2 mM, pH 8.0) for 30 min. Following sonication (10 s, output control 1) lysates were centrifuged (10,000×g, 5 min, 4°C) and stored at −80°C. Protein determinations were performed with the Bradford method (Bradford, 1976). Caspase-3 activity was detected by measuring the proteolytic cleavage of the fluorogenic substrate Ac-DEVD-AMC. Cell lysates (50 μg protein) were incubated in HEPES 100 mM, 10% sucrose, 0.1% CHAPS, pH 7.5, phenymethanesulfonyl fluoride 1 mM, 1 μg ml−1 aprotinin, 10 μg ml−1 leupeptin, DTT 2 mM at 37°C with 12 μM DEVD-AMC in a total volume of 700 μl. Substrate cleavage and AMC accumulation was followed fluorometrically with excitation at 380 nm and emission at 460 nm.
Statistical analysis
Each experiment was performed at least three times and statistical analysis were performed using the two-tailed Student's t-test or ANOVA and for multiple comparison the data were corrected by Dunn's Method.
Results
Effect of dexamethasone on glomerular endothelial cell apoptosis
Treatment of glomerular endothelial cells with 25 ng ml−1 TNF-α or 30 ng ml−1 LPS led to efficient induction of apoptosis as detected by the appearance of a specific sub-G1-peak in cell cycle analysis experiments (Figure 1), the formation of condensed and fragmented nuclei in Hoechst 33258 stainings (Meßmer et al., 1999a), and extensive inter-nucleosomal DNA fragmentation as determined by the diphenylamine assay (Figure 2). As clearly demonstrated previously (Meßmer et al., 1999a) and also documented in Figures 1 and 2, the LPS-mediated response was much more pronounced but qualitatively comparable to that of TNF-α. To characterize the effect of dexamethasone on either TNF-α and LPS-mediated endothelial cell apoptosis, cells were cotreated with 10 nM dexamethasone and 25 ng ml−1 TNF-α or 30 ng ml−1 LPS followed by cell cycle distribution analysis using a FACS calibur. As shown in Figure 1, exposing glomerular endothelial cells for 24 h to TNF-α or LPS revealed roughly 20 or 48% apoptotic cells, respectively, appearing as a relatively distinct sub-G1-peak. Cotreatment of these cells with 10 nM dexamethasone completely suppressed the appearance of apoptotic cells in the case of TNF-α and significantly antagonized apoptosis induction in the case of LPS. In parallel, dexamethasone cotreatment also counteracted the formation of apoptotic nuclei (nuclear condensation and fragmentation) (Table 1). Time kinetic investigations revealed that, starting around 8–10 or 4–6 h after TNF-α or LPS application, respectively, apoptotic DNA fragmentation progressively increased, and 24 h after treatment about 25 or 50% (TNF-α and LPS, respectively) of the cellular DNA was degraded and located in the cytosol (Figure 2). Interestingly, dexamethasone did not affect the initial DNA degradation which appears after 4 h but suppressed the late phase of DNA degradation observed after 4–10 h (Figure 2).
Figure 1.
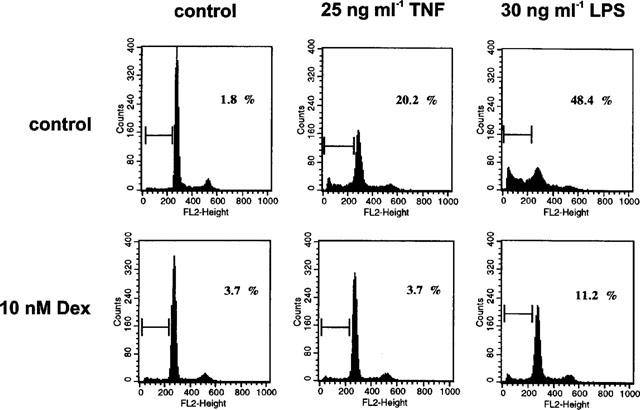
Dexamethasone suppressed apoptosis induced by TNF-α and bacterial LPS. Bovine glomerular endothelial cells were incubated for 24 h with 25 ng ml−1 TNF-α and 30 ng ml−1 LPS in the absence or presence of 10 nM dexamethasone. Cells were harvested and stained with propidium iodide as described in the Methods section. Hypodiploid DNA peaks corresponding to apoptotic nuclei were revealed by FACS analysis and expressed as per cent of those cells exhibiting an intact forward/side scatter ratio. Data are representative of three separate experiments.
Figure 2.
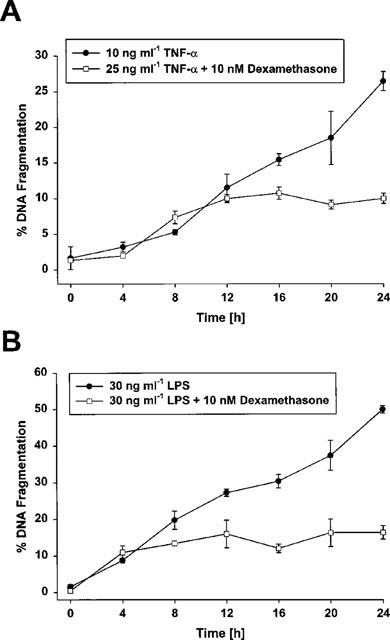
Time-dependent effect of dexamethasone on TNF-α- and LPS-induced apoptosis in bovine glomerular endothelial cells. Cells were simultaneously exposed to 25 ng ml−1 TNF-α or 30 ng ml−1 LPS in the presence of 10 nM dexamethasone for the times indicated. The amount of fragmented DNA was determined with the diphenylamine reaction. Values are means±s.e.mean of four independent experiments.
Table 1.
Dexamethasone suppressed apoptosis induced by TNF-α and bacterial LPS

Time-dependence of the anti-apoptotic effect of dexamethasone
In the experiments described so far, dexamethasone and the apoptotic inducers were administered at the same time and cells were continuously exposed to the agents during the entire incubation period. Next, dexamethasone was administered at various time intervals after the addition of TNF-α or LPS to define the time window required for an effective anti-apoptotic effect. As demonstrated in Figure 3, dexamethasone displayed a maximal anti-apoptotic activity even if it was added 6–8 h after the individual apoptosis inducers TNF-α or LPS compared to simultaneous application. In the time window starting from 6–8 h up to 16–18 h following TNF-α/LPS addition the inhibitory effect of the synthetic glucocorticoid gradually decreased and if dexamethasone was applicated after 16–18 h following the starting point with TNF-α/LPS there was no longer a significant inhibitory effect observable.
Figure 3.
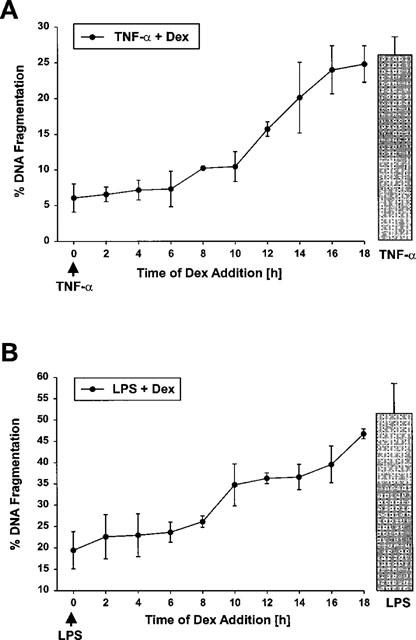
Time-dependency of dexamethasone protection. Bovine glomerular endothelial cells were stimulated for 24 h with 25 ng ml−1 TNF-α (A) or 30 ng ml−1 LPS (B). Ten nM dexamethasone were added at various times in relation to TNF/LPS application starting simultaneously with the apoptotic inducers till 18 h after TNF/LPS application (the arrow indicates the time point of TNF/LPS addition). All experiments were performed for entirely 24 h. The amount of DNA fragmentation was determined with the diphenylamine reaction as outlined in the Methods section. Values are means±s.e.mean of at least four individual experiments.
Effects of dexamethasone on mitochondrial permeability transition and cytochrome c efflux
To characterize the inhibitory mechanism of dexamethasone on cytochrome c efflux, we first measured its effects on mitochondrial permeability transition induced by TNF-α and LPS. The mitochondrial membrane potential was measured by the uptake of the mitochondrial specific dye DiOC6(3). Adherent cells were stimulated with TNF-α or LPS in the presence or absence of dexamethasone for different time periods followed by DiOC6(3) addition to the culture medium 15 min prior to harvesting the cells. As shown in Figure 4A, around 10% of those cells incubated under control conditions exhibited only a low DiOC6(3) uptake capacity reflecting a decreased mitochondrial membrane potential. TNF-α increased the cell fraction with a reduced mitochondrial membrane potential within 10–24 h from 16% to roughly 40%. Coincubation of cells with TNF-α and dexamethasone prevented this effect and only 11–13% of the cells displayed a reduced mitochondrial permeability transition. Comparably, but more pronounced and with a faster time kinetic, LPS strongly induced mitochondrial permeability transition which was partially suppressed by dexamethasone (Figure 5A).
Figure 4.
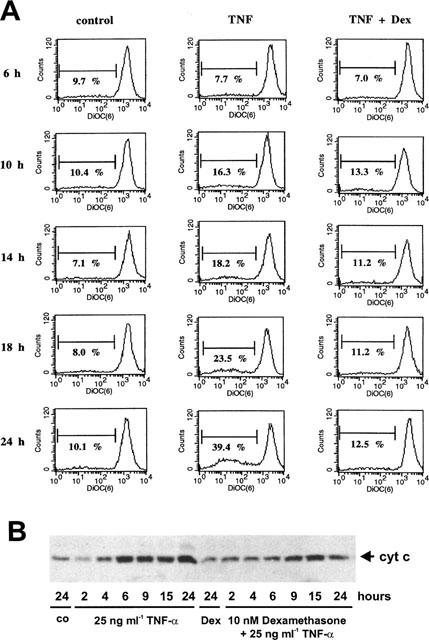
Effect of dexamethasone on TNF-α-induced mitochondrial permeability transition and cytochrome c-release into the cytosol. (A) Glomerular endothelial cells were cultured as described in the Methods section and exposed for the times indicated either to 25 ng ml−1 TNF-α or 25 ng ml−1 TNF-α plus 10 nM dexamethasone. During the last 15 min of the incubation 10 nM DiOC6(3) was added to the culture medium, followed by cell trypsinization and counter staining with 10 μg ml−1 propidium iodide. Cells were analysed with a FACS calibur (Becton Dickinson) and all cells exhibiting an intact forward/side scatter ratio and propidium iodide exclusion were selected. Data are representative of three independent experiments giving similar results. (B) Glomerular endothelial cells were incubated with 25 ng ml−1 TNF-α, 10 nM dexamethasone plus 25 ng ml−1 TNF-α, 10 nM dexamethasone (Dex), or vehicle (control) for the times indicated, harvested by trypsinization, and a cytosolic extract was prepared as described in the Methods section. The samples were then subjected to Western blot analysis for cytochrome c. Values are representative of three individual experiments.
Figure 5.
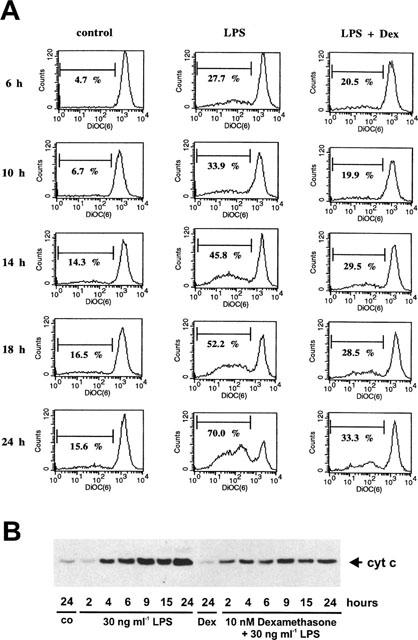
Effect of dexamethasone on LPS-induced mitochondrial permeability transition and cytochrome c-release into the cytosol. (A) Glomerular endothelial cells were exposed for the times indicated either to 30 ng ml−1 LPS or 30 ng ml−1 LPS/10 nM dexamethasone. During the last 15 min of the incubation 10 nM DiOC6(3) was added to the culture medium, followed by cell trypsinization, counter staining with 10 μg/ml propidium iodide, and FACS analysis. Data are representative of three independent experiments giving similar results. (B) Glomerular endothelial cells were incubated with 30 ng ml−1 LPS, 10 nM dexamethasone plus 30 ng ml−1 LPS, 10 nM dexamethasone (Dex), or vehicle (control) for the times indicated, harvested by trypsinization, and a cytosolic extract was prepared as described in the Methods section. The samples were then subjected to Western blot analysis for cytochrome c. Values are representative of three individual experiments.
According to Figures 4B and 5B both TNF-α and LPS induced a dramatic mitochondrial cytochrome c efflux which was first detectable 3–4 h after agonist addition. In both cases, dexamethasone significantly but only partially suppressed mitochondrial cytochrome c release into the cytosol.
Effect of dexamethasone on Bak upregulation and Bcl-xL downregulation
To further characterize the mechanisms by which glucocorticoids interfere with TNF-α/LPS-mediated apoptosis of glomerular endothelial cells, we studied the regulation of the biologically most relevant class of apoptosis-influencing gene products, the family of Bcl-2-related proteins. First, concentrating on gene products that promote apoptosis, Bad and Bax-protein levels were not altered by LPS or TNF-α whereas Bak-protein levels were induced within 10–24 h following either LPS or TNF-α addition. To investigate the effect of dexamethasone on protein levels of these proapoptotic proteins, cells were co-treated with 10 nM dexamethasone and 30 ng ml−1 LPS or 25 ng ml−1 TNF-α and time kinetic studies were performed. As shown in Figure 6, dexamethasone potently blocked Bak-upregulation mediated by either LPS or TNF-α whereas Bax and Bad protein levels did not change. Focusing on the anti-apoptotic sub-family of Bcl-2-related proteins, Bcl-2 protein levels were not altered either by TNF-α or LPS whereas Bcl-xL dramatically declined within 10–24 h following LPS stimulation and significantly declined within 14–24 h after TNF-α addition. Again, exposing glomerular endothelial cells simultaneously to 10 nM dexamethasone and 30 ng ml−1 LPS or 25 ng ml−1 TNF-α revealed that glucocorticoids suppressed Bcl-xL protein downregulation (Figure 6). These data suggest that Bak upregulation and concomitantly Bcl-xL downregulation are involved in both TNF-α and LPS-induced endothelial cell apoptosis and that dexamethasone exerts its antiapoptotic action upstream of Bak/Bcl-xL protein regulation.
Figure 6.
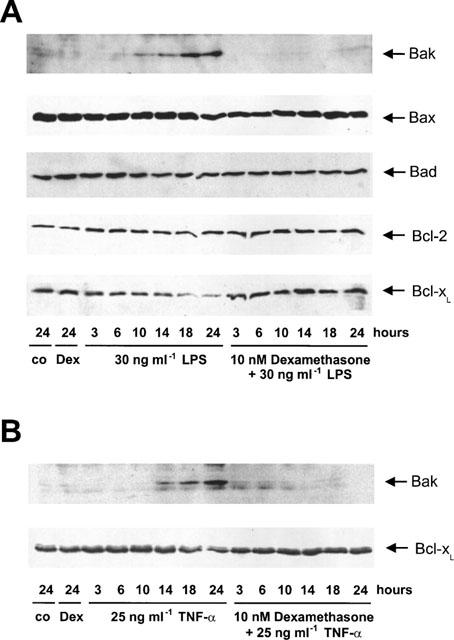
Effect of dexamethasone on TNF-α- and LPS-induced Bak upregulation and Bcl-xL-downregulation. Glomerular endothelial cells were exposed to 30 ng ml−1 LPS, 30 ng ml−1 LPS/10 nM dexamethasone, 10 nM dexamethasone or solvent control (A) or 25 ng ml−1 TNF-α, 25 ng ml−1 TNF-α/10 nM dexamethasone, 10 nM dexamethasone or vehicle (control) (B) for the times indicated. Subsequently, cell lysates were subjected to Western blot analysis for Bad, Bak, Bax, Bcl-xL, and Bcl-2 protein using several antibodies described in the Methods section followed by ECL detection. The blots are representative of at least three independent experiments.
Effects of dexamethasone on caspase-3-like protease activation
To strengthen our suggestion that dexamethasone blocks apoptotic signalling upstream of Bak/Bcl-xL activity and to test how glucocorticoids interfere with the time kinetics of caspase activation we next measured the effects of dexamethasone on caspase-3-like protease activation at several time points. Caspase-3 and caspase-3-like proteases are recognized as the final signalling pathways in apoptosis signal transduction and as the terminal active members of the caspase cascade. Both TNF-α and LPS induced a dramatic activation of caspase-3-like proteases which starts within 14 or 8 h following apogen addition, respectively. In both cases, caspase-3-like protease activity increased continuously up to 24 h (Figure 7). Coincubation of glomerular endothelial cells with TNF-α or LPS and dexamethasone resulted in a highly significant suppression of caspase-3-like protease activity. As shown in Figure 7A,C, the initial activation of caspase-3-like activity was completely blocked by dexamethasone whereas end point measurements after 24 h revealed a proportional inhibition of caspase-3-like activation comparable to the extent of inhibition of apoptosis induction (see also: Meßmer et al., 1999b).
Figure 7.
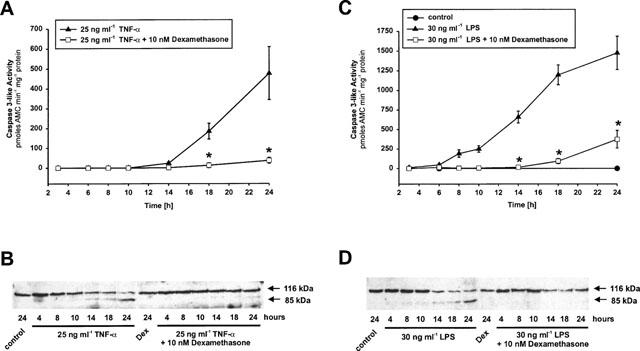
Dexamethasone prevents TNF-α- and LPS-induced caspase-3 activation and PARP cleavage. (A,B) Glomerular endothelial cells were incubated with 25 ng ml−1 TNF-α and 25 ng ml−1 TNF-α plus 10 nM dexamethasone or (C,D) 30 ng ml−1 LPS and 30 ng ml−1 LPS plus 10 nM dexamethasone for the times indicated. Caspase-3-like activity was determined using the fluorogenic substrate DEVD-AMC (A,C) and PARP cleavage (116 kDa holoenzyme and 85 kDa cleavage fragment) was monitored by Western blot analysis using the monoclonal anti-PARP antibody C-II-10 (B,D). Data are means±s.e.mean or are representative of three independent experiments, respectively. *P<0.05 versus corresponding control (ANOVA and for multiple comparison the data were corrected by Dunn's Method).
To verify the effects of dexamethasone on caspase-3-like protease activation we also measured the cleavage of the nuclear enzyme poly(ADP-ribose) polymerase (PARP). In agreement with the activation of caspase-3-like proteases the 116 kDa holoenzyme is cleaved into the 85 kDa subunit stimulated by TNF-α as well as by LPS. Again, dexamethasone potently blocked PARP cleavage in both cases (Figure 7B,D).
Discussion
We have reported that synthetic and natural forms of glucocorticoids are able to block apoptotic cell death in cultured bovine glomerular endothelial cells induced by proinflammatory agents such as the cytokine TNF-α and LPS, the cell wall component of gram negative bacteria. To put our results in the context of an in vivo situation it is important to note that mediators from both immune and non-immune cells play major roles in glomerular disease processes. Of principal importance to the body's response to stress is the secretion of corticotropin from the pituitary and the subsequent release of cortisol from the adrenal cortex. One convincing example for the anti-inflammatory role of endogenous glucocorticoids comes from adrenalectomized rats where the experimental glomerulonephritis elicited by the administration of anti-glomerular basement membrane lgG is drastically amplified (Huang et al., 1997). As apoptosis of glomerular endothelial cells may contribute to the development of glomerulosclerosis during severe forms of glomerulonephritis (Kitamura et al., 1998) we suggest that regulation of glomerular endothelial cell apoptosis in response to cytokines such as TNF-α and bacterial endotoxin may be achieved by endogenous glucocorticoids and this may also form part of the potent anti-inflammatory activity of glucocorticoids used for the treatment of diverse forms of glomerulonephritis.
Besides our results with glomerular endothelial cells several other groups also documented that glucocorticoids suppress apoptosis in various cell types such as human neutrophils (Cox, 1995; Cox & Austin, 1997), L929 mouse fibroblasts activated by TNF-α (Pagliacci et al., 1993), a murine tumorigenic fibroblast cell line (L-M cells) and MCF-7 cells (a human mammary carcinoma cell line) activated by TNF-α (Kull, 1988), a human gastric cancer cell line exposed to actinomycin D (Chang et al., 1997), malignant glioma cells exposed to anticancer drugs (Naumann et al., 1998), lung epithelial cells exposed to IFN-γ and Fas (Wen et al., 1997), rat hepatocytes exposed to aflatoxin B1 (Iida et al., 1998), and serum depletion induced apoptosis in T lymphocytes overexpressing Bcl-2 (Huang & Cidlowski, 1999). Although a protective effect of glucocorticoids was evident in all cell types, the proapoptotic stimuli used and their signalling pathways are quite different and therefore, the mechanisms of the survival effect in general and in glomerular endothelial cells in particular remained to be clarified.
The inhibitory action of glucocorticoids on TNF-α/LPS-mediated endothelial cell death does not require the induction of a long lasting protective signal such as the synthesis of a long-lived anti-apoptotic protein. This suggestion results from our observation that a pretreatment of 2–18 h was not more inhibitory than simultaneous or even a 26 h posttreatment regimen. Moreover, as addition of dexamethasone up to 6 h following TNF-α or LPS revealed the same inhibitory potential as co-treatment we suggested that dexamethasone receptor-dependently (Meßmer et al., 1999b) blocks distinct pro-apoptotic pathways. In contrast to glomerular endothelial cells, in the mouse L-M-fibroblast cell line a pretreatment with dexamethasone was more inhibitory than posttreatment (Kull, 1988). In this cell line the author reported on a reduction of TNF receptor affinity as one potential mechanism of dexamethasone action. For bovine glomerular endothelial cells we can not exclude that the protective effect of dexamethasone against TNF-α and LPS-induced apoptosis partially depends on TNF or LPS receptor downregulation. However, Western blot experiments revealed no significant changes in the expression levels of TNF-RI in bovine glomerular endothelial cells within a 24 h incubation period towards TNF-α or TNF-α plus dexamethasone (data not shown). Moreover, as dexamethasone pretreatment was not more inhibitory than simultaneous treatment, and posttreatment up to several hours showed the same effect, the direct interaction with a downstream pathway seems to be more relevant than receptor downregulation.
Therefore, we further characterized the anti-apoptotic activity of dexamethasone by analysing its effects on TNF-α/LPS-induced activation of caspase-3-like proteases. Caspase-3 and caspase-3-like proteases are recognized as downstream (effector) proteases according to their site of action in the proteolytic caspase cascade (Cryns & Yuan, 1998). We have demonstrated that the activation of caspase-3-like proteases parallels the appearance of distinct DNA fragments in the cytosol (Meßmer et al., 1999a). As the caspase-3-like protease inhibitor Ac-DEVD-fmk only poorly affected TNF-α/LPS-induced apoptosis in glomerular endothelial cells, obviously other caspase family members seem to be required as the final death pathways are blocked by broad spectrum caspase inhibitors (Meßmer et al., 1999a). In this context, it is important to note that dexamethasone blocked caspase-3-like protease activity as well as PARP cleavage. As PARP is not only cleaved by caspase-3 but also by other caspases, we suggest that dexamethasone blocks apoptosis induction upstream of the activation of executioner caspases. Upstream of executioner caspases the Bcl-2 family of proteins exerts important regulatory functions (Reed, 1998). TNF-α as well as LPS potently induce Bak protein expression and concomitantly downregulate Bcl-xL protein levels. Whereas the former one acts in a proapoptotic pathway, the latter one functions to suppress apoptosis (Reed, 1998). Dexamethasone potently interfered with the expression of both proteins. With respect to the suppression of Bak-upregulation the effect of dexamethasone on glomerular endothelial cells is comparable to that of estradiol on MCF-7 cells (Leung et al., 1998) and similar to the downregulation of Bcl-xs in rat thymocytes (Sakamoto et al., 1995). In contrast, in rat hepatoma cells dexamethasone potently induced Bcl-xL expression (Yamamoto et al., 1998; Buchmann et al., 1999). The induction of this anti-apoptotic protein may also be responsible for the high efficiency of glucocorticoid-preexposure in some cells (Kull, 1988). The importance of Bcl-xL for cell survival is demonstrated by the finding that Bcl-xL may modulate cell death by directly interacting with caspases (Clem et al., 1998).
Finally, to investigate upstream pathways, cytochrome c release from the inner-mitochondrial membrane space into the cytosol and mitochondrial permeability transition are a first line apoptotic response shown in many cell death pathways (Mignotte & Vayssiere, 1998). Both events were partially blocked by dexamethasone. This result has important consequences as cytochrome c was identified to form a complex with Apoptosis protease releasing factor (Apaf-1), dATP, and caspase 9 to promote the activation of a caspase cascade (Li et al., 1997). As cytochrome c-release starts within 2–4 h following LPS or TNF-α addition whereas dexamethasone still fully protects cells if it is applied within 6–8 h after TNF-α/LPS application, we suggest that dexamethasone either blocks pathways which are independent of cytochrome c and other amplifying mechanisms which accelerate cytochrome c release.
In summary, TNF/LPS-mediated apoptosis in glomerular endothelial cells is blocked by dexamethasone. The executionary arm of the death machinery, the activation of cysteine proteases called caspases and the shift from anti-apoptotic to pro-apoptotic members of the Bcl-2 family of proteins, are completely blocked by glucocorticoids whereas the release of cytochrome c is only partially affected. Nevertheless, the exact mechanism of the survival effect of glucocorticoids remains to be clarified. Some properties of glucocorticoids are attributed to their effects on transcription factors and gene expression. One prominent mechanism is the inhibitory influence on NF-κB activation which may be achieved by protein–protein interactions between NF-κB and the glucocorticoid receptor (Wissink et al., 1998) and/or induction of the I-κB protein (Auphan et al., 1995; Xie et al., 1997) which, however, is not demonstrable in every cell type (De Bosschner et al., 1997). Apart from its well known effect on NF-κB transcription factor activity, dexamethasone inhibits binding to the lkaros and AP-1 regulatory elements of the granzyme B promoter and downregulates human granzyme B expression (Wargnier et al., 1998). However, also the opposite effect was found. Glucocorticoids were not only known to suppress transcription factor activity but they were also identified to induce expression of the human inhibitor of apoptosis (hlAP-1) also known as clAP-2 (Wen et al., 1997) and the expression of MnSOD in glomerular cells (Yoshioka et al., 1994). Certain cellular IAPs have been shown to repress apoptosis and to selectively inhibit effector caspases such as caspase-3 and caspase-7 (Deveraux & Reed, 1999). Therefore, it is reasonable to assume that dexamethasone elicits cell specific effects either by suppression or activation of certain transcription factors and by simultaneous influencing certain pro- or anti-apoptotic pathways. Similarly, TNF-α is able to elicit pro- and anti-apoptotic signals at the same time (Van Antwerp et al., 1998) where the latter one depends on the activation of NF-κB (Stehlik et al., 1998). Considering the fact that glomerular endothelial cells had to be continuously exposed to TNF-α or LPS to effectively induce an apoptotic response (Meßmer et al., 1999a) in combination with the finding that dexamethasone only partially affects early pro-apoptotic TNF-α and LPS signalling whereas it still exerts full protection when added several hours after TNF-α and LPS we suggest that glucocorticoids receptor-dependently block amplifying pro-apoptotic signals pre- and/or post cytochrome c release and mitochondrial signalling. Whether the dexamethasone/glucocorticoid receptor complex requires DNA binding for its effects or whether it involves rapid, non-genomic activities (Reichardt et al., 1998) remains to be evaluated. Investigations of the participation of transcription factors and in particular non-genomic effects of glucocorticoids are currently in progress.
Acknowledgments
The authors thank Ulrike Müller for expert technical assistance. This study was supported by a grant of the Deutsche Forschungsgemeinschaft (SFB 553), a grant of the Paul and Ursula Klein Stiftung and the Dr med. h.c. Erwin Braun Stiftung.
Abbreviations
- aFGF
acidic fibroblast growth factor
- AP-1
activator protein 1
- ASK-1
apoptosis signal-regulating kinase 1
- CHAPS
3-[(3-chloramidopropyl)-dimethylammonio] propanesulfonate
- DEVD-AMC
N-acetyl-aspartyl-glutamyl-valinyl-aspartyl-7-amino-4-coumarin
- Ac-DEVD-fmk
N-acetyl-aspartyl-glutamyl-valinyl-aspartyl-fluoromethyl ketone
- Dex
dexamethasone
- DTT
dithiothreitol
- EDTA
ethylenediaminetetraacetic acid
- FACS
fluorescence-activated cell sorting
- FADD
Fas associated death domain
- I-κBα
inhibitor of κBα
- I-κBβ
inhibitor of κBβ
- IAP
inhibitor of apoptosis
- IFN-γ
interferon-γ
- LPS
lipopolysaccharide
- MnSOD
manganeous superoxide dismutase
- NF-κB
nuclear factor κB
- NIK
NF-κB inducing kinase
- PARP
poly(ADP-ribose) polymerase
- RIP
receptor interacting protein
- TE
Tris-EDTA
- TNF-α
tumour necrosis factor α
- TNF-RI
tumour necrosis factor receptor
- TRADD
TNF receptor-associated death domain protein
- XIAP
X-chromosome-linked inhibitor of apoptosis
References
- ANDERSON K.L., ANDERSON G., MICHELL R.H., JENKINSON E.J., OWEN J.J.T. Intracellular signaling pathways involved in the induction of apoptosis in immature thymic T lymphocytes. J. Immunol. 1996;156:4083–4091. [PubMed] [Google Scholar]
- AUPHAN N., DIDONATO J.A., ROSETTE C., HELMBERG A., KARIN M. Immunosuppression by glucocorticoids: inhibition of NF-κB activity through induction of IκB synthesis. Science. 1995;270:286–290. doi: 10.1126/science.270.5234.286. [DOI] [PubMed] [Google Scholar]
- BAKER S.J., REDDY E.P. Modulation of life and death by the TNF receptor superfamily. Oncogene. 1998;17:3261–3270. doi: 10.1038/sj.onc.1202568. [DOI] [PubMed] [Google Scholar]
- BALLERMANN B.J.Regulation of bovine glomerular endothelial cell growth in vitro Am. J. Physiol. 1989256C182–C189.(Cell. Physiol.25) [DOI] [PubMed] [Google Scholar]
- BRADFORD M.M. A rapid and sensitive method for the quantification of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- BRINER V.A., KERN F. ATP stimulates Ca2+ mobilization by a nucleotide receptor in glomerular endothelial cells. Am. J. Physiol. Renal Fluid Electrol. Physiol. 1994;26635:F210–F217. doi: 10.1152/ajprenal.1994.266.2.F210. [DOI] [PubMed] [Google Scholar]
- BUCHMANN A., WILLY C., BUENEMANN C.L., STROH C., SCHMIECHEN A., SCHWARZ M. Inhibition of transforming growth factor beta1-induced hepatoma cell apoptosis by liver tumour promoters: characterization of primary signalling events and effects on CPP32-like caspase activity. Cell Death Differ. 1999;6:190–200. doi: 10.1038/sj.cdd.4400475. [DOI] [PubMed] [Google Scholar]
- BURTON K. A study of the conditions and mechanisms of the diphenylamine reaction for the estimation of deoxyribonucleic acid. Biochem. J. 1956;62:315–323. doi: 10.1042/bj0620315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANG T.-C., HUNG M.-W., JIANG S.-Y., CHU J.-T., CHU L.-L., TSAI L.-C. Dexamethasone suppress apoptosis in a human gastric cancer cell line through modulation of bcl-x gene expression. FEBS Lett. 1997;415:11–15. doi: 10.1016/s0014-5793(97)01083-1. [DOI] [PubMed] [Google Scholar]
- CLEM R.J., CHENG E.H., KARP C.L., KIRSCH D.G., UENO K., TAKAHASHI A., KASTAN M.B., GRIFFIN D.E., EARNSHAW W.C., VELIUONA M.A., HARDWICK J.M. Modulation of cell death by Bcl-xL through caspase interaction. Proc. Natl. Acad. Sci. U.S.A. 1998;95:554–559. doi: 10.1073/pnas.95.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COLE T.J., BLENDY J.A., MONAGHAN A.P., SCHMID W., AGUZZI A., SCHUTZ G. Molecular genetic analysis of glucocorticoid signaling during mouse development. Steroids. 1995;60:93–96. doi: 10.1016/0039-128x(94)00009-2. [DOI] [PubMed] [Google Scholar]
- COX G. Glucocorticoid treatment inhibits apoptosis in human neutrophils. Separation of survival and activation outcomes. J. Immunol. 1995;154:4719–4725. [PubMed] [Google Scholar]
- COX G., AUSTIN R.C. Dexamethasone-induced suppression of apoptosis in human neutrophils requires continuous stimulation of new protein synthesis. J. Leuc. Biol. 1997;61:224–230. doi: 10.1002/jlb.61.2.224. [DOI] [PubMed] [Google Scholar]
- CRYNS V., YUAN J. Proteases to die for. Genes Dev. 1998;12:1551–1570. doi: 10.1101/gad.12.11.1551. [DOI] [PubMed] [Google Scholar]
- DA SILVA J.A. Sex hormones, glucocorticoids and autoimmunity: facts and hypotheses. Ann. Rheum. Dis. 1995;54:6–16. doi: 10.1136/ard.54.1.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DARNAY B.G., AGGARWAL B.B. Early events in TNF signaling: a story of associations and dissociations. J. Leuc. Biol. 1997;61:559–566. doi: 10.1002/jlb.61.5.559. [DOI] [PubMed] [Google Scholar]
- DE BOSSCHNER K., SCHMITZ M.L., VAN DEN BERGHE W., PLAISANCE S., FIERS W., HAEGEMAN G. Glucocorticoid-mediated repression of nuclear factor-kappaB-dependent transcription involves direct interference with transactivation. Proc. Natl. Acad. Sci. U.S.A. 1997;94:13504–13509. doi: 10.1073/pnas.94.25.13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEVERAUX Q.L., REED J.C. IAP family proteins – suppressors of apoptosis. Genes Dev. 1999;13:239–252. doi: 10.1101/gad.13.3.239. [DOI] [PubMed] [Google Scholar]
- EDELSTEIN C.L., LING H., SCHRIER R.W. The nature of renal cell injury. Kidney Int. 1997;51:1341–1351. doi: 10.1038/ki.1997.183. [DOI] [PubMed] [Google Scholar]
- GRUBER J., SGONC R., HU Y.H., BEUG H., WICK G. Thymocyte apoptosis induced by elevated endogenous corticosterone levels. Eur. J. Immunol. 1994;24:1115–1121. doi: 10.1002/eji.1830240516. [DOI] [PubMed] [Google Scholar]
- HUANG S.J., CIDLOWSKI J.A. Glucocorticoids inhibit serum depletion-induced apoptosis in T lymphocytes expressing Bcl-2. FASEB J. 1999;13:467–476. doi: 10.1096/fasebj.13.3.467. [DOI] [PubMed] [Google Scholar]
- HUANG X.R., LEECH M., TIPPING P.G., HOLDSWORTH S.R. Endogenous corticosteroids inhibit acute neutrophil mediated glomerulonephritis in rats. Nephrology. 1997;1 Suppl.:S486. [Google Scholar]
- IIDA N., SUGIYAMA A., MYOUBUDANI H., INOUE K., SUGAMATA M., IHARA T., UENO Y., TASHIRO F. Suppression of arachidonic acid cascade-mediated apoptosis in aflatoxin B1-induced rat hepatoma cells by glucocorticoids. Carcinogenesis. 1998;19:191–202. doi: 10.1093/carcin/19.7.1191. [DOI] [PubMed] [Google Scholar]
- KITAMURA H., SHIMIZU A., MASUDA Y., ISHIZAKI M., SUGISAKI Y., YAMANAKA N. Apoptosis in glomerular endothelial cells during the development of glomerulosclerosis in the remnant-kidney model. Exp. Nephrol. 1998;6:328–336. doi: 10.1159/000020540. [DOI] [PubMed] [Google Scholar]
- KULL F.C. Reduction in tumor necrosis factor receptor affinity and cytotoxicity by glucocorticoids. Biochem. Biophys. Res. Commun. 1988;153:402–409. doi: 10.1016/s0006-291x(88)81238-5. [DOI] [PubMed] [Google Scholar]
- LEUNG L.K., DO L., WANG T.T.Y. Regulation of death promoter Bak expression by cell density and 17β-estradiol in MCF-7 cells. Cancer Lett. 1998;124:47–52. doi: 10.1016/s0304-3835(97)00430-8. [DOI] [PubMed] [Google Scholar]
- LI P., NIJHAWAN D., BUDIHARDJO I., SRINIVASULA S.M., AHMAD M., ALNEMRI E.S., WANG X. Cytochrome c and dATP-dependent formation of Apaf1/caspase-9 complex initiates an apoptotic protease cascade. Cell. 1997;91:479–489. doi: 10.1016/s0092-8674(00)80434-1. [DOI] [PubMed] [Google Scholar]
- LIU Y., COUSIN J.M., HUGHES J., VAN DAMME J., SECKL J.R., HASLETT C., DRANSFIELD I., SAVILL J., ROSSI A.G. Glucocorticoids promote nonphlogistic phagocytosis of apoptotic leukocytes. J. Immunol. 1999;15:3639–3646. [PubMed] [Google Scholar]
- LIPWORTH B.J. Systemic adverse effects of inhaled corticosteroid therapy: A systematic review and meta-analysis. Arch. Intern. Med. 1999;10:941–955. doi: 10.1001/archinte.159.9.941. [DOI] [PubMed] [Google Scholar]
- MEßMER U.K., BRINER V.A., PFEILSCHIFTER J. Tumor necrosis factor-α and lipopolysaccharide potently induce apoptotic cell death in bovine glomerular endothelial cells. Kidney Int. 1999a;55:2322–2337. doi: 10.1046/j.1523-1755.1999.00473.x. [DOI] [PubMed] [Google Scholar]
- MEßMER U.K., REED J.C., BRÜNE B. Bcl-2 protects macrophages from nitric oxide-induced apoptosis. J. Biol. Chem. 1998;271:20192–20197. doi: 10.1074/jbc.271.33.20192. [DOI] [PubMed] [Google Scholar]
- MEßMER U.K., WINKEL G., BRINER V.A., PFEILSCHIFTER J. Glucocorticoids potently block tumour necrosis factor-α- and lipopolysaccharide-induced apoptotic cell death in bovine glomerular endothelial cells upstream of caspase 3 activation. Br. J. Pharmacol. 1999b;127:1633–1640. doi: 10.1038/sj.bjp.0702726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIGNOTTE B., VAYSSIERE J.-L. Mitochondria and apoptosis. Eur. J. Biochem. 1998;252:1–15. doi: 10.1046/j.1432-1327.1998.2520001.x. [DOI] [PubMed] [Google Scholar]
- NAUMANN U., DURKA S., WELLER M. Dexamethasone-mediated protection from drug cytotoxicity: association with p21WAF1/CIP1 protein accumulation. Oncogene. 1998;17:1567–1575. doi: 10.1038/sj.onc.1202071. [DOI] [PubMed] [Google Scholar]
- NITTOH T., FUJIMORI H., KOZUMI Y., ISHIHARA K., MUE S., OHUCHI K. Effects of glucocorticoids on apoptosis of infiltrated eosinophils and neutrophils in rats. Eur. J. Pharmacol. 1998;31:73–81. doi: 10.1016/s0014-2999(98)00426-9. [DOI] [PubMed] [Google Scholar]
- OSBORNE B.A., SMITH S.W., MCLAUGHLIN K.A., GRIMM L., KALLINCH T., LIU Z., SCHWARTZ L.M. Genes that regulate apoptosis in the mouse thymus. J. Cell. Biochem. 1996;60:18–22. doi: 10.1002/(sici)1097-4644(19960101)60:1<18::aid-jcb4>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- PAGLIACCI M.C., MIGLIORATI G., SMACCHIA M., GRIGNANI F., RICCARDI C., NICOLETTI I. Cellular stress and glucocorticoid hormones protect L929 mouse fibroblasts from tumor necrosis factor alpha cytotoxicity. J. Endocrinol. Invest. 1993;16:591–599. doi: 10.1007/BF03347677. [DOI] [PubMed] [Google Scholar]
- PETIT P.X., O'CONNOR J.E., GRUNWALD D., BROWN S.C. Analysis of the membrane potential of rat- and mouse-liver mitochondria by flow cytometry and possible applications. Eur. J. Biochem. 1990;194:389–397. doi: 10.1111/j.1432-1033.1990.tb15632.x. [DOI] [PubMed] [Google Scholar]
- PFEILSCHIFTER J., MÜHL H. Immunopharmacology: anti-inflammatory therapy targeting transcription factors. Eur. J. Pharmacol. 1999;375:237–245. doi: 10.1016/s0014-2999(99)00361-1. [DOI] [PubMed] [Google Scholar]
- REED J.C. Bcl-2 family proteins. Oncogene. 1998;17:3225–3236. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- REICHARDT H.M., KAESTNER K.H., TUCKERMANN J., KRETZ O., WESSELY O., BOCK R., GASS P., SCHMID W., HERRLICH P., ANGEL P., SCHÜTZ G. DNA binding of the glucocorticoid receptor is not essential for survivial. Cell. 1998;93:531–541. doi: 10.1016/s0092-8674(00)81183-6. [DOI] [PubMed] [Google Scholar]
- SAKAMOTO T., REPASKY W.T., CHEN J., HIRATA A., HIRATA F. Down-regulation of bcl-xs gene expression in rat thymocytes by dexamethasone. Biochem. Biophys. Res. Commun. 1995;215:511–516. doi: 10.1006/bbrc.1995.2494. [DOI] [PubMed] [Google Scholar]
- SHIMIZU A., KITAMURA H., MASUDA Y., ISHIZAKI M., SUGISAKI Y., YAMANAKA N. Rare capillary regeneration and subsequent capillary regression with endothelial cell apoptosis in progressive glomerulonephritis. Am. J. Pathol. 1997;151:1231–1239. [PMC free article] [PubMed] [Google Scholar]
- SHIMIZU A., MASUDA Y., KITAMURA H., ISHIZAKI M., SUGISAKI Y., YAMANAKA N. Recovery of damaged glomerular capillary network with endothelial cell apoptosis in experimental proliferative glomerulonephritis. Nephron. 1998;79:206–214. doi: 10.1159/000045026. [DOI] [PubMed] [Google Scholar]
- STEHLIK C., DE MARTIN R., KUMABASHIRI I., SCHMID J.A., BINDER B.R., LIPP J. Nuclear factor (NF)-κB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor alpha-induced apoptosis. J. Exp. Med. 1998;188:211–216. doi: 10.1084/jem.188.1.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STERZEL R.B., LOVETT D.M.Interactions of inflammatory and glomerular cells in the response to glomerular injury Immunopathology of Renal Disease 1988Churchill Livingston, New York; 137–173.Wilson C.B. (ed) [Google Scholar]
- VAN ANTWERP D.J., MARTIN S.J., VERMA I.M., GREEN D.R. Inhibition of TNF-induced apoptosis by NF-κB. TICS. 1998;8:107–111. doi: 10.1016/s0962-8924(97)01215-4. [DOI] [PubMed] [Google Scholar]
- WARGNIER A., LAFAURIE C., LEGROS-MAIDA S., BOURGE J.-F., SIGAUX F., SASPORTES M., PAUL P. Down-regulation of human granzyme B expression by glucocorticoids. J. Biol. Chem. 1998;273:35326–35331. doi: 10.1074/jbc.273.52.35326. [DOI] [PubMed] [Google Scholar]
- WEN L.P., MADANI K., FAHRNI J.A., DUNCAN S.R., ROSEN G.D. Dexamethasone inhibits lung epithelial cell apoptosis induced by IFN-γ and Fas. Am. J. Physiol. 1997;273:921–929. doi: 10.1152/ajplung.1997.273.5.L921. [DOI] [PubMed] [Google Scholar]
- WISSINK S., VAN HEERDE E.C., VAN DER BURG B., VAN DER SAAG P.T. A dual mechanism mediates repression of NF-κB activity by glucocorticoids. Mol. Endocrinol. 1998;12:355–363. doi: 10.1210/mend.12.3.0081. [DOI] [PubMed] [Google Scholar]
- XIE H., SEWARD R.J., HUBER B.T. Cytokine rescue from glucocorticoid induced apoptosis in T cells is mediated through inhibition of IκBα. Mol. Immunol. 1997;34:987–994. doi: 10.1016/s0161-5890(97)00128-4. [DOI] [PubMed] [Google Scholar]
- YAMAMOTO M., FUKUDA K., MIURA N., SUZUKI R., KIDO T., KOMATSU Y. Inhibition by dexamethasone of transforming growth factor β1-induced apoptosis in rat hepatoma cells: a possible association with Bcl-xL induction. Hepatology. 1998;27:959–966. doi: 10.1002/hep.510270410. [DOI] [PubMed] [Google Scholar]
- YOSHIOKA T., KAWAMURA T., MEYRICK B.O., BECKMAN J.K., HOOVER R.L., YOSHIDA H., ICHIKAWA I. Induction of manganese superoxide dismutase by glucocorticoids in glomerular cells. Kidney Int. 1994;45:211–219. doi: 10.1038/ki.1994.25. [DOI] [PubMed] [Google Scholar]