

Abstract
We have investigated the contribution of specific PLA2s to eicosanoid release from A549 cells by using specific inhibitors of secretory PLA2 (ONO-RS-82 and oleyloxyethylphosphocholine), cytosolic PLA2 (AACOCF3 and MAFP) and calcium-independent PLA2 (HELSS, MAFP and PACOCF3). Similarly, by using specific inhibitors of p38 MAPK (SB 203580), ERK1/2 MAPK (Apigenin) and MEK1/2 (PD 98059) we have further evaluated potential pathways of AA release in this cell line.
ONO-RS-82 and oleyloxyethylphosphocholine had no significant effect on EGF or IL-1β stimulated 3H-AA or PGE2 release or cell proliferation. AACOCF3, HELSS, MAFP and PACOCF3 significantly inhibited both EGF and IL-1β stimulated 3H-AA and PGE2 release as well as cell proliferation. Apigenin and PD 98509 significantly inhibited both EGF and IL-1β stimulated 3H-AA and PGE2 release and cell proliferation whereas, SB 203580 had no significant effect on EGF or IL-1β stimulated 3H-AA release, or cell proliferation but significantly suppressed EGF or IL-1β stimulated PGE2 release.
These results confirm that the liberation of AA release, generation of PGE2 and cell proliferation is mediated largely through the actions of cPLA2 whereas, sPLA2 plays no significant role. We now also report a hitherto unsuspected contribution of iPLA2 to this process and demonstrate that the stimulating action of EGF and IL-1β in AA release and cell proliferation is mediated in part via a MEK and ERK-dependent pathway (but not through p38MAPK). We therefore propose that selective inhibitors of MEK and MAPK pathways may be useful in controlling AA release, eicosanoid production and cell proliferation.
Keywords: EGF, IL-1β, PGE2, cell proliferation, arachidonic acid, MAPK, PLA2 enzymes
Introduction
Prostaglandins (PGs) act as local hormones to regulate both physiological and pathological responses within the host. They have been implicated as causative factors in the pathogenesis of many diseases, including, asthma, atherosclerosis and cancer (Vane, 1978; Cuthbert, 1973; Borg et al., 1994; Earnest et al., 1992). Additionally, PGs can also act as autocrine modulators of cell proliferation in several cell lines (Karmali et al., 1979; Qiao et al., 1995). We have identified PGE2 as an autocrine mediator of cell growth in the human lung adenocarcinoma A549 cell line (Croxtall & Flower, 1992). PGE2 production is regulated both by the release of AA from cellular phospholipids by PLA2 enzymes (Mukherjee et al., 1994) and by the conversion of AA to PGH2, the common substrate for all PGs, by the enzyme cyclo-oxygenase (COX) (Smith & Marnett, 1991).
AA, the precursor for PGs biosynthesis, does not exist in free form but is released from cellular phospholipids by the rate-limiting PLA2 enzymes in response to a variety of extracellular stimuli. PLA2 enzymes are generally characterized according to their cellular localization, calcium dependency and molecular weight. The two main forms are the secretory PLA2 (sPLA2) and the cytosolic PLA2 (cPLA2) (reviewed by Leslie, 1997; Kramer & Sharp, 1997; Dennis, 1997). However, another calcium independent form of PLA2 has also been identified, referred to as iPLA2 (reviewed by Ackermann & Dennis, 1995; Balsinde & Dennis, 1997).
The key differences between the PLA2 enzymes are that sPLA2 is released extracellularly, has a low molecular weight (14–18 kDa), requires milli-molar calcium concentrations for catalysis and shows little fatty acid specificity. sPLA2 is thought to be involved in the digestion of dietary lipids rather than in cell signalling (Murakami et al., 1997) since it fails to selectively hydrolyse arachidonyl containing phospholipids suggesting that its primary function is not to initiate the biosynthesis of PGs. In contrast, cPLA2 functions intracellularly, has a high molecular weight (40–110 kDa), requires micro-molar levels of calcium for membrane translocation, exhibits a definite preference for the sn-2 AA position, and is phosphorylated by mitogen-activated protein kinases (MAPK). Thus, cPLA2 is more likely to be involved in agonist induced AA release mediated by MAPK-dependent pathways.
Although iPLA2 also occurs intracellularly and has high molecular weight (45–80 kDa) its activity appears to be independent of calcium-concentrations. Unlike the calcium-dependent PLA2s, very little is known about its catalytic mechanism or intracellular role. iPLA2 has no acyl specificity and has been suggested to function in steady state membrane remodelling and the control of intracellular AA levels rather than agonist induced AA release (Balsinde et al., 1995).
To investigate which PLA2 enzymes are involved in AA release, PGE2 generation and cell growth in A549 cells, the following PLA2 inhibitors were used. For sPLA2 inhibition, ONO-RS-82 or [2-(p-amylacinnamoyl)-amino-p-chlorobenzoic acid] and oleyloxyethylphosphocholine were used. ONO-RS-82 is a 14 kDa sPLA2 inhibitor which inhibits 5-HT stimulated thromboxane production in human platelets (Konrad et al., 1992). Oleyloxyethylphosphocholine is a site specific inhibitor of porcine pancreatic PLA2 (Augert et al, 1989). For cPLA2 inhibition, a triflouromethyl ketone analogue of AA, arachidonyl triflouromethyl ketone (AACOCF3) was used. This is a potent, reversible and selective slow-binding inhibitor of human 85 kDa cPLA2 (Street et al., 1993). It is active in intact platelets (Bartoli et al., 1994) and U937 cells (Riendeau et al., 1994) and is 1000-fold more selective for cPLA2 than for sPLA2 (Gelb et al., 1994). For iPLA2 inhibition, both HELSS (haloenol lactone suicide substrate) or [E-6-(bromomethylene)tetrahydro-3-(1-napthalenyl)-2H-pyran-2-one haloenol lactone suicide substrate] and PACOCF3 (palmitoyl trifluoromethyl ketone) were used. HELSS is a potent, irreversible iPLA2 inhibitor (Ackermann et al., 1995) that binds covalently to the active site of the iPLA2 enzyme and possesses a 1000-fold selectivity for iPLA2 over cPLA2 (Hazen et al., 1991). PACOCF3 is also a selective inhibitor of iPLA2 at concentrations up to 10 μM (Ackermann et al, 1995). Finally, for comparative purposes, MAFP (methyl arachidonyl fluorophosphate) a mixed inhibitor of both cPLA2 and iPLA2 (Balsinde & Dennis, 1997) was also tested.
It is known that cPLA2 is catalytically activated by phosphorylation on serine-505 by a serine/threonine specific kinase, mitogen activated protein kinase (MAPK) (Lin et al., 1992). MAPKs are a group of protein kinases that are activated in response to a variety of extracellular stimuli including growth factors, cytokines and hormones. They mediate signal transduction from the cell surface to the nucleus. In mammalian cells, five distinct MAPK signalling cascades have been identified so far, each having several isoforms that respond synergistically to different upstream signals (Robinson & Cobb, 1997). These include the mitogen or extracellular signal regulated protein kinases (MAPK/ERKs) which include ERK1 (p44mapk) and ERK2 (p42mapk) which are activated by mitogenic stimuli from growth factor receptors and phorbol esters (Seger et al., 1991); stress activated protein kinases (SAPKs) or c-jun NH2-terminal kinases (JNKs), which include JNK1 (p45sapks γ) and JNK2 (p54sapks α/β) which are activated by stress inducing signals such as osmotic and heat shock, UV and γ-irradiation, protein synthesis inhibitors, metabolic poisons or pro-inflammatory cytokines IL-1β (Kyriakis & Avruch, 1996); the p38 MAPKs (α, β, βII, γ and δ) which are activated by UV and pro-inflammatory cytokines IL-1β (Brunet & Pouyssegur, 1996); as well as ERK3 and ERK5 that as yet, represent two poorly understood pathways. These kinases are in turn activated by distinct upstream MAPK/ERK kinases (MEKs, MKKs) which phosphorylate threonine and tyrosine residues. Several distinct MAPK kinases or MEKs have been identified in mammalian cells. MEK1/2 does not phosphorylate or activate p38 MAPK or JNKs whilst MEK1/2 is a strong activator of ERK1/2 (Derijard et al., 1995; Crews et al., 1992). The existence of all these various kinases presumably allows input into MAPK pathways from many different stimuli.
To identify the MAPK pathways involved in A549 cell signalling, the following MAPK and MEK inhibitors were used. SB 203580 ([4-(4-flourophenyl)-2-(4-methylsulphinylphenyl)-5-(4-pyridyl)imidazole]) is a member of a novel class of pyridinyl imidazoles. It is potent and specific inhibitor of p38 MAPK and p38β MAPK (Cuenda et al., 1997) but does not inhibit any other protein kinases including ERK1/2 MAPK or JNK (Goedert et al., 1997). The ERK1/2 MAPK inhibitor, apigenin (4′,5,7-trihydroxyflavone) is a plant flavonoid that reverses the transformed phenotypes of various rastransformed cells due to inhibition of MAPK associated signal transduction pathways (Kuo & Young, 1995). It also induces the dephosphorylation of ERK1/2 and thereby reduces ERK1/2 activity (Sato et al., 1994). The MEK1/2 inhibitor PD 98059 or ([2-(2′-amino-3′-methoxyphenyl)-oxanaphthalen-4-one]) is a potent and reversible inhibitor that binds to the inactive form of MEK1/2 thereby inhibiting the phosphorylation and activation of ERK/MAPK (Dudley et al., 1995; Alessi et al., 1995). This highly selective MEK1/2 inhibitor was used to further evaluate the role of ERK1/2 in the control of A549 cell signalling.
EGF and IL-1β are known to stimulate AA release, PGE2 production and increase cell growth in a variety of cell types including A549 cells (Carpenter & Cohen, 1979; Croxtall et al., 1993; Dinarello, 1996) and are therefore used as stimuli in this study. Here we attempt to evaluate the effects of PLA2 and MAPK enzyme inhibitors on the AA release, PGE2 generation and increase cell growth of A549 cells stimulated by EGF and IL-1β.
Methods
Cell culture
A549 cells (Flow) were maintained in continuous log phase growth in Dulbecco's modified Eagle medium/F-12 (DMEM/F-12) containing phenol red, 10% foetal calf serum (FCS) and 1% penicillin/streptomycin (PS) in T-150 flasks (Greiner) at 37°C, 5% CO2. The cells were not allowed to reach confluence at any time as this diminishes their response to growth factors, stimulators and inhibitors.
Measurement of arachidonic acid release
Sub-confluent A549 cells were seeded into 12-place multi-well plates (Falcon) at a density of 3×105 cells ml−1 well−1 in DMEM/F-12, 10% FCS, 1% PS and incubated overnight. [3H]-arachidonic acid [3H]-AA in ethanol was evaporated to dryness under N2 and resuspended in an appropriate volume of DMEM/F-12 (without phenol red) and after after vortex mixing left at 37°C for 1 h. After the cells had been washed with PBS, 9.25 KBq of [3H]-AA in 0.5 ml DMEM/F-12 (without phenol red or FCS) was added to each well and incubated overnight. The media containing free [3H]-AA was then removed and the cells washed three times with 1 ml DMEM/F-12 containing 1 mg ml−1 BSA. The cells thus labelled with [3H]-AA were then treated for 3 h with various inhibitors and vehicle control (DMSO). Then either 10 nM EGF was added for 30 min or 1 ng ml−1 IL-1β was added for 3 h. After incubation, 0.4 ml of medium was removed from each well for scintillation counting.
Measurements of PGE2 release
Sub-confluent A549 cells seeded into 12-place multi-well plates at a density of 5×104 cells ml−1 well−1 in DMEM/F-12, 10% FCS and 1% PS. Following incubation overnight, the cells were washed in 2 mls of sterile PBS and 1 ml of fresh DMEM/F-12 (without the phenol red) containing 1% PS plus various inhibitors, EGF, IL-1β or vehicle control added. On day 2, cells were replenished with fresh experimental media containing test agents or vehicle control. On day 3, 0.5 mls of experimental media was removed and PGE2 was measured in the samples using an enzymeimmunoassay (EIA) kit.
Cell proliferation experiments
Sub-confluent A549 cells were seeded into 12-place multi-well plates at a density of 5×104 cells ml−1 well−1 in DMEM/F-12, 10% FCS and 1% PS. Following incubation overnight, the cells were washed in 2 mls of sterile PBS and 1 ml of fresh DMEM/F-12 (without the phenol red) containing 1% PS plus various inhibitors, EGF, IL-1β or vehicle control added. On day 2, cells were replenished with fresh experimental media containing test agents or vehicle control. On day 3, media was removed from each well and 1 ml of PBS containing 0.05% trypsin and 0.02% EDTA was added to each well. The dispersed cells then counted with a Coulter Multisizer II counter. The percentage inhibition of cell proliferation for PLA2 inhibitor treated culture was calculated compared to control well. Trypan Blue was used to determine cell viability. The data reported are due to inhibition of cell proliferation and not cell death due to toxicity of the compounds tested.
Determination of the activation of MAPK's and cPLA2
Following experimental treatment, the medium was aspirated and the A549 cell monolayer washed with PBS, 1 mM EDTA to remove adherent surface-bound proteins. The monolayer was dispersed with 0.05% trypsin in PBS, 10 mM EDTA and cell pellets were snap-frozen in 3 ml of PBS, 10 mM EDTA containing 1 mg ml−1 soyabean trypsin inhibitor, 0.01% leupeptin, 1 mM PMSF and 1 mM sodium orthovanadate. Once thawed, cell lysates were clarified by centrifugation at 13,000×g for 20 min. Protein concentrations were measured by Bradford assay and identical concentrations were used in each immunoprecipitation. 1 ml of cell lysate was incubated with 5 μg of precipitating monoclonal antibody for 16 h with continuous rocking. Then 20 mg Protein A-Sepharose was added for a further 2 h. The Protein A-Sepharose bound immunocomplexes were washed three times in PBS, 10 mM EDTA and then incubated with 250 μl sample buffer for 5 min at 90°C prior to SDS–PAGE analysis by Western blotting with the appropriate anti-phospho specfic monoclonal antibody (10 μg ml−1) and detection by DAB. Western blots were scanned using a ScanMaker (Microtek, CA, U.S.A.) and the image composite transferred into Power Point (Micosoft, WA, U.S.A.) running on an Apple Macintosh. Densitometric analysis was performed with NIH Image 1.54 and relative band intensities reported as % changes within each blot. The values given are semiquantitative and are only meant to give some numerical guide to the ratio of band intensities. The blots presented are typical examples of at least three such experiments. Although overall band intensities varied between experiments, the ratio of band intensities remained the same.
Materials
EGF, ONO-RS-82, anti-phospho specific antibodies, DAB, DMEM/F-12, FCS and trypsin were purchased from Sigma Chemical Company (Poole, U.K.). [5,6,8,9,11,12,14,15-3H-(N)]-arachidonic acid from NEN, Du Pont (Belgium). AACOCF3, PD 98059 and HELSS compounds from Biomol Research Lab., Inc. (Plymouth, PA, U.S.A.). Apigenin, oleyloxyethylphosphocholine, MAFP, PACOCF3 and SB 203580 compounds from Calbiochem-Novabiochem Ltd. (Nottingham, U.K.). Antibodies to COX2 (goat), ERK1/2 (rabbit), p38MAPK (mouse), JNK1 (rabbit) and cPLA2 (mouse) were from Autogen Bioclear (Wilts, U.K.). PGE2 (EIA) kit from Amersham (Buckinghamshire, U.K.). hrIL-1β was a generous gift from Dr Mauro Perretti (Dept of Biochemical Pharmacology, The William Harvey Research Institute, London, U.K.).
Statistical analysis
Each experiment was performed in triplicate (n=3) and each experiment is a typical example of at least three such experiments. Results were calculated as the mean±s.e.mean and are presented as the per cent inhibition±s.e.mean. Statistical differences were calculated on raw data using the ANOVA test with post analysis Bonferroni correction. A threshold value of P<0.05 was taken as significant.
Results
Effects of PLA2 inhibitors, MEK, ERK and p38 MAPK inhibitors on EGF and IL-1β stimulated 3H-AA release from A549 cells
Treatment of A549 cells with 10 nM EGF for 30 min typically stimulated release of 3H-AA by 40% above control values. Such EGF stimulated AA release has been observed in several other different cell lines (Hack et al., 1991; Spaargaren et al., 1992). To investigate the isoforms of PLA2 involved in the liberation of EGF stimulated AA release, A549 cells were pretreated with the specific inhibitors for each kind of PLA2. Pre-treatment for 3 h with the sPLA2 inhibitor ONO-RS-82 up to concentrations of 20 μM had no significant effect on EGF stimulated release (Figure 1). Similarly, oleyloxyethylphosphocholine up to concentrations of 20 μM was also without any significant effect (data not shown). However, pre-treatment with the iPLA2 inhibitor, HELSS, at concentrations of 5 μM and above, significantly inhibited EGF stimulated 3H-AA release. This inhibition was maximal (40%) at 10 μM with an IC50 of 5 μM (Figure 1). Further, PACOCF3 also inhibited EGF stimulated release with an IC50 of 5 μM and a maximal effect at 10 μM (60%, data not shown). Similarly, the cPLA2 inhibitor, AACOCF3 at concentrations of 0.1 μM and above significantly inhibited EGF stimulated 3H-AA release. This inhibition was maximal (80%) at 20 μM with an IC50 of 1 μM (Figure 1). Using MAFP, an inhibitor of both iPLA2 and cPLA2, resulted in the greatest inhibition of EGF stimulated 3H-AA release with 45% inhibition at 0.1 μM and a maximal effect of 88% at 10 μM (data not shown). These results imply that the production of EGF stimulated AA release in A549 cells is apparently not mediated through the action of sPLA2, but rather through the activation of both iPLA2 and cPLA2. This is in keeping with our previous findings that A549 cells appear to express undetectable levels of sPLA2, but rather levels of the cytosolic isoform predominate instead (Tokumoto et al., 1993). Furthermore, the inhibition by the iPLA2 inhibitor was surprising. The expression of this form of PLA2 has not yet been characterized in A549 cells, however these results would appear to indicate its participation in AA release.
Figure 1.
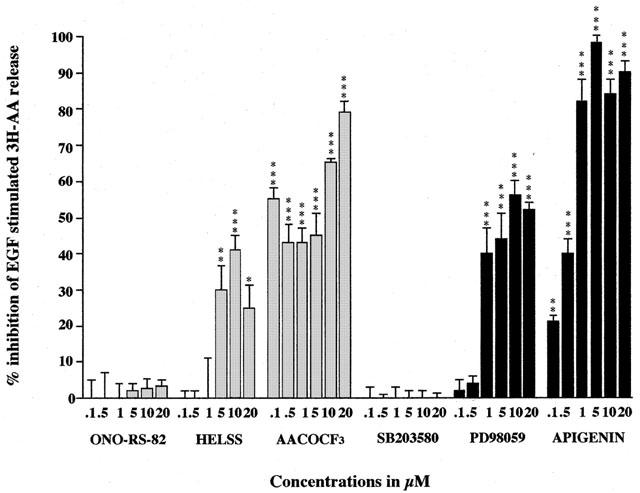
The effect of PLA2, MEK and MAPK inhibitors on EGF stimulated 3H-AA release from A549 cells. Cells were pre-incubated either with sPLA2 inhibitor ONO-RS-82, iPLA2 inhibitor HELSS, cPLA2 inhibitor AACOCF3, p38 MAPK inhibitor SB 203580, MEK inhibitor PD 98059 or with ERK inhibitor apigenin for 3 h, prior to stimulation by EGF (10 nM) for 30 min. The data are presented as mean±s.e.mean, expressed as per cent inhibition of 3H-AA release relative to EGF stimulation of 41%±4% above control values. *P<0.05, **P<0.01, ***P<0.001 inhibition compared to EGF stimulation.
The MEK inhibitor PD 98059 and ERK inhibitor apigenin both inhibited EGF stimulated 3H-AA release (Figure 1). Pre-treatment for 3 h with PD 98059 significantly inhibited EGF stimulated AA release, with maximal inhibition (56%) at 10 μM PD 98059 and an IC50 of 0.75 μM (Figure 1). This result confirms the involvement of MEK in the EGF signal transduction pathway leading to AA release and also indicates that the ERK pathway may also be involved since PD 98059 is also known to inhibit the MEK activation of ERK (Alessi et al., 1995).
Indeed, pre-treatment of A549 cells with the ERK inhibitor apigenin for 3 h also significantly suppressed EGF stimulated AA release, with maximal inhibition (98%) at 5 μM apigenin and an IC50 of 0.75 μM (Figure 1). Apigenin induces dephosphorylation of ERK1 and a reduction of MAPK activity in v-H-ras-transformed NIH-3T3 cells (Sato et al., 1994), and also inhibits MAPK-associated signal transduction pathways in various ras-transformed cells. Our results suggest that ERK is also involved in the EGF signal transduction pathway leading to the activation of PLA2 and liberation of AA in A549 cells. In contrast, pretreatment for 3 h with the p38 MAPK inhibitor SB 203580 up to concentrations of 20 μM had no significant effect on EGF stimulated 3H-AA release and therefore suggests that p38 MAPK does not play a part in the EGF-dependent activation of AA release in A549 cells.
IL-1β has also been shown to stimulate AA release from A549 cells (Newman et al., 1994). Typically, 1 ng ml−1 IL-1β stimulated AA release 110% above control values and pre-treatment for 3 h with the sPLA2 inhibitor ONO-RS-82 up to concentrations of 20 μM had no significant effect on this stimulated 3H-AA release (Figure 2). Likewise, oleyloxyethylphosphocholine also had no significance at concentrations up to 20 μM (data not shown). However, pre-treatment for 3 h with the iPLA2 inhibitor, HELSS at concentrations 0.1 μM and above significantly inhibited IL-1β stimulated 3H-AA release. This inhibition was maximal (40%) at 5 μM with an approximate IC50 of 0.75 μM (Figure 2). Further, PACOCF3 also inhibited IL-1β stimulated release with an IC50 of 10 μM and a maximal effect of 57% inhibition at 20 μM (data not shown). Similarly, the cPLA2 inhibitor, AACOCF3 at concentrations of 0.1 μM and above significantly inhibited IL-1β stimulated 3H-AA release. This inhibition was maximal (66%) at 20 μM (Figure 2). Using the mixed inhibitor MAFP produced the greatest inhibitory effect with an IC50 of 0.5 μM and a maximal effect of 98% at 10 μM (data not shown). This data shows the lack of involvement of sPLA2 in IL-1β stimulated AA release and also again that iPLA2 and cPLA2 both appear to play a contributory role in A549 cells.
Figure 2.
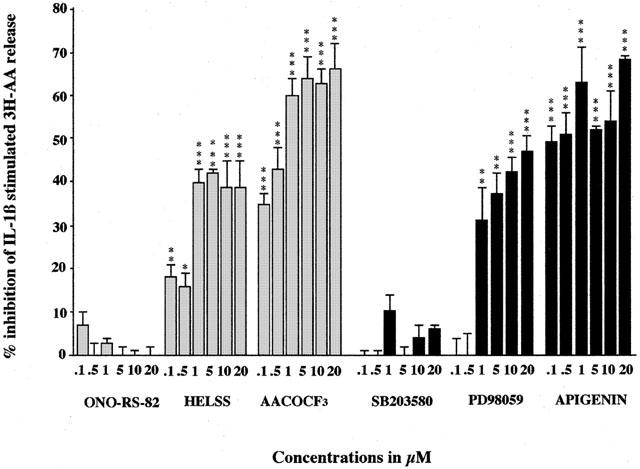
The effect of PLA2, MEK and MAPK inhibitors on IL-1β stimulated 3H-AA release from A549 cells. Cells were pre-incubated either with sPLA2 inhibitor ONO-RS-82, iPLA2 inhibitor HELSS, cPLA2 inhibitor AACOCF3, p38 MAPK inhibitor SB 203580, MEK inhibitor PD 98059 or with ERK inhibitor apigenin for 3 h, prior to stimulation by IL-1β (1 ng ml−1) for 3 h. The data are presented as mean±s.e.mean, expressed as per cent inhibition of 3H-AA release relative to IL-1β stimulation of 110%±6% above control values. *P<0.05, **P<0.01, ***P<0.001 inhibition compared to IL-1β stimulation.
Likewise, the MEK inhibitor PD 98059 and ERK inhibitor apigenin both significantly inhibited IL-1β stimulated AA release, whereas, p38 MAPK inhibitor SB 203580 upto concentrations of 20 μM had no effect (Figure 2). Pre-treatment for 3 h with PD 98059 at concentrations of 1 μM and above significantly inhibited IL-1β stimulated AA release, with maximal inhibition (47%) at a 20 μM concentration (Figure 2). This suggests that MEK plays a role in the signal transduction pathway. Furthermore, pre-treatment for 3 h with ERK inhibitor apigenin at concentrations of 0.1 μM and above significantly inhibited IL-1β stimulated AA release, with maximal inhibition (68%) at a concentration of 20 μM (Figure 2), suggesting that ERK pathways are also involved in IL-1β stimulated AA release. All this appears to indicates that MEK and ERK pathways are involved in IL-1β stimulated AA release in A549 cells, whereas the p38 MAPK pathway does not appear to play a significant part.
The use of PLA2 inhibitors, MEK, ERK and p38 MAPK inhibitors on EGF and IL-1β stimulated PGE2 release from A549 cells
Previously we have demonstrated that PGE2 release plays an important role in the control of cell growth in A549 cells (Croxtall & Flower, 1992). To further investigate which isoforms of PLA2 are involved in the generation of PGE2, we have used the panel of PLA2 inhibitors in a PGE2 release assay. Figure 3 shows that untreated control A549 cells after 72 h in culture, typically release between 50 and 150 pg ml−1 of PGE2 to the media. 10 nM EGF typically stimulated this PGE2 release by between 2–3-fold pre-treatment for 3 h with sPLA2 inhibitor ONO-RS-82 up to concentrations of 1 μM had no significant effect on EGF-stimulated PGE2 release. Similarly, oleyloxyethylphosphocholine had no effect on significant effect on PGE2 release (data not shown). However, pre-treatment for 3 h with the iPLA2 inhibitor HELSS at concentrations ranging from 0.1 μM and 1 μM inhibited EGF-stimulated PGE2 release (Figure 3) as did PACOCF2 (IC50 1 μM, data not shown). Concentrations above 1 μM for both ONO-RS-82 and HELSS appears to have a toxic effect on the cells probably due to longer contact times. Similarly, the cPLA2 inhibitor AACOCF3 at concentrations of 5 μM and above significantly inhibited EGF-stimulated PGE2 release (Figure 3). Moreover, the mixed inhibitor MAFP significantly inhibited PGE2 release at all concentrations tested (0.1 μM and above, P<0.001 all points, data not shown). This reinforces the concept that the level of PGE2 release from A549 cells stimulated by EGF is mediated largely through the concerted actions of iPLA2 and cPLA2 whereas sPLA2 plays no role.
Figure 3.
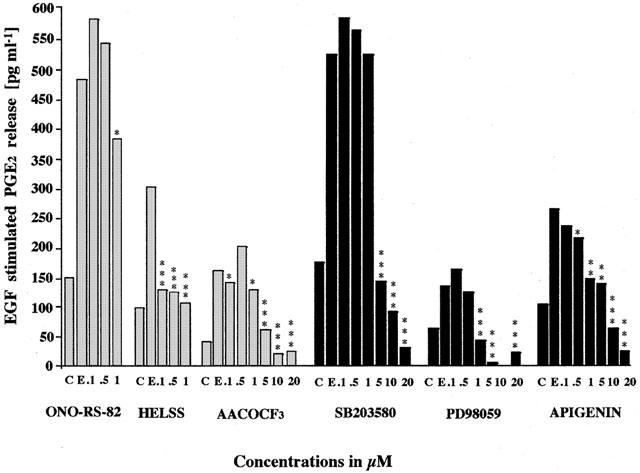
The effect of PLA2 inhibitors, MEK and MAPK inhibitors on EGF stimulated PGE2 release from A549 cells. Cells were treated with EGF (E, 10 nM) plus either with sPLA2 inhibitor ONO-RS-82, iPLA2 inhibitor HELSS, cPLA2 inhibitor AACOCF3, p38 MAPK inhibitor SB 203580, MEK inhibitor PD 98059, ERK inhibitor apigenin or untreated as control (C) for 72 h. The data are presented as mean±s.e.mean, the concentrations of PGE2 released in the media in pg ml−1.
The EGF stimulated PGE2 release was also significantly inhibited by treatment with SB 203580 at concentrations of 5 μM and above, PD 98059 at concentrations of 1 μM and above and by apigenin at concentrations of 10 μM and above (Figure 3). This indicates not only the involvement of MEK and ERK signalling pathways in EGF stimulated PGE2 generation in A549 cells but also that the p38 MAPK pathway may play a role.
We have also shown that IL-1β induced A549 cell growth is also mediated, at least in part, by elevated PGE2 levels (Newman et al., 1994). Figure 4 illustrates that 1 ng ml−1 IL-1β typically stimulated PGE2 release by 1500–5000% above control values and this was not affected by ONO-RS-82 (and oleyloxyethylphosphocholine not shown), whereas co-treatment with HELSS (and PACOCF3 and MAFP not shown) at concentrations 0.1 μM and above and AACOCF3 at concentrations of 5 μM and above significantly inhibited PGE2 release. Concentrations of ONO-RS-82 and HELSS above 1 μM appeared to be toxic. Therefore, again the active forms of PLA2 which are involved in IL-1β stimulated PGE2 generation appear to be iPLA2 and cPLA2 and not sPLA2.
Figure 4.
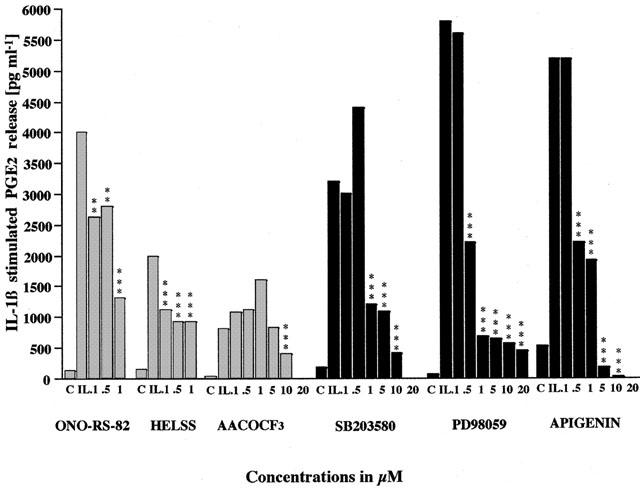
The effect of PLA2 inhibitors, MEK and MAPK inhibitors on IL-1β stimulated PGE2 release from A549 cells. Cells were treated with IL-1β (IL, 1 ng ml−1) plus with either sPLA2 inhibitor ONO-RS-82, iPLA2 inhibitor HELSS, cPLA2 inhibitor AACOCF3, p38 MAPK inhibitor SB 203580, MEK inhibitor PD 98059, ERK inhibitor apigenin or untreated as control (C) for 72 h. The data are presented as mean±s.e.mean, the concentrations of PGE2 released in the media in pg ml−1.
IL-1β stimulated PGE2 release from A549 cells was also significantly inhibited by co-incubation with SB 203580 at concentrations 1 μM and above, PD 98059 at concentrations 1 μM and above and apigenin at concentrations 0.5 μM and above (Figure 4). This implies that not only MEK and ERK signalling pathways play a role but also that p38 MAPK is also involved in IL-1β stimulated PGE2 generation in A549 cells.
Cell growth and the effect of PLA2 inhibitors, MEK, ERK and p38 MAPK inhibitors on EGF and IL-1β stimulated A549 cells
Following 72 h in culture, basal cell numbers increased from 50,000 (day 0) to 81,000, which is an increase of 62%±2%. Typically, 10 nM EGF stimulated this cell growth by 70%±2% over control values at 72 h. Our previous work has shown that this EGF-stimulated proliferation of A549 cells is dependent (at least in part) upon elevated PGE2 levels (Croxtall & Flower, 1992). The data presented here show that increased cell proliferation of A549 cells by EGF was inhibited by HELSS, PACOCF3 (not shown) and AACOCF3 at concentrations of 0.5 μM and above and by MAFP (0.1 μM and above, not shown), whereas ONO-RS-82 (and oleyloxyethylphosphocholine, not shown) was without effect (Figure 5). This again shows the contributory effect of both iPLA2 and cPLA2 to EGF-induced cell growth, whereas sPLA2 plays no role.
Figure 5.
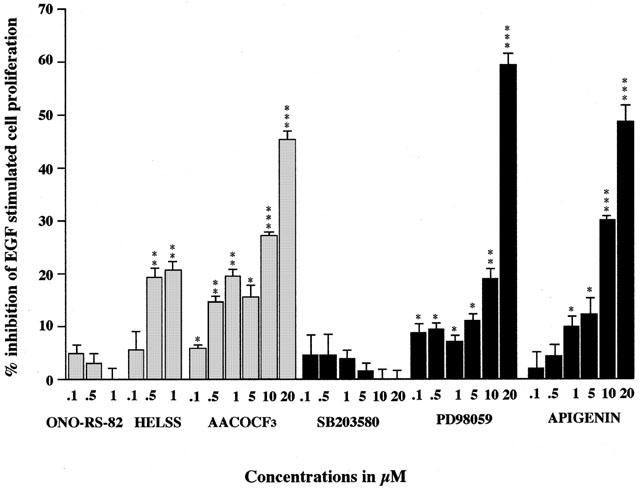
The effect of PLA2 inhibitors, MEK and MAPK inhibitors on EGF stimulated A549 cell growth. Cells were treated with EGF (10 nM) plus with either sPLA2 inhibitor ONO-RS-82, iPLA2 inhibitor HELSS, cPLA2 inhibitor AACOCF3, p38 MAPK inhibitor SB 203580, MEK inhibitor PD 98059, ERK inhibitor apigenin or untreated as control (C) for 72 h. The data are presented as mean±s.e.mean, expressed as per cent inhibition of cell growth with respect to EGF stimulation of 72%±3% above control values. *P<0.05, **P<0.01, ***P<0.001 inhibition compared to EGF stimulation.
Co-incubation with SB 203580 up to concentrations of 20 μM had no significant effect on EGF-stimulated A549 cell growth. However, both PD 98059 and apigenin at concentrations 5 μM and above significantly inhibited EGF stimulated A549 cell growth. Therefore, it seems that both MEK and ERK pathways appear to be involved in the control of EGF induced A549 cell growth. However, inhibition of the p38 MAPK pathway does not appear to inhibit cell growth despite inhibiting EGF-stimulated PGE2 release.
IL-1β (1 ng ml−1) also typically stimulated cell growth by 35%±4% over control values at 72 h (Figure 6). This IL-1β induced cell proliferation was significantly inhibited by co-incubation with HELSS at concentrations 0.5 μM and above and by AACOCF3 at concentrations of 10 μM and above. Similarly, PACOCF3 at 0.1 μM and above and MAFP at 0.1 μM also inhibited IL-1β stimulation (data not shown). Whereas ONO-RS-82 at concentrations up to 1 μM had no effect (and oleyloxyethylphosphocholine had no effect up to 0.5 μM, data not shown). Concentrations above 1 μM of ONO-RS-82 and HELSS appear to be toxic for these cells. This again shows that IL-1β stimulated A549 cell proliferation is mediated through the actions of both iPLA2 and cPLA2, whereas sPLA2 plays no role.
Figure 6.
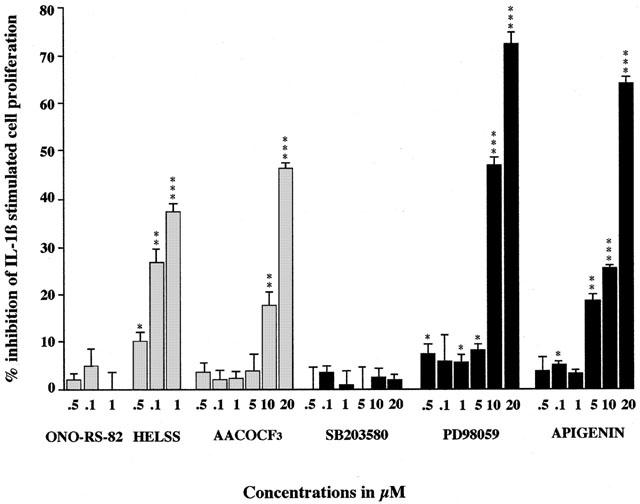
The effect of PLA2 inhibitors, MEK and MAPK inhibitors on IL-1β stimulated A549 cell growth. Cells were treated with IL-1β (1 ng ml−1) plus with either sPLA2 inhibitor ONO-RS-82, iPLA2 inhibitor HELSS, cPLA2 inhibitor AACOCF3, p38 MAPK inhibitor SB 203580, MEK inhibitor PD 98059, ERK inhibitor apigenin or untreated as control (C) for 72 h. The data are presented as mean±s.e.mean, expressed as per cent inhibition of cell growth with respect to IL-1β stimulation of 35%±2% above control values. *P<0.05, **P<0.01, ***P<0.001 inhibition compared to IL-1β stimulation.
Co-incubation with SB 203580 up to concentrations of 20 μM had no significant effect on IL-1β stimulated A549 cell proliferation. However, both PD 98059 and apigenin at concentrations 5 μM and above significantly inhibited IL-1β stimulated A549 cell growth (Figure 6). This data again suggests that inhibition of the p38 MAPK signalling pathway does not appear to inhibit cell proliferation stimulated by IL-1β. Whereas it seems that both MEK and ERK pathways are involved in this process.
Effect of EGF and IL-1β on MAPK activation
Lysates of A549 cells treated either with 10 nM EGF or 1 ng ml−1 IL-1β for the indicated times were immunoprecipitated with specific monoclonal antibodies to several members of the MAPK family. Western blotting of the selected immunoprecipitates with anti-phospho specific antibodies enabled the activation status of each MAPK to be determined. Figure 7 shows increased activation of ERK1, p38 and JNK after 5 min of treatment with EGF (ERK2 remained unchanged, not shown). Generally, longer contact times were required for IL-1β to activate these kinases with maximal activation occuring between 1–3 h. These results are in accord with our previous observations on the activation of cPLA2 (Croxtall et al., 1996a,1996b) and arachidonic acid release (Croxtall et al., 1995) by these factors.
Figure 7.
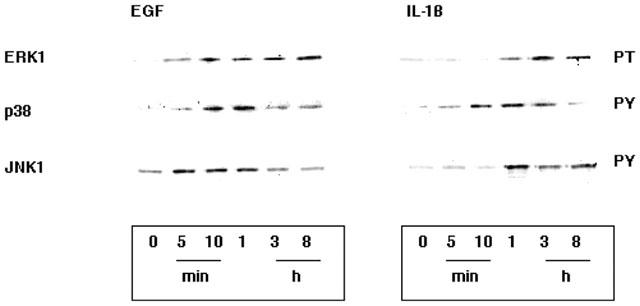
Time course of the activation of ERK1, p38MAPK and JNK1. A549 cells were treated for the times indicated by either 10 nM EGF or 1 ng ml−1 IL-1β. Immunoprecipitates of the selected MAPK's were examined for activation status by Western blotting with antibodies to either phosphothreonine (PT) or phosphotyrosine (PY). EGF induced a 20 fold activation of ERK1 from 5 min, a 10-fold activation of p38MAPK from 10 min and a 2 fold activation of JNK1 from 5 min. Whereas, IL-1β induced a 5 fold activation of ERK1 at 1 h, a 10 fold activation of p38MAPK at 1 h and a 30 fold activation of JNK1 at 1 h.
Effect of MAPK inhibitors on cPLA2 activation and COX2 expression
Results shown in Figure 8 demonstrate that A549 cells pre-incubated for 3 h with either PD 98059 or apigenin fail to activate ERK1 and cPLA2 in response to EGF or IL-1β. Whereas, the induction of COX2 remains unaffected. However, in contrast, pre-incubation with SB 203580 does not effect activation of ERK1 or cPLA2 but significantly attenuates the induction of COX2. These results reinforce the premise of this study that EGF and IL-1β stimulate proliferation of A549 cells by activation of the ERK1 pathway and subsequently cPLA2 resulting in enhanced arachidonic acid release. Inhibition of this pathway results in a significant attenuation of cell growth. However, it appears that COX2 is differentially activated by an alternative but parallel pathway involving p38MAPK. Inhibition of this pathway attenuates PGE2 release but fails to arrest cell proliferation.
Figure 8.
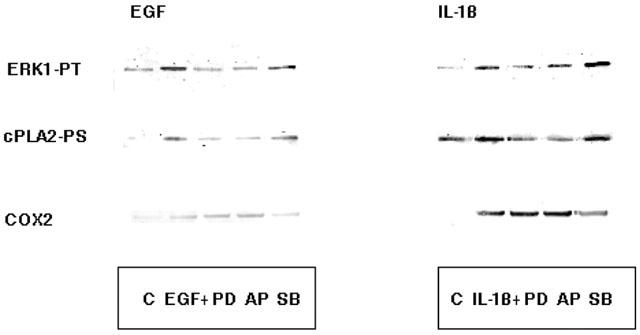
The effect of MAPK inhibitors on cPLA2 activation and COX2 induction. Cells were pre-treated with either 10 μM PD 98059 (PD), 10 μM apigenin (AP) or 10 μM SB 203580 (SB) for 3 h prior to treatment with 10 nM EGF for 30 min or 1 ng ml−1 IL-1β for 3 h. Immunoprecipitates of ERK1 and cPLA2 were examined for activation status by Western blotting with antibodies to phosphothreonine (PT) and phosphoserine (PS) respectively. COX2 expression was assessed Western blotting the total cell lysate with COX2 antibody. The activation of ERK1 and cPLA2 by either EGF or IL-1β was prevented by both PD and AP but unaffected by SB. Whereas the induction of COX2 was unaffected by PD and AP but reveresed by SB.
Discussion
From the results obtained in this study it is apparent that both EGF and IL-1β increase AA release, up-regulate PGE2 production and increase cell proliferation in A549 cells. EGF and IL-1β stimulated AA release was followed by an increase in PGE2 production and a subsequent increase in cell proliferation. This demonstrates the close association of enhanced PGE2 release with elevated A549 cell growth. Data obtained showed that EGF stimulates AA release by 40% over control values, up-regulates PGE2 production by 450% over control values and increases cell proliferation by 70% over control values, whereas IL-1β stimulates AA release by 110% over control values, up-regulates PGE2 production by 4000% over control values and increases cell proliferation by 35% over control values in A549 cells. However, the extent of EGF stimulated cell growth was much greater than IL-1β, despite a much lower increase in PGE2. This perhaps indicates that PGE2 may not be the only mediator involved and that other PG's may have a role to play in A549 cell proliferation. This is supported by our previous work (Croxtall & Flower, 1992) where dexamethasone suppression of cell growth was only partially reversed by the addition of exogenous PGE2, implying that other mediators are necessary.
The PLA2 inhibitor studies revealed that cPLA2 inhibitor AACOCF3 has a profound inhibitory effect on both EGF and IL-1β stimulated AA release on, EGF or IL-1β stimulated PGE2 release, and also on cell proliferation of A549 cells. The crucial role of cPLA2 in the rapid release of AA after stimulus-coupled cell activation is now well established. cPLA2 preferentially liberates AA following translocation from the cytosol to the membrane fraction in response to stimulus-initiated transient increase in cytoplasmic Ca2+ concentrations and phosphorylation by MAPK (Wissing et al., 1997). This increase in AA release results in up-regulation of PGE2 release with concomitant enhanced cell growth. AACOCF3 has been used by several workers to inhibit cPLA2 activity in platelets, U937 monocytes and messengial cells (Riendeau et al., 1994; Bartoli, et al., 1994). It also potently depressed the cPLA2 activity and eicosanoid production in rat corpora lutea (Kurusu et al., 1997). Consistently, results obtained from our study also showed that EGF and IL-1β stimulation of AA release, up-regulation of PGE2 release and enhanced cell growth are all mediated, in part, by the activation of cPLA2, whereas sPLA2 plays no significant role.
Data obtained from this study also demonstrate the ability of the iPLA2 inhibitors HELSS and PACOCF3 to significantly inhibit either EGF or IL-1β stimulated cell proliferation, PGE2 generation and AA release from A549 cells, albeit to a lesser extent than AACOCF3. This raises the question of the significance of iPLA2 mediated cell proliferation in this cell line. Recent reports suggest its presence in most cell lines and is crucial for membrane remodelling and controlling the amount of AA released (Balsinde & Dennis, 1997). Normally, small amounts of free AA are constantly generated in the cell and this step is thought to be mediated by iPLA2 (Balsinde et al., 1995). Surette and colleagues (1999) have shown using HL-60 cells that iPLA2 plays a significant role in the control of cellular AA levels, cell proliferation and survival. Therefore, iPLA2 may also act as a housekeeping enzyme in regulating phospholipid turnover and cell growth in A549 cells. The lack of inhibition of EGF or IL-1β stimulated cell proliferation, basal PGE2 generation and EGF or IL-1β stimulated AA release from A549 cells by the sPLA2 inhibitor ONO-RS-82 supports data from many groups that this isoform appears to play little role in these processes.
MAPKs play a pivotal role in the phosphorylation and activation of cPLA2 (Lin et al., 1992). Components of MAPK pathways have also been implicated as mediators of phosphorylation of intracellular substrates such as protein kinases and transcription factors as well as regulators of cell growth and differentiation (Johnson et al., 1996). So far, all known mitogens including EGF exert intracellular responses through the induction of MAPK cascades (Seger & Krebs, 1995). To investigate the involvement of MAPK cascades by EGF and IL-1β in stimulating AA release, PGE2 generation and cell growth in A549 cells, the ERK inhibitor apigenin, the MEK inhibitor PD 98059 and the p38 MAPK inhibitor SB 203580 were used in these assays.
Results obtained from this study showed that ERK inhibitor apigenin significantly inhibited in a concentration dependent manner EGF or IL-1β stimulated cell proliferation, PGE2 generation and AA release from A549 cells and also blocked the activation of cPLA2. This is highly suggestive of a role for the ERK pathway in controlling the acivity of cPLA2 and thus the release of AA and PGE2 as a means of regulating cell growth. Many groups have reported the concomitant activation of MAPK and cPLA2 in a variety of stimulated cells (Lin et al., 1993; Qiu & Leslie, 1994; Durstin et al., 1994; Xing et al., 1994; Sa et al., 1995; Ambs et al., 1995) and we can now add the concomitant activation of PGE2 release and cell growth.
To further evaluate the role of ERK in the control of A549 cell signalling, the MAPK kinase (MEK) inhibitor PD 98059 was used. PD 98059 also significantly inhibited in a concentration dependent manner EGF or IL-1β stimulated cell proliferation, PGE2 generation and AA release from A549 cells and again also blocked the activation of cPLA2. This suggests that EGF and IL-1β actions are mediated through MAPK-activating enzyme MEK and, which thereby regulates acivity of the MAPK cascades and thus the phosphorylation and activation of cPLA2 resulting in AA release, PGE2 generation and enhanced cell growth.
Zhang and colleagues (1997) have reported that aggregation of the high affinity IgE receptor in the RBL-2H3 mast cell line resulted in the release of AA. Treatment of these cells with PD 98059 inhibits activation of ERK and the release of AA. Cybulsky and McTavish (1997) also reported that EGF stimulated the activity and tyrosine phosphorylation of ERK leading to cell proliferation in a rat glomerular epithelial cell line. Likewise, Dudley and coworkers (1995) demonstrated that PD 98059 prevented activation of MAPK and inhibited stimulation of cell growth when stimulated by EGF or PDGF in BALB-3T3 mouse fibroblasts and rat kidney cells. Furthermore, dominant negative or constitutively active mutants of MEK inhibit or accelerate cell proliferation of NIH-3T3 cells respectively (Seger et al., 1994). Also mutants of ERK and its antisense cDNA cause an inhibition of cell proliferation (Pages et al., 1993). All these data implicate the importance of MEK/ERK cascades in the control of cell proliferation. Our data also supports the importance of this pathway and suggests that one key feature of their action may be the activation of AA release by these kinases.
The p38 MAPK inhibitor SB 203580 had no significant effect on EGF and IL-1β stimulated AA release, and on EGF or IL-1β stimulated cell proliferation in A549 cells. However, SB 203580 significantly inhibited EGF and IL-1β stimulated PGE2 production and blocked the induction of COX2 (Figure 8). This perhaps suggest that PGE2 production in this A549 cell line is under the dual control of ERK1 and p38 MAPK through the concerted regulation of AA release and COX-2 expression respectively and that cell growth arrest is accomplished by blocking both pathways. Both EGF and IL-1β induce COX-2 and a role for p38 MAPK in COX-2 induction explains the effect of SB 203580 compound in EGF and IL-1β stimulated PGE2 release.
Recently, it has been shown that EGF also activates the SAPK/JNK pathway which is mediated by Rac 1 and CDC42 following Ras stimulation (Coso et al., 1995; Minden et al., 1995). The Raf/ERK pathway is well known in EGF induced cell proliferation and intracellular signalling and the role of SAPK/JNK pathway as a mediator of EGF stimulated cell growth in A549 cells has been recently suggested by Bost and colleagues (1997). Furthermore, EGF induced stimulation of JNK activity has also been observed in two other lung cancer cell lines, M103 cells and H1011 cells (Bost et al., 1997). All this indicates that SAPK/JNK may be another important growth promoting pathway in human non-small lung cancer cells. However, to date, there is no evidence to suggest that p38 MAPK pathways are involved in EGF stimulated cell proliferation in A549 cells as yet or in any other cell lines. Nevertheless, since this pathway shares many similar stimuli with the SAPK/JNK pathway it cannot be completely ruled out as a possible role in EGF or IL-1β stimulated cell growth under some circumstances.
In summary, we show that both EGF and IL-1β stimulate proliferation of A549 cells by a mechanism including the activation of MAPK pathways which in turn leads to the activation of cPLA2 and not sPLA2. However, the involvement of iPLA2 also appears to play an important role. It is evident that MEK and ERK pathways are involved in the modulation of EGF and IL-1β induced AA release A549 cell growth in A549 cells. The MEK and ERK pathways appear to be more active in EGF than IL-1β stimulated A549 cells in controlling arachidonic acid release rather than the p38 MAPK pathway. Whereas, in EGF and IL-1β stimulated PGE2 generation and COX2 expression, it appears that p38 MAPK, ERK and MEK all participate in the regulation of PGE2 release. A better understanding of the signalling pathways that lead to activation of AA release may help in the application of selective kinase inhibitors to control abberant cell proliferation and inflammatory diseases.
Acknowledgments
This project was funded by The Wellcome Trust of which R.J. Flower is a Principal Research Fellow and by the Joint Research Board of the St. Barthlomew's Trust with a studentship to D.T. McKay.
Abbreviations
- AA
arachidonic acid
- AACOF3
arachidonyl triflouromethyl ketone
- AP or apigenin
4′,5,7-trihydroxyflavone
- COX
cyclo-oxygenase
- cPLA2
cytosolic phospholipase A2
- DMEM
Dulbecco's modified Eagle medium
- EGF
epidermal growth factor
- FCS
foetal calf serum
- HELSS
E-6-(bromomethylene)tetrahydro-3(1-napthalenyl)-2H-pyran-2-one haloenol lactone
- IL-1β
interleukin-1β
- iPLA2
calcium independent phospholipase A2
- MAPK
mitogen acitivated protein kinase
- ONO-RS-82
2-(p-amylacinnamoyl)-amino-p-chlorobenzoic acid
- PD 98059′ 2-(2′-amino-3′-methoxyphenyl)-oxanaphthalen-4-one; PGE2
prostaglandin E2
- PS
phoshoserine
- PT phosphotreonine; PY
phosphotyrosine
- SB 203580
4-(4-flourophenyl)-2-(4-methylsulphinylphenyl)-5-(4-pyridyl)imidazole
- sPLA2
secretory phospholipase A2.
References
- ACKERMANN E.J., CONDE-FRIEBOES K., DENNIS E.A. Inhibition of macrophage Ca2+-independent phospholipase A2 by bromoenol lactone and trifluoromethyl ketones. J. Biol. Chem. 1995;270:445–450. doi: 10.1074/jbc.270.1.445. [DOI] [PubMed] [Google Scholar]
- ACKERMANN E.J., DENNIS E.A. Mammalian calcium-independent phospholipase A2. Biochim. Biophys. Acta. 1995;1259:125–136. doi: 10.1016/0005-2760(95)00143-z. [DOI] [PubMed] [Google Scholar]
- ALESSI D.R., CUENDA A., COHEN P., DUDLEY D.T., SALTIEL A.R. PD 098059 Is a Specific Inhibitor of the Activation of Mitogen-activated Protein Kinase in Vitro and in Vivo. J. Biol. Chem. 1995;270:27489–27494. doi: 10.1074/jbc.270.46.27489. [DOI] [PubMed] [Google Scholar]
- AMBS P., BACCARINI M., FITZKE E., DIETER P. Role of cytosolic phospholipase A2 in arachidonic acid release of ratliver macrophages: regulation by Ca2+ and phosphorylation. Biochem. J. 1995;311:189–195. doi: 10.1042/bj3110189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AUGERT G., BOCCKINO S.B., BLACKMORE P.F., EXTON J.H. Hormonal stimulation of diacylglycerol formation in hepatocytes. J. Biol. Chem. 1989;264:21689–21698. [PubMed] [Google Scholar]
- BALSINDE J., BIANCO I.D., ACKERMAN E.J., CONDE-FRIEBOES K., DENNIS E.A. Inhibition of calcium-independent phospholipase A2 prevents arachidonic acid incorporation and phospholipid remodeling in P388D1 macrophages. Proc. Natl. Acad. Sci. U.S.A. 1995;92:8527–8531. doi: 10.1073/pnas.92.18.8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BALSINDE J., DENNIS E.A. Function and Inhibition of Intracellular Calcium-independent Phospholipase A2. J. Biol. Chem. 1997;272:16069–16072. doi: 10.1074/jbc.272.26.16069. [DOI] [PubMed] [Google Scholar]
- BARTOLI F., LIN H-K., GHOMASHCHI F., GELB M.H., JAIN M.K., APITZ-CASTRO R. Tight binding inhibitors of 85-kDa phospholipase A2 but not 14-kDa phospholipase A2 inhibit release of free arachidonate in thrombin-stimulated human platelets. J. Biol. Chem. 1994;269:15625–15630. [PubMed] [Google Scholar]
- BORG C., LIM C.T., YEOMANS D.C., DIETER J.P., KOMIOTIS D., ANDERSON E.G., LE BRETON C.L. Purification of the rat aorta and Human platelet thromboxane A2/prostaglandin receptors by immunoaffinity chromatography employing antipeptide and anti-receptor antibodies. J. Biol. Chem. 1994;269:6109–6116. [PubMed] [Google Scholar]
- BOST F., MCKAY R., DEAN N., MERCOLA D. The JUN Kinase/Stress-activated Protein Kinase Pathway Is Required for Epidermal Growth Factor Stimulation of Growth of Human A549 Lung Carcinoma Cells. J. Biol. Chem. 1997;272:34322–33429. doi: 10.1074/jbc.272.52.33422. [DOI] [PubMed] [Google Scholar]
- BRUNET A., POUYSSEGUR J. Identification of MAP kinase domains by redirecting stress signals into growth factor responses. Science. 1996;272:1652–1655. doi: 10.1126/science.272.5268.1652. [DOI] [PubMed] [Google Scholar]
- CARPENTER G., COHEN S. Epidermal growth factor. Ann. Review of Biochem. 1979;48:193–216. doi: 10.1146/annurev.bi.48.070179.001205. [DOI] [PubMed] [Google Scholar]
- COSO O.A., CHIEAREILLO M., YU J., TERAMOTO H., CRESPO P., XU N., MIKI T., GUTKIND J.S. The small GTP-binding proteins Rac1 and Cdc42 regulate the activity of the JNK/SAPK signalling pathway. Cell. 1995;81:1137–1146. doi: 10.1016/s0092-8674(05)80018-2. [DOI] [PubMed] [Google Scholar]
- CREWS C.M., ALESSANDRANI A., EUKONI R.L. The primary structure of MEK, a protein kinase that phosphorylates the ERK gene product. Science. 1992;258:478–480. doi: 10.1126/science.1411546. [DOI] [PubMed] [Google Scholar]
- CROXTALL J.D., FLOWER R.J. Lipocortin 1 mediates dexamethasone-induced growth arrest of the A549 lung adenocarcinoma cell line. Proc. Natl. Acad. Sci. U.S.A. 1992;89:3571–3575. doi: 10.1073/pnas.89.8.3571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CROXTALL J.D., CHOUDHURY Q., NEWMAN S., FLOWER R.J. Lipocortin 1 and the control of cPLA2 activity in A549 cells. Biochem. Pharmacol. 1996a;52:351–356. doi: 10.1016/0006-2952(95)02442-5. [DOI] [PubMed] [Google Scholar]
- CROXTALL J.D., CHOUDHURY Q., TOKUMOTO H., FLOWER R.J. Lipocortin-1 and the control of arachidonic acid release in cell signalling. Biochem. Pharmacol. 1995;50:465–474. doi: 10.1016/0006-2952(95)00156-t. [DOI] [PubMed] [Google Scholar]
- CROXTALL J.D., NEWMAN S.P., CHOUDHURY Q., FLOWER R.J. The concerted regulation of cPLA2, COX2, and lipocortin 1expression by IL-1β in A549 cells. Biochem. Biophys. Res Comm. 1996b;220:491–495. doi: 10.1006/bbrc.1996.0432. [DOI] [PubMed] [Google Scholar]
- CROXTALL J.D., WAHEED S., CHOUDHURY Q., ANAND R., FLOWER R.J. N-terminal fragments of lipocortin 1 inhibit A549 cell growth and block EGF-induced stimulation of proliferation. Int. J. Cancer. 1993;54:153–158. doi: 10.1002/ijc.2910540124. [DOI] [PubMed] [Google Scholar]
- CUENDA A., COHEN P., BUEE-SCHERRER V., GOEDERT M. Activation of stress-activated protein kinase-3 (SAPK 3) by cytokine and cellular stresses is mediated via SAPKK3 (MKK6); comparison of the specification of SAPK3 and SAPK2 (RK/p38) EMBO J. 1997;16:295–305. doi: 10.1093/emboj/16.2.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CUTHBERT M.F.Prostaglandins and respiratory smooth muscle. The Prostaglandins and respiratory smooth muscle The Prostaglandins: Pharmacological and Therapeutic Advances 1973Philadelphia: J.B. Lippincott Co; 253–286.ed. Cuthbert, M.F. pp [Google Scholar]
- CYBULSKY A.V., MCTAVISH A.J. Extracellular matrix is required for MAP kinase activation and proliferation of rat glomerular epithelial cells. Biochem. Biophys. Res. Commun. 1997;231:160–166. doi: 10.1006/bbrc.1997.6064. [DOI] [PubMed] [Google Scholar]
- DENNIS E.A. The growing phospholipase A2 superfamily of signal transduction enzymes. Trends Biochem. Sci. 1997;22:1–2. doi: 10.1016/s0968-0004(96)20031-3. [DOI] [PubMed] [Google Scholar]
- DERIJARD B., RAINGEAUD J., BARRETT T., WU I.H., HAN J., ULEVITCH R.J., DAVIS R.J. Independent human MAP-kinase signal transduction pathways defined by MEK and MKK isoforms. Science. 1995;267:682–685. doi: 10.1126/science.7839144. [DOI] [PubMed] [Google Scholar]
- DINARELLO C.A. Biological Basis for Interleukin−1 in Disease. Blood. 1996;87:2095–2147. [PubMed] [Google Scholar]
- DUDLEY D.T., PANG L., DECKER S.J., BRIDGES A.J., SALTIEL A.R. A synthetic inhibitor of the mitogen-activated protein kinase cascade. Proc. Natl. Acad. Sci. U.S.A. 1995;92:7686–7689. doi: 10.1073/pnas.92.17.7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DURSTIN M., DURSTIN S., MOLSKI T.F., BEEKU E.L., SHA'AFI R.I. Cytoplasmic phospholipase A2 translocates to membrane fraction in human neutrophils activated by stimuli that phosphorylate mitogen-activated protein kinase. Proc. Natl. Acad. Sci. U.S.A. 1994;91:3142–3146. doi: 10.1073/pnas.91.8.3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EARNEST D.L., HIXSON L.J., ALBERTS D.S. Piroxicam and other cyclooxygenase inhibitors: potential for cancer chemoprevention. J. Cell Biochem. 1992;161:156–166. doi: 10.1002/jcb.240501330. [DOI] [PubMed] [Google Scholar]
- GELB M.H., JAIN M.K., BERG O.G. Inhibition of phospholipase A2. FASEB J. 1994;8:916–992. doi: 10.1096/fasebj.8.12.8088457. [DOI] [PubMed] [Google Scholar]
- GOEDERT M., CUENDA A., CRAXTON M., JAKES R., CONEN P. Activation of the novel stress-activated protein kinase SAPK4 by cytokine and cellular stress is mediated by SKK3 (MKK6): comparison of its substrate specificity with that of other SAP kinase. EMBO J. 1997;16:3663–3671. doi: 10.1093/emboj/16.12.3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HACK N., MARGOLIS B.L., ULLRICH A., SCHLESSINGER J., SKORECKI K.L. Distinct structural specificities for functional coupling of the epidermal growth factor receptor to calcium-signaling versus phospholipase A2 responses. Biochem. J. 1991;275:563–567. doi: 10.1042/bj2750563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAZEN S.L., ZUPAN L.A., WEISS R.H., GETMAN D.P., GROSS R.W. Suicide inhibition of canine myocardial cytosolic calcium-independent phospholipase A2. Mechanism-based discrimination between calcium-dependent and -independent phospholipases A2. J. Biol. Chem. 1991;266:7227–7232. [PubMed] [Google Scholar]
- JOHNSON G.L., GARDNER A.M., DIENER K.M., LANGE-CARTER C.A., GLEAVY J., JARPE M.B., MINDEN A. Signal transduction pathways regulated by mitogen-activated/extracellular response kinase kinase induces cell death. J. Biol. Chem. 1996;271:3229–3237. doi: 10.1074/jbc.271.6.3229. [DOI] [PubMed] [Google Scholar]
- KARMALI R.A., HORROBIN D.F., MENEGES J., PATEL P. The relationship between concentrations of prostaglandins A1, E1, E2 and E2 alpha and rates of cell proliferation. Pharmacol. Res. Commun. 1979;11:69–75. doi: 10.1016/s0031-6989(79)80100-9. [DOI] [PubMed] [Google Scholar]
- KONRAD R.J., JOLLY Y.C., MAYOR C., WOLF B.A. Inhibition of phospholipase A2 and insulin secretion in pancreatic islets. Biochim. Biophys. Acta. 1992;1135:215–220. doi: 10.1016/0167-4889(92)90139-3. [DOI] [PubMed] [Google Scholar]
- KRAMER R.M., SHARP J.D. Structure, function and regulation of Ca2+ sensitive cytosolic phospholipase A2 (cPLA2) FEBS Letters. 1997;410:49–53. doi: 10.1016/s0014-5793(97)00322-0. [DOI] [PubMed] [Google Scholar]
- KUO M.L., YOUNG N.C. Reversion of v-H-ras transformed NIH 3T3 cells by apigenin through inhibiting mitogen-activated protein kinase and its downstream oncogenes. Biochem. Biophys. Res. Commun. 1995;212:767–775. doi: 10.1006/bbrc.1995.2035. [DOI] [PubMed] [Google Scholar]
- KURUSU S., NOGUCHI T., KAWAMINAMI M., HASHIMOTO I. Role of cytosolic phospholipase A2 in eicosanoid generation by corpora lutea of pseudopregnant rats: effects of its specific inhibitor. Prostaglandins, Leukotrienes and Essential Fatty Acids. 1997;57:119–124. doi: 10.1016/s0952-3278(97)90001-6. [DOI] [PubMed] [Google Scholar]
- KYRIAKIS J.M., AVRUCH J. Protein kinase cascades activated by stress and inflammatory cytokines. BioEssays. 1996;18:567–577. doi: 10.1002/bies.950180708. [DOI] [PubMed] [Google Scholar]
- LESLIE C.C. Properties and regulation of cytosolic phospholipase A2. J. Biol. Chem. 1997;272:16709–16712. doi: 10.1074/jbc.272.27.16709. [DOI] [PubMed] [Google Scholar]
- LIN L-L., LIN A.Y., KNOPF J.L. Cytosolic phospholipase A2 is coupled to hormonally regulated release of arachidonic acid. Proc. Natl. Acad. Sci. U.S.A. 1992;89:6147–6151. doi: 10.1073/pnas.89.13.6147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIN L-L., WARTMANN M., LIN A.Y., KNOPF J.L., SETH A., DAVIS R.J. cPLA2 is phosphorylaed and activated by MAP kinase. Cell. 1993;72:269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- MINDEN A., LIN A., CLARET F., ABO A., KARIN M. Selective activation of the JNK signaling cascade and c-Jun transcriptional activity by the small GTPases Rac and Cdc42Hs. Cell. 1995;81:1147–1157. doi: 10.1016/s0092-8674(05)80019-4. [DOI] [PubMed] [Google Scholar]
- MUKHERJEE A.B., MIELE L., PATTABIRAMAN N. Phospholipase A2 Enzymes: Regulation and Physiological Role. Biochem. Pharm. 1994;48:1–10. doi: 10.1016/0006-2952(94)90216-x. [DOI] [PubMed] [Google Scholar]
- MURAKAMI M., NAKATANI Y., ATSUMI GI., INOUE K., KUDO I. Regulatory functions of phospholipase A2. Crit. Rev. Immunol. 1997;17:225–283. doi: 10.1615/critrevimmunol.v17.i3-4.10. [DOI] [PubMed] [Google Scholar]
- NEWMAN S.P., FLOWER R.J., CROXTALL J.D. Dexamethasone suppression of IL-1β induced cyclooxygenase 2 expression is not mediated by lipocortin-1 in A549 cells. Biochem. Biophys. Res. Commun. 1994;202:931–939. doi: 10.1006/bbrc.1994.2019. [DOI] [PubMed] [Google Scholar]
- PAGES G., LENORMAND P., L'ALLEMAIN G., CHAMBARD J.C., MELOCHE S., POUYSSEGUR J. Mitogen-activated protein kinase p42mapk and p44mapk are required for fibroblasts proliferation. Proc. Natl. Acad. Sci. U.S.A. 1993;90:8319–8323. doi: 10.1073/pnas.90.18.8319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QIAO L., KOZONI V., TSIOULIAS G.J., KOUTSOS M.I., HANIF R., SHIFF S.J., RIGAS B. Selected eicosanoids increase the proliferation rate of human colon carcinoma cell lines and mouse colonocytes in vivo. Biochima et Biophysica Acta. 1995;1258:215–223. doi: 10.1016/0005-2760(95)00100-q. [DOI] [PubMed] [Google Scholar]
- QIU Z-H., LESLIE C.C. Protein kinase C-dependent and -independent pathways of mitogen-activated protein kinase activation in macrophages by stimuli that activate phospholipase A2. J. Biol. Chem. 1994;269:19480–19487. [PubMed] [Google Scholar]
- RIENDEAU D., GUAY J., WEECH P.K., LALIBERTE F., YERGEY J., LI C., DESMARAIS S., PERRIER H., LIU S., NICOLL-GRIFFITH D., STREET I.P. Arachidonyl trifluoromethyl ketone, a potent inhibitor of 85 kDa phospholipase A2, blocks production of arachidonate and 12-hydroxyeicosatetraenoic acid by calcium ionophore-challanged platelets. J. Biol. Chem. 1994;269:15619–15624. [PubMed] [Google Scholar]
- ROBINSON M.J., COBB M.H. Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol. 1997;9:180–186. doi: 10.1016/s0955-0674(97)80061-0. [DOI] [PubMed] [Google Scholar]
- SA G., MURUGESAN G., JAYE M., IVASHCHENKO Y., FOX P.L. Activation of cytosolic phospholipase A2 by basic fibroblast growth factor via a p42 mitogen-activated protein kinase-dependent phosphorylation pathway in endothelial cells. J. Biol. Chem. 1995;270:2360–2366. doi: 10.1074/jbc.270.5.2360. [DOI] [PubMed] [Google Scholar]
- SATO F., MATSUKAWA Y., MATSUMOTO K., NISHINO H., SAKAI T. Apigenin induces morphological differentiation and G2-M arrest in rat neuronal cells. Biochem. Biophys. Res. Commun. 1994;204:578–584. doi: 10.1006/bbrc.1994.2498. [DOI] [PubMed] [Google Scholar]
- SEGER R., AHN N.G., BOULTON T.G., YANCOPOULOS G.D., PANAYOTATOS N., RADZIEJEWSKA E., ERICSSON L., BRATLIEN R.L., COBB M.H., KREBS E.G. Microtubule-associated protein 2 kinases, ERK1 and ERK2, undergo autophosphorylation on both tyrosine and threonine residues: implication for their mechanism of activation. Proc. Natl. Acad. Sci. U.S.A. 1991;88:6142–6146. doi: 10.1073/pnas.88.14.6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEGER R., KREBS E.G. The MAPK signaling cascade. FASEB J. 1995;9:726–735. [PubMed] [Google Scholar]
- SEGER R., SEGER D., RESZKA A.A., MUNAR E.S., ELDAR-FINKELMAN H., DOBROWOLSKA G., JENSEN A.M., CAMPBELL J.S., FISHER E.H., KREBS E.G. Overexpression of mitogen-activated protein kinase kinase (MAPKK) and its mutants in NIH 3T3 cells. J. Biol.Chem. 1994;269:25699–25709. [PubMed] [Google Scholar]
- SMITH W.L., MARNETT L.J. Prostaglandin endoperoxidase synthase: Structure and catalysis. Biochim. Biophys. Acta. 1991;1083:1–17. doi: 10.1016/0005-2760(91)90119-3. [DOI] [PubMed] [Google Scholar]
- SPAARGAREN M., WISSINK S., DEFIZE L.H.K., DE LAAT S.W., BOONSTRA J. Characterization and identification of an epidermal growth factor activated phospholipase A2. Biochem. J. 1992;287:37–43. doi: 10.1042/bj2870037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STREET I.P., LIN H-K., LALIBERTE R., GHOMASCHI F., WANG Z., PERRIER H., TREMBLAY N.M., HUANG Z., WEECH P.K., GELB M.H. Slow and tight-binding inhibitor of the 85 kDa human phospholipase A2. Biochemistry. 1993;32:5935–5940. doi: 10.1021/bi00074a003. [DOI] [PubMed] [Google Scholar]
- SURETTE M.E., FONTEH A.N., BERNATCHEL C., CHILTON F.H. Pertabutions in the control of cellular arachidonate levels block cell growth and induce apoptosis in HL-60 cells. Carcinogenesis. 1999;20:757–763. doi: 10.1093/carcin/20.5.757. [DOI] [PubMed] [Google Scholar]
- TOKUMOTO H., CROXTALL J.D., CHOUDHURY Q., FLOWER R.J. Phospholipase A2-induced stimulation of A549 lung adenocarcinoma cell line proliferation. Biochim. Biophys. Acta. 1993;1164:236–242. doi: 10.1016/0005-2760(93)90246-6. [DOI] [PubMed] [Google Scholar]
- VANE J.R. The mode of action of asprin-like drugs. Agents Actions. 1978;8:430–431. doi: 10.1007/BF01968671. [DOI] [PubMed] [Google Scholar]
- WISSING D., MOURITZEN H., EGEBLAD M., POIRIER G.G., JAATTELA M. Involvement of caspase-dependent activation of cytosolic phospholipase A2 in tumor necrosis factor induced apoptosis. Proc. Natl. Acad. Sci. U.S.A. 1997;94:5073–5077. doi: 10.1073/pnas.94.10.5073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XING M., ZHAO J., MATTERA R. Regulation of phospholipase A2 activity in undifferentiated and neutrophil-like HL-60 cells. J. Biol. Chem. 1994;269:3117–3124. [PubMed] [Google Scholar]
- ZHANG C., BAUMGARTNER R.A., YAMADAS K., BEAVEN M.A. Mitogen-activated Protein (MAP) Kinase Regulates Production of Tumor Necrosis Factor-α and Release of Arachidonic Acid in Mast Cells. J. Biol. Chem. 1997;272:13397–13402. doi: 10.1074/jbc.272.20.13397. [DOI] [PubMed] [Google Scholar]