

Abstract
The ability of 19 agonists to elevate Ca2+ and inhibit forskolin-induced cyclic AMP elevation through α2A-adrenoceptors in HEL 92.1.7 cells was investigated. Ligands of catecholamine-like- (five), imidazoline- (nine) and non-catecholamine-non-imidazoline-type (five) were included.
The relative maximum responses were similar in both assays. Five ligands were full or nearly full agonists, six produced 20 – 70% of the response to a full agonist and the remaining eight gave lower responses (<20%) so that their potencies were difficult to evaluate.
Marked differences in the potencies of the agonists with respect to the two measured responses were seen. The catecholamines were several times less potent in decreasing cyclic AMP than in increasing Ca2+, whereas the other, both imidazoline and ox-/thiazoloazepine ligands, were several times more potent with respect to the former than the latter response. For instance, UK14,304 was more potent than adrenaline with respect to the cyclic AMP response but less potent than adrenaline with respect to the Ca2+ response.
All the responses were sensitive to pertussis toxin-pretreatment. Also the possible role of PLA2, β-adrenoceptors or ligand transport or metabolism as a source of error could be excluded. The results suggest that the active receptor states produced by catecholamines and the other agonists are markedly different and therefore have different abilities to activate different signalling pathways.
Keywords: α2-adrenoceptors, Ca2+, cyclic AMP, HEL 92.1.7 cells, agonist trafficking, G protein, Gi/o, fura-2
Introduction
G protein-coupled receptors are able to couple to a multitude of signal transduction pathways. Some diversity arises from the specific coupling of certain G proteins to certain responses, e.g. Gαq to stimulation of phospholipase Cβ, Gαs to stimulation of adenylyl cyclase and Gαi to inhibition of adenylyl cyclase. Additional diversity is dependent on the coupling of the same G protein to several responses either through the same subunit (e.g. coupling of Gβγ to stimulation of phospholipase Cβ and inhibition of Ca2+ channels) or through the different subunits released in equal amounts by the G protein activation (e.g. coupling of Gαi to inhibition of adenylyl cyclase and Gβγ to stimulation of phospholipase Cβ). The prevailing division of G protein-coupled receptors to Gq-, Gs- and Gi/o-coupled receptors is based on the signalling through G protein α-subunits, though great differences in the ability of different G protein-coupled receptors to couple to different subfamilies of G proteins have become apparent (reviewed in Gudermann et al., 1996): for instance, human thyrotropin receptor interacts with at least 10 different G proteins from all four subfamilies (Gs, Gi, Gq, G12) (Laugwitz et al., 1996) whereas for most other receptors the spectrum is much more limited. There are several subtypes of G proteins in all the subfamilies, e.g. in the most diverse Gi/o-subfamily nine α-subunits, six of which are more or less ubiquitous and three tissue specific, have been cloned. Additional functional diversity is created by the coupling of one Gα-subunit to different Gβγ subunits. The ability of the receptors to activate different members of this G protein subfamily has recently received increasing attention. Direct and indirect evidence indicates that α2A/D-adrenoceptors can couple to at least Gi2, Gi3 and Go1 (Simonds et al., 1989; Gerhardt & Neubig, 1991; McClue et al., 1992; Remaury et al., 1993; Yang & Lanier, 1999) whereas α2C-adrenoceptors may couple to Gi1, Go1 or Go2 (Duzic et al., 1992). When reconstituted in phospholipid vesicles together with G proteins, both α2A and α2C activated members of the Gi/o subfamily with the efficacy order of Gi3>Gi1⩾Gi2>Go1 (Kurose et al., 1991).
It has previously been observed that different agonists can couple to separate signalling pathways with different efficacy via the same receptors (reviewed in Kenakin, 1995). Such behaviour can often be explained by differences in stoichiometries at the receptor-G protein or G protein-effector level. In such cases the order of efficacy for the different ligands should be the same irrespective of the response measured, and only dictated by differences in the respective ligands' intrinsic efficacy (Kenakin, 1995). However, it has sometimes been observed that the order of efficacy between ligands is reversed for different responses (Boddeke, 1991; Krumins & Barber, 1997; Berg et al., 1998). This cannot be explained by the reasoning above. On the contrary, it has been taken as an indication that different agonists can either induce (induction theory) or stabilize (selection theory) different active receptor conformations (Kenakin, 1995; Leff et al., 1997). If these enriched different active conformations will preferentially activate specific G proteins, a distinct activation of different signalling pathways by different agonists will be observed. This phenomenon has been termed ‘agonist trafficking' (of receptor signals) (Kenakin, 1995).
The natural ligands for α2-adrenoceptors are the catecholamines noradrenaline and adrenaline. They usually behave as full agonists, but their potencies (EC50) are often lower than the potencies of some common synthetic imidazolines, like UK14,304 (5-bromo-N-[4,5-dihydro-1H-imidazol-2-yl]-6-quinoxalinamine), D-medetomidine ([+]-[S]-4-[1-(2,3-dimethylphenyl)ethyl]-1H-imidazole HCl) and clonidine (2-[2,6-dichloroaniline]-2-imidazoline HCl). This has been seen with respect to the most often measured response, inhibition of stimulated cyclic AMP production (Jones et al., 1987; Voigt et al., 1991; Jansson et al., 1994; Pohjanoksa et al., 1997), but also with respect to GTPγS binding as well as cyclic AMP and Ca2+ elevation (Jansson et al., 1995; Holmberg et al., 1998; Jasper et al., 1998; Kukkonen et al., 1998). The reason for this may simply be the higher binding affinity of the imidazolines (Jansson et al., 1994; 1995; Pohjanoksa et al., 1997; Jasper et al., 1998). The other groups of synthetic ligands include dichlorophenylguanidines guanabenz (1-[2,6-dichlorobenzylideneamino]guanidine) and guanfacine (N-[aminoiminomethyl]-2,6-dichlorobenzenacetamide), ox-/thiazoloazepines B-HT 920 (2-amino-6-allyl-5,6,7,8-tetrahydro-4H-thiazolo-[5,4-d]-azepine di-HCl) and B-HT 933 (2-amino-6-ethyl-4,5,7,8-tetrahydro-6H-oxazolo-[5,4-d]-azepine di-HCl) and the thiazine xylazine (N-[2,6-dimethylphenyl]-5,6-dihydro-4H-1,3-thiazin-2-amine HCl), which all usually behave as partial agonists, though the dichlorophenylguanidines can be very potent (Jasper et al., 1998).
The HEL 92.1.7 human erythroleukaemia cell line expresses α2A-adrenoceptors which elevate intracellular free [Ca2+] ([Ca2+]i) (Michel et al., 1989; Musgrave & Seifert, 1995; Kukkonen et al., 1997; Jansson et al., 1998) and inhibit stimulated adenylyl cyclase activity (McKernan et al., 1987; Jansson et al., 1998) through Gi/o-type G proteins. In this study we address the issue of possible differential signalling through different active receptor states produced by different agonists. This is performed by measurement of Ca2+ and cyclic AMP responses to 17 different α2-adrenoceptor agonists belonging to five different chemical classes.
Methods
Cell culture
HEL 92.1.7 cells, obtained from the ATCC (Rockville, MD, U.S.A), were grown in suspension culture in RPMI-1640 medium supplemented with 7.5% heat-inactivated foetal calf serum (FCS; Gibco, Paisley, U.K.), 100 u ml−1 penicillin (Sigma, St Louis, MO, U.S.A.) and 80 u ml−1 streptomycin (Sigma) as described previously (Kukkonen et al., 1997). Cells were harvested by centrifugation for 5 min at 250×g. When the effect of pertussis toxin-pretreatment was investigated, the cells were treated with 100 ng ml−1 pertussis toxin for 24 h. As a control for cell viability, the lack of the inhibitory effect of pertussis toxin-pretreatment on the Ca2+ elevation induced by P2Y-purinoceptor stimulation with 10 μM ATP was used.
Drugs
[3H]-Adenine and [14C]-cyclic AMP were from Amersham (Buckinghamshire, U.K.). (−)-Adrenaline, clonidine, desipramine (10,11-dihydro-N-methyl-5H-dibenz[b,f]azepine-5-propanamine), guanabenz, guanfacine, 3-isobutyl-1-methylxanthine, (−)-isoproterenol ([−]-1-[3′,4′-dihydroxyphenyl]-2-isopropylaminoethanol), naphazoline (4,5-dihydro-2-[1-naphthalenylmethyl]-1H-imidazole), nialamide (N-isonicotinoyl-N′-[β-(N-benzylcarboxamido)ethyl]hydrazine), (−)-noradrenaline, (±)-p-octopamine, oxymetazoline (3-[(4,5-di-hydro-1H-imidazol-2-yl-)-methyl]-6-[1,1-dimethylethyl]-2,4-dimethylphenol HCl), pertussis toxin, phentolamine, (S)-(−)-propranolol ([S]-1-[isopropylamino]-3-[1-naphthyloxy]-2-propanol HCl), quinacrine (6-chloro-9-[(4-diethylamino)-1-methylbutyl]amino-2-methoxy-acridine), tizanidine (5-chloro-4-[2-imidazolin-2-yl-amino]-2,1,3-benzothiadiazole) and xylazine were from Sigma. α-Methyl-noradrenaline, B-HT 920, B-HT 933, dopamine, p-I-clonidine (2-[(2,6-dichloro-4-iodophenyl)imino]imidazoline HCl), rauwolscine and UK14,304 were from RBI (Natick, MA, U.S.A.) and digitonin from Merck AG (Darmstadt, Germany). Fura-2 acetoxymethylester was from Molecular Probes (Eugene, OR, U.S.A.). D-Medetomidine and detomidine (4[5]-[2,3-dimethylbenzyl]imidazole HCl) were from Orion-Corporation Orion-Pharma (Turku, Finland). Moxonidine (4-chloro-N-[4,5-dihydro-1H-imidazol-2-yl]-6-methoxy-2-methyl-5-pyrimidinamine) and RX821002 (2-[2-methoxy-1,4-benzodioxan-2-yl]-2-imidazoline [methoxy idazoxan]) were kind gifts from Dr Birgit Brueggemann (Beiersdorf-Lily GmbH, Hamburg, Germany) and Dr Corinne Gelhay (Pierre Fabre, Castres, France), respectively.
Media
The TES buffered medium (TBM) consisted of (mM): NaCl 137, KCl 5, CaCl2 1, glucose 10, MgCl2 1.2, KH2PO4 0.44, NaHCO3 4.2 and 2-([2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino) ethane sulfonic acid (TES) 20 adjusted to pH 7.4 with NaOH.
Measurement of [Ca2+]i
The fluorescent Ca2+-indicator fura-2 was used to monitor changes in [Ca2+]i as described previously for HEL 92.1.7 cells (Kukkonen et al., 1997; Jansson et al., 1998) and for CHO cells (Kukkonen et al., 1998). Fluorescence measurements were performed with a Hitachi F-4000 fluorescence spectrophotometer at the wavelengths 340 nm (excitation) and 505 nm (emission), with a Hitachi F-2000 fluorescence spectrophotometer at the wavelengths 340/380 nm (excitation) and 505 nm (emission) or with a PTI QuantaMaster QM1 fluorescence spectrophotometer at the wavelengths 340/360/380 nm (excitation) and 505 nm (emission). The dye responses were calibrated by sequential addition of digitonin (60 μg ml−1) and EGTA (10 mM) at the end of the experiment to obtain the maximum (Fmax) and minimum (Fmin) fluorescence values, respectively. The extracellular fura-2 concentration was measured by first adding EGTA and then digitonin. The intracellular free Ca2+ concentration was calculated from the fluorescence values (F) obtained at 340 nm as in Kukkonen et al. (1997) or from the 340/360 nm data.
Measurement of intracellular cyclic AMP
The growth medium of confluent cultures was replaced with fresh medium supplemented with 2.5 μCi ml−1 of [3H]-adenine. After incubation for 2 h the cells were collected, spun down and resuspended in TBM supplemented with 0.5 mM 3-isobutyl-1-methylxanthine (a phosphodiesterase inhibitor) and incubated for 10 min at 37°C. When the effect of α2- or β-adrenoceptor antagonists, the phospholipase A2 inhibitor quinacrine, the monoamine oxidase inhibitor nialamide and catecholamine reuptake inhibitor desipramine were tested, they were preincubated together with 3-isobutyl-1-methylxanthine. The reactions were started by pipetting the cell suspension (≈amp;8×106 cells ml−1) in 96-well plates (Nunc 269620, Nunc A/S, Roskilde, Denmark). The compounds to be investigated (α2-adrenoceptor agonists and forskolin) had previously been diluted in TBM and dispensed in a volume of 100 μl; this, together with 50 μl of cell suspension per well, gave a total reaction volume of 150 μl. The reactions were allowed to proceed for 10 min at 37°C, after which they were terminated by centrifugation at 1100×g for 1 min, rapid decanting of the supernatants and addition of 200 μl ice-cold 0.33 M perchloric acid per well. The plates were frozen down to −20°C, thawed, and the cell debris were spun down (1100×g, 15 min). The extent of conversion of [3H]-ATP to [3H]-cyclic AMP was determined by sequential Dowex/alumina chromatography of the supernatants. [14C]-cyclic AMP tracer in 0.75 ml 0.33 M perchloric acid (about 1000 c.p.m.) was added into each Dowex column together with the samples. Radioactivity was determined by liquid scintillation counting (Wallac 1410, Wallac, Turku, Finland) in Optiphase HiSafe 3. Conversion to [3H]-cyclic AMP was calculated as a percentage of total eluted [3H]-ATP and was normalized to the recovery of [14C]-cyclic AMP tracer (generally 70%).
Data analysis
Values are given as mean±s.e.mean; n refers to the number of batches of cells on which the measurements were performed. Non-linear curve-fitting was performed using SigmaPlot for Windows 4.00 (Jandel Scientific, Corte Madera, CA, U.S.A.). The difference in the ability of each agonist to activate Ca2+ and cyclic AMP response was evaluated by calculating the group averages of EC50-Ca2+/EC50-cyclic AMP and performing between-group comparisons (catecholamines versus imidazolines and ox-/thiazoloazepines) using the Student's two-tailed non-paired t-test.
Results
Ca2+
The basal intracellular Ca2+ concentration ([Ca2+]i) was 97±7 nM (n=25). As also reported before, α2-adrenoceptor activation resulted in Ca2+ elevations that consisted of a transient release and a sustained influx (data not shown; see also Michel et al., 1989; Jansson et al., 1998). Nineteen agonists were tested for their ability to induce Ca2+ elevation, and for 14 of them, a good estimate of the EC50 for the maximum ‘spike' response was obtained (Table 1; Figure 1). Adrenaline was clearly the most efficacious of the agonists with an approximately 4.6 fold elevation of basal [Ca2+]i. HEL 92.1.7 cells have previously been reported to express β-adrenoceptors with high affinity for the β-antagonist propranolol (Michel et al., 1989). These receptors did not seem to interfere with the α2A-mediated Ca2+ elevation since all the agonist-mediated Ca2+ elevations were abolished with 10 μM RX821002, 10 μM rauwolscine or by pertussis toxin-pretreatment. Propranolol (10 or 100 μM) caused a slight right shift in the concentration-response curves of the ligands, probably due to its affinity for α2A-adrenoceptors (Gerhardt et al., 1990; see also below). We also tested the effect of the phosphodiesterase inhibitor 3-isobutyl-1-methylxanthine in the Ca2+ assay: 150 μM 3-isobutyl-1-methylxanthine had no effect on the EC50 value or the maximum response.
Table 1.
The maximum responses (Emax) and EC50 values for Ca2+ elevation and cyclic AMP decrease elicited by different α2-adrenoceptor ligands
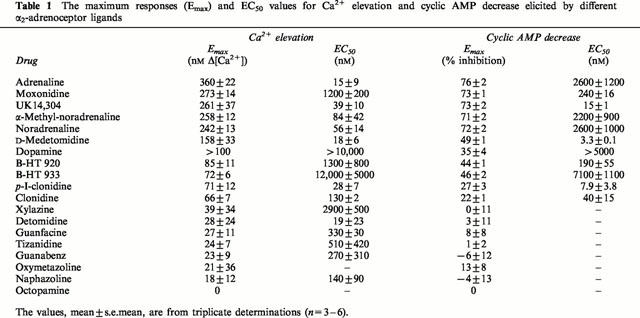
Figure 1.
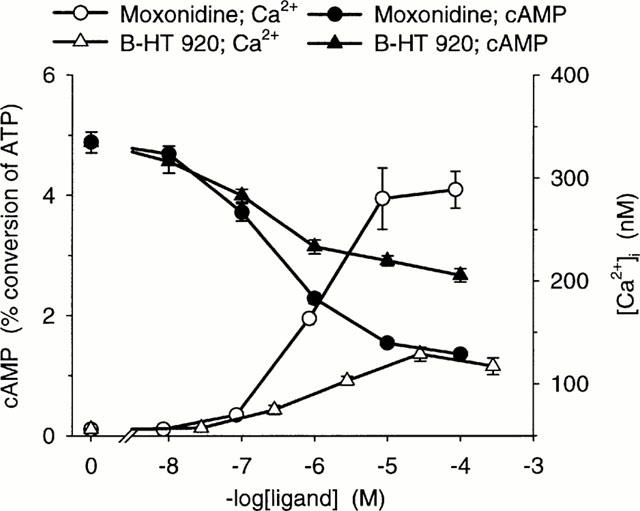
Ca2+ and cyclic AMP responses to two ligands, moxonidine (an imidazoline) and B-HT 920 (an oxazoloazepine), as mean±s.e.mean of three determinations in triplicate.
Cyclic AMP
The basal conversion of [3H]-ATP to [3H]-cyclic AMP was 0.17±0.04% (n=3). Forskolin (10 μM) increased the basal cyclic AMP production to 5.16±0.31% (n=3). The same 19 α2-adrenoceptor agonists were tested for the inhibition of forskolin-induced cyclic AMP production. Ten of them gave responses that were large enough to allow determination of the EC50 (Table 1; Figure 1). The most efficacious agonist was once again adrenaline, although this response was only slightly higher than the response to the other strong agonists, noradrenaline, α-methyl-noradrenaline, moxonidine and UK14,304. There was a good correlation with the maximum responses measured in the Ca2+ assay (Table 1; Figure 2A). On the contrary, the EC50 values were clearly different. When EC50 values for the Ca2+ and cyclic AMP responses were compared graphically (Figure 2B) the agonists were clearly divided into two groups. Linear regression of the data from catecholamine agonists, which are consistently more potent for inducing Ca2+ elevation compared to cyclic AMP inhibition, yields a potency ratio (Ca2+ versus cyclic AMP) of 59, whereas the corresponding ratio for imidazoline and ox-/thiazoloazepine agonists, which are more potent against the cyclic AMP response, is 0.27. The difference in EC50-Calcium/EC50-cyclic AMP between the groups is highly significant (P<0.01). Thus, low concentrations of catecholamines couple exclusively to Ca2+ elevation whereas low concentrations of imidazolines and ox-/thiazolazepines preferentially couple to inhibition of cyclic AMP production.
Figure 2.
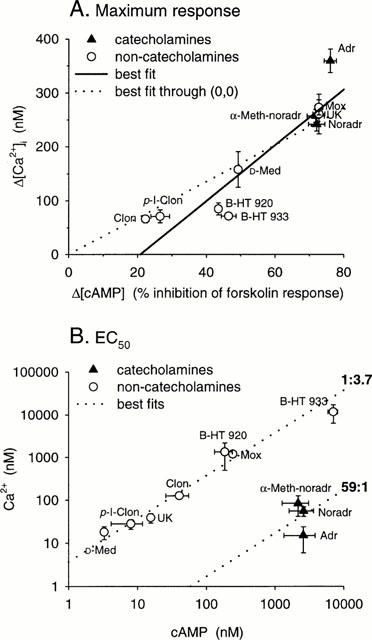
Correlation of the abilities of α2-adrenoceptor agonists to induce cyclic AMP decrease and Ca2+ elevation. (A) The maximum responses; (B) the EC50 values. Also drawn are the best linear fit (solid line) and the best linear fit forced to origo (dotted line) in (A) and the relationships 1 : 3.7 and 59 : 1 in (B). The drawn relationships of indicated value are based on the average of logarithms of EC50-Ca2+/EC50-cyclic AMP for the corresponding ligand groups. The overall r (calculated for the logarithmic data) for (A) is 0.903 and for (B) 0.437. The overall EC50-Ca2+/EC50-cyclic AMP in (B) is 2.84±0.79. Catecholamines alone have EC50-Ca2+/EC50-cyclic AMP of 0.0219± 0.0095 and the imidazolines and ox-/thiazoloazepines 4.05±0.72 (significance for difference between groups: P<0.01).
All the responses were inhibited with pertussis toxin-treatment, as they also were inhibited by 10 μM RX 821002. β-adrenoceptors could once again be excluded: 10 μM propranolol, which should have a prominent effect on the previously reported high affinity β-adrenoceptors in HEL cells (Michel et al., 1989), caused a similar dextral shift in the concentration-response curves (average of 10 agonists 1.6±0.6 times) to that in the Ca2+ measurements. The Ki calculated from this, 16 μM, is in the agreement with the Ki of 23±7 μM for propranolol reported for human α2A-adrenoceptors (Gerhardt et al., 1990). Thus no β-adrenoceptors interfered with the α2A-adrenoceptor-mediated responses in the HEL cells used in this study. No response was obtained to isoproterenol (β-adrenoceptor agonist), suggesting that functional β-adrenoceptors are scarce in the HEL cells used in this study. Catecholamine breakdown (monoamine oxidase) and uptake inhibitors nialamide and desipramine (10 μM each), respectively, did not have any effect on the concentration-response curves. To remove possible phospholipase A2-dependent generation of other messengers we included 150 μM of the cell permeable phospholipase A2 inhibitor quinacrine (see, for example, Fraser et al., 1989) in the assay. This treatment did not change the conclusion of agonist trafficking of α2-adrenoceptor responses.
Discussion
The results of this study show a difference in the ability of chemically different α2-adrenoceptor agonists to activate two signalling pathways. The catecholamine ligands are more potent in the Ca2+ assay than in the cyclic AMP assay, whereas the imidazoline and ox-/thiazoloazepine ligands are more potent in the cyclic AMP than in the Ca2+ assay. Unfortunately, the catecholamine-like ligands possessing a lower number of hydroxyl groups (dopamine and octopamine) did not give any useful information due to low potency or lack of efficacy, respectively. Altogether, there is a 180 fold difference between the groups in the relative abilities to cause these responses. The greater ability of non-catecholamine ligands to reduce cyclic AMP is also seen in the markedly higher efficacy of the weak partial ox-/thiazoloazepine ligands B-HT 920 and B-HT 933 with respect to the cyclic AMP response, though the same is not seen with imidazolines (clonidine, p-I-clonidine, D-medetomidine, UK14,304, moxonidine) which follow rather well the 1 : 1 relationship. This suggests that there might be differences even between imidazolines and ox-/thiazoloazepines.
To ensure that the results obtained corresponded to a true agonist trafficking of receptor responses, we performed a series of control experiments. In these experiments we could show that neither the previously reported β-adrenoceptors (Michel et al., 1989) nor any putative catecholamine uptake or breakdown mechanisms affected the responses measured. We also excluded the possibility that the activation of an intracellular system, phospholipase A2, could be causing the observed effect by using a phospholipase A2 inhibitor, quinacrine. Nor was any agonist concentration-curve biphasic or shallow, which would have been indicative of differential coupling to several pathways.
The difference in the efficiency of different agonists towards Ca2+ and cyclic AMP responses shows that different agonists can preferentially activate particular signalling pathways. The difference in the chemical structure of the agonists suggests that this is caused by a stabilization/induction of partially different receptor conformations.
Altogether, the results of the present study show that chemically different ligands can activate different signalling pathways with different efficacy. Our present results, together with our previous results as well as results from many other groups with HEL 92.1.7 cells, indicate that all the responses are mediated (i) by α2- and not by other adrenoceptors or any other receptors and (ii) by only the α2A-subtype (McKernan et al., 1987; Michel et al., 1989; Musgrave & Seifert, 1995; Jansson et al., 1998). Therefore, our results demonstrate that ligand-specific trafficking of signals is seen with this receptor. The situation is most obvious with ligands like adrenaline and clonidine, the former of which is 8.7 times more potent than the latter with respect to the Ca2+ elevation, whereas clonidine decreases cyclic AMP 65 times more potently than adrenaline. The obvious hypothesis is that the diverging signalling for the chemically different ligands is caused by enrichment of different active receptor states, which have different ability to activate different G proteins. The possibilities for divergence are schematically illustrated in Figure 3. The receptor (R) exists in two interconvertible active conformations (R* and R*), which can be differently activated by chemically different ligands. These active receptor conformations activate signal transducers (T1 and T2; e.g. G proteins), the activation of which finally leads to activation of separate second messenger-generating pathways (E1 and E2). The agonist trafficking of the receptor signals is accomplished at the different levels of signal transduction cascade: agonist – receptor-state-coupling (A), receptor-state – transducer-coupling (B) or transducer – second-messenger-cascade-coupling (C). All of these schemes are possible in the light of the results. The nature of the signal transducers cannot be directly resolved. However, a major part could be played by the G proteins. The previously mentioned results with the partial agonists D-medetomidine and B-HT 920 suggest that not only are catecholamines and synthetic ligands trafficking the signals in a different way, but that there may also be differences in the trafficking properties between the chemically different synthetic ligands. This is also indicated in previous studies. Two imidazoline ligands, UK14,304 and p-I-clonidine, may activate Gαi2 and Gαi3 to a different extent via α2A/D (Gerhardt & Neubig, 1991); p-I-clonidine is also usually found to be a partial agonist whereas UK 14,304 is a full agonist for α2A/D (Gerhardt et al., 1990; Jansson et al., 1998). Noradrenaline, UK14,304, oxymetazoline and clonidine are full agonists with respect to the Gαo1 but only the two first are full agonists with respect to the Gαi1 activation (Yang & Lanier, 1999). Thus the puzzling results in the present study, where responses putatively mediated by a single G protein subfamily (Gi/o) show ligand-dependent trafficking, may be explained by differential activation of different members of this family, playing different roles in intracellular signal transduction. Previous results have shown that α2A-adrenoceptors can couple to two different G protein families, namely Gi/o and Gs (Eason et al., 1992; Pepperl & Regan, 1993). Even agonist trafficking has been shown, although no general conclusions of the activation profile of the ligands of different chemical classes on these responses can be drawn (Eason et al., 1994; Airriess et al., 1997; Brink et al., 2000). This shows that α2A-adrenoceptors are able to adopt different conformations to activate different G proteins; in some cases distinct intracellular domains of the receptor have been shown to be responsible for activation of different G proteins (Eason & Liggett, 1996).
Figure 3.
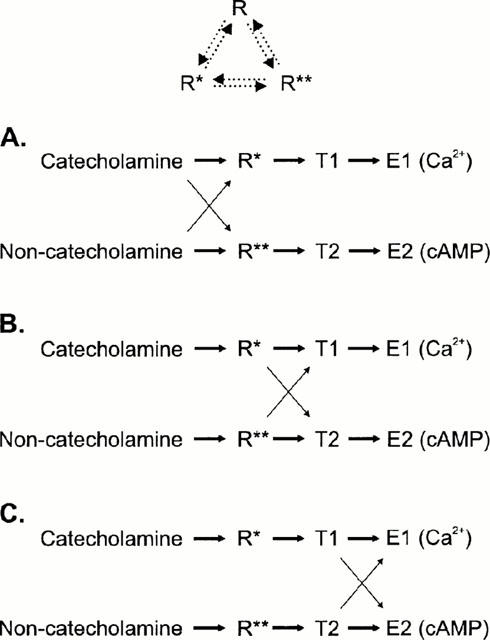
A schematic representation of the hypothesis with three somewhat different models, which are all possible based on the results. R is the inactive and R* and R** the two active receptor conformations, T1 and T2 are two signal transducers (e.g. G proteins) and E1 and E2 are the separate signal cascades which finally lead to a generation of a second messenger response (Ca2+ and cyclic AMP, respectively). The solid arrows indicate the direction of the response (the thicker the arrow, the stronger the coupling) and the dotted arrows in the top scheme the interconvertibility of the receptor conformations. In (A) a single receptor conformation only couples to a single transducer, which then couples to a single second messenger cascade. Catecholamines bind more strongly to R* whereas the other ligands bind more strongly to R** leading to a stronger activation of E1 (Ca2+) or E2 (cyclic AMP) response, respectively. In (B) the agonist trafficking of the receptor signals is accomplished at the level of receptor-transducer-coupling: the catecholamine-activated receptor (R*) is more effective in coupling to T1 (and thus to E1) whereas the non-catecholamine-activated receptor (R*) is more effective in its coupling to T2 (and thus to E2). In the third scheme, (C), a single receptor conformation only couples to a single transducer (as in A), which then couples to both second messenger cascades. The agonist trafficking of the receptor signals is brought about by the different ability of transducers to activate different cascades: the catecholamine-activated transducer T1 is more effective in coupling to E1 whereas the non-catecholamine-activated transducer T2 is more effective in its coupling to E2. The schemes (B) and (C) are essentially similar to the scheme presented in Berg et al. (1998), though somewhat more complex.
The significance of expression of several members of the Gi/o subfamily is rather unclear. In particular, there has been much speculation on the role of Go. Gαo does not inhibit adenylyl cyclase (except for type 1), whereas Gαi1, Gαi2 and Gαi3 do (Taussig et al., 1994). Some effects on ion channels seem to rely on Go (Diverse-Pierluissi et al., 1995; Valenzuela et al., 1997) and novel forms of regulation of intracellular signalling have been suggested (Hajdu-Cronin et al., 1999; Jordan et al., 1999; Posner et al., 1999). Also, different Gαi subtypes may differ in signalling. In Rat1 cells the α2A-mediated cyclic AMP decrease is completely dependent on Gαi2 although both Gαi2 and Gαi3 are activated by the receptor (McClue et al., 1992). Furthermore, the regulation of different members of the Gi/o family by RGS proteins may be different (Diverse-Pierluissi et al., 1999; Posner et al., 1999; Cavalli et al., 2000). Differential Gβγ coupling of these two subtypes has been suggested by, for example, Diverse-Pierluissi et al. (1995).
The role of Ca2+ as an intracellular mediator regulated by α2-adrenoceptors is at the moment unclear. It contradicts the traditional role of α2-adrenoceptors as inhibitory receptors. Under normal circumstances, α2-adrenoceptor mediated Ca2+ elevations have been measured in smooth muscle preparations (Aburto et al., 1993; Lepretre & Mironneau, 1994). Otherwise, α2-receptor signalling may be directed towards Ca2+ elevation under special conditions. For instance, we have shown that treatment of rat cerebral astrocytes with cyclic AMP, which mimics the ‘reactive astrocyte'-phenotype, increases the α2A-adrenoceptor expression and α2-mediated Ca2+ elevations (Enkvist et al., 1996). More recently, our results have suggested that the α2-adrenoceptor response can be redirected from the inhibitory, cyclic AMP-decreasing response to a stimulatory response, Ca2+ elevation, by the Gq-coupled P2Y-purinoceptors (Åkerman et al., 1998). α2-adrenoceptors may thus be switched to stimulatory responses as part of normal cellular signalling but putatively also under pathological conditions. In this context the observed difference in the ligands' ability to activate the inhibitory (cyclic AMP decrease) and stimulatory responses (Ca2+ increase) may also be interesting from the therapeutic point of view.
The pharmacotherapeutic aim of the receptor research is usually to obtain a ligand with a good receptor subtype selectivity. This is evidently not always enough: for instance, in the case of α2-adrenoceptors, the α2A subtype is responsible for most of the α2-receptors' very diverse biological functions. In this study we are suggesting that additional selectivity for certain responses can be obtained provided (i) that they are mediated by distinct pathways (i.e. G proteins) and (ii) that these distinct pathways can be activated by pathway-selective ligands. Selective activation of signalling cascades is a new therapeutic principle in line with the other novel ideas of pharmacological intervention of the intracellular signalling pathways.
Acknowledgments
This study was funded by The Lars Hierta Foundation, The Åke Wiberg Foundation, The Magnus Ehrnrooth Foundation, Oy Veikkaus Ab, The Medical Research Council of Sweden and The Cancer Research Fund of Sweden. We acknowledge Drs Johnny Näsman and Tomas Holmqvist for critical comments during the manuscript preparation, and Drs Birgit Brueggemann (Beiersdorf-Lily GmbH, Hamburg, Germany) and Corinne Gelhay (Pierre Fabre, Castres, France) for the supply of moxonidine and RX821002, respectively.
Abbreviations
- B-HT 920
2-amino-6-allyl-5,6,7,8-tetrahydro-4H-thiazolo-(5,4-d)-azepine
- B-HT 933
2-amino-6-ethyl-4,5,7,8-te-trahydro-6H-oxazolo-[5,4-d]-azepine
- [Ca2+]i
intracellular Ca2+ concentration
- Δ[Ca2+]i
change in intracellular Ca2+ concentration
- clonidine
2-(2,6-dichloroaniline)-2-imidazoline
- p-I-clonidine
2-([2,6-dichloro-4-iodophenyl]imino)imidazoline
- desipramine
10,11-dihydro-N-methyl-5H-dibenz(b,f)azepine-5-propanamine
- detomidine
4(5)-(2,3-dimethylbenzyl)imidazole
- FCS
foetal calf serum
- guanabenz
1-(2,6-dichlorobenzylideneamino)guanidine
- guanfacine
N-(aminoiminomethyl)-2,6-dichlorobenzenacetamide
- (−)-isoproterenol
(−)-1-(3′,4′-dihydroxyphenyl)-2-isopropylaminoethanol
- D-medetomidine
(+)-(S)-4-(1-[2,3-dimethylphenyl]ethyl)-1H-imidazole
- moxonidine
4-chloro-N-(4,5-dihydro-1H-imidazol-2-yl)-6-methoxy-2-methyl-5-pyrimidinamine
- naphazoline
4,5-dihydro-2-(1-naphthalenylmethyl)-1H-imidazole
- nialamide
N-isonicotinoyl-N′-(β-[N-benzylcarboxamido]ethyl)hydrazine
- oxymetazoline
(3-[(4,5-dihydro-1H-imidazol-2-yl-)methyl]-6-[1,1-dimethylethyl]-2,4-dimethylphenol
- (S)-(−)-propranolol
(S)-1-(isopropylamino)-3-(1-naphthyloxy)-2-propanol
- quinacrine
6-chloro-9-([4-diethylamino]-1-methylbutyl)amino-2-methoxy-acridine
- RX821002
2-(2-methoxy-1,4-benzodioxan-2-yl)-2-imidazoline
- TBM
TES buffered medium
- TES
2-([2-hydroxy-1,1-bis(hydroxymethyl)ethyl]amino) ethane sulphonic acid
- tizanidine
5-chloro-4-(2-imidazolin-2-yl-amino)-2,1,3-benzothiadiazole
- UK14,304
5-bromo-N-(4,5-dihydro-1H-imidazol-2-yl)-6-quinoxalinamine
- xylazine
N-(2,6-dimethylphenyl)-5,6-dihydro-4H-1,3-thiazin-2-amine
References
- ABURTO T.K., LAJOIE C., MORGAN K.G. Mechanisms of signal transduction during alpha 2-adrenergic receptor-mediated contraction of vascular smooth muscle. Circ. Res. 1993;72:778–785. doi: 10.1161/01.res.72.4.778. [DOI] [PubMed] [Google Scholar]
- AIRRIESS C.N., RUDLING J.E., MIDGLEY J.M., EVANS P.D. Selective inhibition of adenylyl cyclase by octopamine via a human cloned alpha 2A-adrenoceptor. Br. J. Pharmacol. 1997;122:191–198. doi: 10.1038/sj.bjp.0701348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ÅKERMAN K.E.O., NÄSMAN J., LUND P.E., SHARIATMADARI R., KUKKONEN J.P. Endogenous extracellular purine nucleotides redirect alpha2-adrenoceptor signaling. FEBS Lett. 1998;430:209–212. doi: 10.1016/s0014-5793(98)00664-4. [DOI] [PubMed] [Google Scholar]
- BERG K.A., MAAYANI S., GOLDFARB J., SCARMELLINI C., LEFF P., CLARKE W.P. Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol. Pharmacol. 1998;54:94–104. [PubMed] [Google Scholar]
- BODDEKE H.W. Different effects of muscarinic agonists in rat superior cervical ganglion and hippocampal slices. Eur. J. Pharmacol. 1991;201:191–197. doi: 10.1016/0014-2999(91)90344-p. [DOI] [PubMed] [Google Scholar]
- BRINK C.B., WADE S.M., NEUBIG R.R. Agonist-directed trafficking of porcine alpha(2A)-adrenergic receptor signaling in Chinese hamster ovary cells: l-isoproterenol selectively activates G(s) J. Pharmacol. Exp. Ther. 2000;294:539–547. [PubMed] [Google Scholar]
- CAVALLI A., DRUEY K.M., MILLIGAN G. The regulator of G protein signaling RGS4 selectively enhances alpha 2A-adrenoreceptor stimulation of the GTPase activity of Go1alpha and Gi2alpha. J. Biol. Chem. 2000;275:23693–23699. doi: 10.1074/jbc.M910395199. [DOI] [PubMed] [Google Scholar]
- DIVERSE-PIERLUISSI M., GOLDSMITH P.K., DUNLAP K. Transmitter-mediated inhibition of N-type calcium channels in sensory neurons involves multiple GTP-binding proteins and subunits. Neuron. 1995;14:191–200. doi: 10.1016/0896-6273(95)90254-6. [DOI] [PubMed] [Google Scholar]
- DIVERSE-PIERLUISSI M.A., FISCHER T., JORDAN J.D., SCHIFF M., ORTIZ D.F., FARQUHAR M.G., DE VRIES L. Regulators of G protein signaling proteins as determinants of the rate of desensitization of presynaptic calcium channels. J. Biol. Chem. 1999;274:14490–14494. doi: 10.1074/jbc.274.20.14490. [DOI] [PubMed] [Google Scholar]
- DUZIC E., COUPRY I., DOWNING S., LANIER S.M. Factors determining the specificity of signal transduction by guanine nucleotide-binding protein-coupled receptors. I. Coupling of alpha 2-adrenergic receptor subtypes to distinct G-proteins. J. Biol. Chem. 1992;267:9844–9851. [PubMed] [Google Scholar]
- EASON M.G., JACINTO M.T., LIGGETT S.B. Contribution of ligand structure to activation of alpha 2-adrenergic receptor subtype coupling to Gs. Mol. Pharmacol. 1994;45:696–702. [PubMed] [Google Scholar]
- EASON M.G., KUROSE H., HOLT B.D., RAYMOND J.R., LIGGETT S.B. Simultaneous coupling of alpha 2-adrenergic receptors to two G-proteins with opposing effects. Subtype-selective coupling of alpha 2C10, alpha 2C4, and alpha 2C2 adrenergic receptors to Gi and Gs. J. Biol. Chem. 1992;267:15795–15801. [PubMed] [Google Scholar]
- EASON M.G., LIGGETT S.B. Chimeric mutagenesis of putative G-protein coupling domains of the alpha2A-adrenergic receptor. Localization of two redundant and fully competent gi coupling domains. J. Biol. Chem. 1996;271:12826–12832. doi: 10.1074/jbc.271.22.12826. [DOI] [PubMed] [Google Scholar]
- ENKVIST M.O., HÄMÄLÄINEN H., JANSSON C.C., KUKKONEN J.P., HAUTALA R., COURTNEY M.J., ÅKERMAN K.E. Coupling of astroglial alpha 2-adrenoreceptors to second messenger pathways. J. Neurochem. 1996;66:2394–2401. doi: 10.1046/j.1471-4159.1996.66062394.x. [DOI] [PubMed] [Google Scholar]
- FRASER C.M., ARAKAWA S., MCCOMBIE W.R., VENTER J.C. Cloning, sequence analysis, and permanent expression of a human alpha 2-adrenergic receptor in Chinese hamster ovary cells. Evidence for independent pathways of receptor coupling to adenylate cyclase attenuation and activation. J. Biol. Chem. 1989;264:11754–11761. [PubMed] [Google Scholar]
- GERHARDT M.A., NEUBIG R.R. Multiple Gi protein subtypes regulate a single effector mechanism. Mol. Pharmacol. 1991;40:707–711. [PubMed] [Google Scholar]
- GERHARDT M.A., WADE S.M., NEUBIG R.R. p-[125I]iodoclonidine is a partial agonist at the alpha 2-adrenergic receptor. Mol. Pharmacol. 1990;38:214–221. [PubMed] [Google Scholar]
- GUDERMANN T., KALKBRENNER F., SCHULTZ G. Diversity and selectivity of receptor-G protein interaction. Annu. Rev. Pharmacol. Toxicol. 1996;36:429–459. doi: 10.1146/annurev.pa.36.040196.002241. [DOI] [PubMed] [Google Scholar]
- HAJDU-CRONIN Y.M., CHEN W.J., PATIKOGLOU G., KOELLE M.R., STERNBERG P.W. Antagonism between G(o)alpha and G(q)alpha in Caenorhabditis elegans: the RGS protein EAT-16 is necessary for G(o)alpha signaling and regulates G(q)alpha activity. Genes Dev. 1999;13:1780–1793. doi: 10.1101/gad.13.14.1780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOLMBERG C.I., KUKKONEN J.P., BISCHOFF A., NÄSMAN J., COURTNEY M.J., MICHEL M.C., ÅKERMAN K.E. Alpha2B-adrenoceptors couple to Ca2+ increase in both endogenous and recombinant expression systems. Eur. J. Pharmacol. 1998;363:65–74. doi: 10.1016/s0014-2999(98)00780-8. [DOI] [PubMed] [Google Scholar]
- JANSSON C.C., KARP M., OKER-BLOM C., NÄSMAN J., SAVOLA J.M., ÅKERMAN K.E. Two human alpha 2-adrenoceptor subtypes alpha 2A-C10 and alpha 2B-C2 expressed in Sf9 cells couple to transduction pathway resulting in opposite effects on cAMP production. Eur. J. Pharmacol. 1995;290:75–83. doi: 10.1016/0922-4106(95)90019-5. [DOI] [PubMed] [Google Scholar]
- JANSSON C.C., KUKKONEN J.P., NÄSMAN J., HUIFANG G., WURSTER S., VIRTANEN R., SAVOLA J.-M., COCKCROFT V., ÅKERMAN K.E. Protean agonism at alpha2A-adrenoceptors. Mol. Pharmacol. 1998;53:963–968. [PubMed] [Google Scholar]
- JANSSON C.C., SAVOLA J.M., ÅKERMAN K.E. Different sensitivity of alpha 2A-C10 and alpha 2C-C4 receptor subtypes in coupling to inhibition of cAMP accumulation. Biochem. Biophys. Res. Commun. 1994;199:869–875. doi: 10.1006/bbrc.1994.1309. [DOI] [PubMed] [Google Scholar]
- JASPER J.R., LESNICK J.D., CHANG L.K., YAMANISHI S.S., CHANG T.K., HSU S.A., DAUNT D.A., BONHAUS D.W., EGLEN R.M. Ligand efficacy and potency at recombinant alpha2 adrenergic receptors: agonist-mediated [35S]GTPgammaS binding. Biochem. Pharmacol. 1998;55:1035–1043. doi: 10.1016/s0006-2952(97)00631-x. [DOI] [PubMed] [Google Scholar]
- JONES S.B., TOEWS M.L., TURNER J.T., BYLUND D.B. Alpha 2-adrenergic receptor-mediated sensitization of forskolin-stimulated cyclic AMP production. Proc. Natl. Acad. Sci. U.S.A. 1987;84:1294–1298. doi: 10.1073/pnas.84.5.1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JORDAN J.D., CAREY K.D., STORK P.J., IYENGAR R. Modulation of rap activity by direct interaction of Galpha(o) with Rap1 GTPase-activating protein. J. Biol. Chem. 1999;274:21507–21510. doi: 10.1074/jbc.274.31.21507. [DOI] [PubMed] [Google Scholar]
- KENAKIN T. Agonist-receptor efficacy. II. Agonist trafficking of receptor signals. Trends Pharmacol. Sci. 1995;16:232–238. doi: 10.1016/s0165-6147(00)89032-x. [DOI] [PubMed] [Google Scholar]
- KRUMINS A.M., BARBER R. The stability of the agonist beta2-adrenergic receptor-Gs complex: evidence for agonist-specific states. Mol. Pharmacol. 1997;52:144–154. doi: 10.1124/mol.52.1.144. [DOI] [PubMed] [Google Scholar]
- KUKKONEN J.P., HUIFANG G., JANSSON C.C., WURSTER S., COCKCROFT V., SAVOLA J.M., ÅKERMAN K.E.O. Different apparent modes of inhibition of alpha-2A-adrenoceptor by alpha-2-antagonists. Eur. J. Pharmacol. 1997;335:99–105. doi: 10.1016/s0014-2999(97)01180-1. [DOI] [PubMed] [Google Scholar]
- KUKKONEN J.P., RENVAKTAR A., SHARIATMADARI R., ÅKERMAN K.E. Ligand- and subtype-selective coupling of human alpha-2 adrenoceptors to Ca++ elevation in Chinese hamster ovary cells. J. Pharmacol. Exp. Ther. 1998;287:667–671. [PubMed] [Google Scholar]
- KUROSE H., REGAN J.W., CARON M.G., LEFKOWITZ R.J. Functional interactions of recombinant alpha 2 adrenergic receptor subtypes and G proteins in reconstituted phospholipid vesicles. Biochemistry. 1991;30:3335–3341. doi: 10.1021/bi00227a024. [DOI] [PubMed] [Google Scholar]
- LAUGWITZ K.L., ALLGEIER A., OFFMANNS S., SPICHER K., VAN SANDE J., DUMONT J.E., SCHULTZ G. The human thyrotropin receptor: a heptahelical receptor capable of stimulating members of all four G protein families. Proc. Natl. Acad. Sci. U.S.A. 1996;93:116–120. doi: 10.1073/pnas.93.1.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEFF P., SCARAMELLINI C., LAW C., MCKECHNIE K. A three-state receptor model of agonist action. Trends Pharmacol. Sci. 1997;18:355–362. doi: 10.1016/s0165-6147(97)01105-x. [DOI] [PubMed] [Google Scholar]
- LEPRETRE N., MIRONNEAU J. Alpha 2-adrenoceptors activate dihydropyridine-sensitive calcium channels via Gi-proteins and protein kinase C in rat portal vein myocytes. Pflügers Arch. 1994;429:253–261. doi: 10.1007/BF00374320. [DOI] [PubMed] [Google Scholar]
- MCCLUE S.J., SELZER E., FREISSMUTH M., MILLIGAN G. Gi3 does not contribute to the inhibition of adenylate cyclase when stimulation of an alpha 2-adrenergic receptor causes activation of both Gi2 and Gi3. Biochem. J. 1992;284:565–568. doi: 10.1042/bj2840565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCKERNAN R.M., HOWARD M.J., MOTULSKY H.J., INSEL P.A. Compartmentation of alpha 2-adrenergic receptors in human erythroleukemia (HEL) cells. Mol. Pharmacol. 1987;32:258–265. [PubMed] [Google Scholar]
- MICHEL M.C., BRASS L.F., WILLIAMS A., BOKOCH G. M., LAMORTE V.J., MOTULSKY H.J. Alpha 2-adrenergic receptor stimulation mobilizes intracellular Ca2+ in human erythroleukemia cells. J. Biol. Chem. 1989;264:4986–4991. [PubMed] [Google Scholar]
- MUSGRAVE I.F., SEIFERT R. Alpha 2A-adrenoceptors mediate activation of non-selective cation channels via Gi-proteins in human erythroleukaemia (HEL) cells. No evidence for a functional role of imidazoline receptors in modulating calcium. Biochem. Pharmacol. 1995;49:187–196. doi: 10.1016/s0006-2952(94)00432-3. [DOI] [PubMed] [Google Scholar]
- PEPPERL D.J., REGAN J.W. Selective coupling of alpha 2-adrenergic receptor subtypes to cyclic AMP-dependent reporter gene expression in transiently transfected JEG-3 cells. Mol. Pharmacol. 1993;44:802–809. [PubMed] [Google Scholar]
- POHJANOKSA K., JANSSON C.C., LUOMALA K., MARJAMÄKI A., SAVOLA J., SCHEININ M. Alpha2-adrenoceptor regulation of adenylyl cyclase in CHO cells: dependence on receptor density, receptor subtype and current activity of adenylyl cyclase. Eur. J. Pharmacol. 1997;335:53–63. doi: 10.1016/s0014-2999(97)01154-0. [DOI] [PubMed] [Google Scholar]
- POSNER B.A., GILMAN A.G., HARRIS B.A. Regulators of G protein signaling 6 and 7. Purification of complexes with gbeta5 and assessment of their effects on g protein-mediated signaling pathways. J. Biol. Chem. 1999;274:31087–31093. doi: 10.1074/jbc.274.43.31087. [DOI] [PubMed] [Google Scholar]
- REMAURY A., LARROUY D., DAVIAUD D., ROUOT B., PARIS H. Coupling of the alpha 2-adrenergic receptor to the inhibitory G-protein Gi and adenylate cyclase in HT29 cells. Biochem. J. 1993;292:283–288. doi: 10.1042/bj2920283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIMONDS W.F., GOLDSMITH P.K., CODINA J., UNSON C.G., SPIEGEL A.M. Gi2 mediates alpha 2-adrenergic inhibition of adenylyl cyclase in platelet membranes: in situ identification with G alpha C-terminal antibodies. Proc. Natl. Acad. Sci. U.S.A. 1989;86:7809–7813. doi: 10.1073/pnas.86.20.7809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAUSSIG R., TANG W.J., HEPLER J.R., GILMAN A.G. Distinct patterns of bidirectional regulation of mammalian adenylyl cyclases. J. Biol. Chem. 1994;269:6093–6100. [PubMed] [Google Scholar]
- VALENZUELA D., HAN X., MENDE U., FANKHAUSER C., MASHIMO H., HUANG P., PFEFFER J., NEER E.J., FISHMAN M.C. G alpha(o) is necessary for muscarinic regulation of Ca2+ channels in mouse heart. Proc. Natl. Acad. Sci. U.S.A. 1997;94:1727–1732. doi: 10.1073/pnas.94.5.1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VOIGT M.M., MCCUNE S.K., KANTERMAN R.Y., FELDER C.C. The rat alpha 2-C4 adrenergic receptor gene encodes a novel pharmacological subtype. FEBS Lett. 1991;278:45–50. doi: 10.1016/0014-5793(91)80080-m. [DOI] [PubMed] [Google Scholar]
- YANG Q., LANIER S. M. Influence of G protein type on agonist efficacy. Mol. Pharmacol. 1999;56:651–656. doi: 10.1124/mol.56.3.651. [DOI] [PubMed] [Google Scholar]