

Abstract
Two sodium channel toxins, BgII and BgIII, have been isolated and purified from the sea anemone Bunodosoma granulifera. Combining different techniques, we have investigated the electrophysiological properties of these toxins.
We examined the effect of BgII and BgIII on rat ventricular strips. These toxins prolong action potentials with EC50 values of 60 and 660 nM and modify the resting potentials.
The effect on Na+ currents in rat cardiomyocytes was studied using the patch-clamp technique. BgII and BgIII slow the rapid inactivation process and increase the current density with EC50 values of 58 and 78 nM, respectively.
On the cloned hH1 cardiac Na+ channel expressed in Xenopus laevis oocytes, BgII and BgIII slow the inactivation process of Na+ currents (respective EC50 values of 0.38 and 7.8 μM), shift the steady-state activation and inactivation parameters to more positive potentials and the reversal potential to more negative potentials.
The amino acid sequences of these toxins are almost identical except for an asparagine at position 16 in BgII which is replaced by an aspartic acid in BgIII. In all experiments, BgII was more potent than BgIII suggesting that this conservative residue is important for the toxicity of sea anemone toxins.
We conclude that BgII and BgIII, generally known as neurotoxins, are also cardiotoxic and combine the classical effects of sea anemone Na+ channels toxins (slowing of inactivation kinetics, shift of steady-state activation and inactivation parameters) with a striking decrease on the ionic selectivity of Na+ channels.
Keywords: Sea anemone toxin, voltage-gated sodium channels, inactivation, ionic selectivity, cardiotoxin, neurotoxin
Introduction
As key elements of signal transduction, voltage-gated sodium channels (VGSCs) are the target of toxins of various origins and chemical structures. The different effects of these toxins range from blocking the pore of the channel (e.g. tetrodotoxin, μ-conotoxin) to modifying its gating and permeation characteristics (e.g. sea anemone toxins, scorpions α- and β-toxins, batrachotoxin). These natural compounds are powerful tools for understanding the physiological contribution of VGSCs to cell and organ behaviour and for probing and correlating ion channel structure and function (Catterall, 1995; 2000; Cestele & Catterall, 2000; Denac et al., 2000; Lazdunski et al., 1986; Renaud et al., 1986). Moreover, elucidation of the mechanisms of action of toxins, knowledge of their 3D-structures and the discovery of common scaffolds between toxins open wide perspectives in designing various drugs (Ménez, 1998).
VGSCs are composed of a pore-forming α subunit of approximately 260 kDa flanked by two auxiliary β subunits, a non covalently associated β1 subunit of 36 kDa and a β2 subunit of 33 kDa linked by a disulphide bond. In heterologous systems, expression of the α subunit alone is sufficient to form a functional Na+ channel regulated by voltage. Membrane depolarization causes a voltage-dependent conformational change of the channel that induces an increase of its permeability to Na+. This is followed by an inactivation process, wherein the channel closes and the permeability to Na+ is shut off (Hille, 1992). The β subunits, which do not form the pore, modulate the level of expression, channel gating properties, and interaction with cytoskeleton proteins (Catterall, 2000). Sodium channels can be differentiated by their primary structure, current kinetics and relative sensitivity to the neurotoxin tetrodotoxin (TTX). They have been cloned in different tissues of various species. The human cardiac isoform (hH1) of the VSGC (Gellens et al., 1992), or Nav 1.5 according to the recent nomenclature of VGSCs (Goldin et al., 2000), is a TTX-resistant channel expressed in cardiomyocytes where it plays a key role in the generation and propagation of the cardiac action potential (Balser, 1999).
Sea anemones produce several polypeptide toxins, mainly active on ionic channels of excitable membranes, to capture their prey. Sea anemone toxins acting on Na+ channel have been intensively studied over the past years. They are both cardio- and neurotoxins and their main effects are to slow the inactivation process of Na+ currents and to prolong the action potentials. These polypeptides cross-linked by three disulphide bridges and of molecular masses of approximately 5 kDa have been broadly categorized as type 1 (e.g. ATXI and ATXII from Anemonia sulcata (Wunderer & Eulitz, 1978; Wunderer et al., 1976), ApA and ApB from Anthopleura xanthogrammica (Norton, 1981; Reimer et al., 1985; Tanaka et al., 1977)) and type 2 (e.g. ShI from Stichodactyla helianthus (Kem et al., 1989; Wilcox et al., 1993)), according to amino acid sequence similarity and immunological cross-reactivity (Figure 2; see (Norton, 1991; Norton, 1998) for reviews). Toxins acting on Na+ channels bind to at least six receptor sites (Catterall, 2000; Denac et al., 2000). Sea anemone toxins bind to receptor site 3, located in the extracellular linker between segments S3 and S4, in the fourth domain (D4). They share their receptor site with scorpion α-toxins (Gordon et al., 1998; Possani et al., 1999; Rogers et al., 1996) and funnel web spider toxins (Nicholson et al., 1994; 1998). The common point between these toxins is that they induce a slowing of the inactivation process of Na+ channels. From a mechanical point of view, the Na+ channel inactivation process derives mainly from the voltage-dependent coupling of activation driven by the transmembrane movement of the voltage sensor (S4 segments). By binding to site 3, toxins prevent the normal gating movements of the IVS4 segment and thus uncouple Na+ channel activation from inactivation (Catterall, 2000).
Figure 2.

Comparison of the amino acid sequences of BgII, BgIII with other sea anemone toxins. BgII and BgIII have been purified from the sea anemone Bunodosoma granulifera (Loret et al., 1994), ATXII from Anemonia sulcata (Wunderer et al., 1976), ApA and ApB from Anthopleura xanthogrammica (Norton, 1981; Reimer et al., 1985; Tanaka et al., 1977) and ShI from Stichodactyla helianthus (Kem et al., 1989; Wilcox et al., 1993). Identical amino acids are indicated with a black background, homologous amino acids are indicated with a gray background and ‘%id' stands for the percentage of identity in comparison to BgII. Dashes represent gaps.
Bunodosoma granulifera is a common anemone of Cuban sea shores. Several active compounds with different pharmacological actions have been isolated from its secretions (Aneiros et al., 1993; Loret et al., 1994; Salinas et al., 1997). BgII and BgIII are two toxins that have been purified and sequenced from this sea anemone originally by Loret et al. (1994). They consist in two peptides of 48 amino acids each containing six cysteine residues that form three disulphide bridges. The amino acid sequences of these two toxins possess a higher similarity with type 1 sea anemone toxins, like ApA and ApB or ATX II, than with the toxins of type 2, like ShI (Figure 2). The sequences of BgII and BgIII are almost identical, only differing by a single amino acid. In BgIII, at position 16, an aspartic acid replaces the asparagine of BgII (Figure 2). Despite their resemblance, the toxicity and the binding to rat brain synaptosomes of BgII is interestingly significantly higher than that of BgIII. However, up to now, electrophysiological characterization of these two toxins was still completely lacking.
In the present study, combining different approaches, we have therefore investigated the electrophysiological properties of BgII and BgIII toxins. Firstly, we have examined their effect on rat ventricular strips and observed that both toxins are able to prolong action potentials. Then, using the patch-clamp technique, we have studied the effect of BgII and BgIII on Na+ currents in cardiomyocytes revealing that these toxins slowed the rapid inactivation process of Na+ currents and increased the current density. Finally, in order to refine our study, we have investigated the influence of these toxins on the cloned cardiac sodium channel hH1 expressed in Xenopus laevis oocytes, using the two-electrode voltage clamp technique. These experiments confirmed that BgII and BgIII markedly slow the inactivation process of Na+ currents in a dose-dependent way and showed that both steady-state inactivation and activation are shifted to more positive potentials. It was also revealed that toxin-modified channels displayed a decreased ionic selectivity. Moreover, effects of BgII are more marked than effects of BgIII emphasizing the importance of the amino acid in position 16 for the biological effects of these two Bunodosoma granulifera toxins.
Methods
Toxin extraction and isolation
Sea anemone Bunodosoma granulifera was collected along the north coast of Havana province, Cuba and transported to the laboratory in containers filled with sea water.
To prepare crude extract, in brief, 1 kg of B. granulifera was homogenized in 1 l of ethanol adjusted to pH 5.4 with acetic acid. After centrifugation (30 min, 4°C, 15,000×g), the supernatant was separated and precipitated with 10 volumes of acetone (4°C, overnigth). The precipitate was recovered, diluted in water, concentrated under reduced pressure to eliminate acetone and freeze dried later. Lyophilized crude extract was dissolved in 0.10 M ammonium acetate pH 6.7 and gel filtered on Sephadex G-50 m (4.5×132 cm) as previously described (Aneiros et al., 1993; Salinas et al., 1997).
The toxic peak was submitted to chromatography on the cation-exchanger Fractogel EMD SO3-650 M (1.2×32 cm) eluted with a linear gradient of ammonium acetate (pH 5.4) from 0.01 to 1.0 M. Toxins were rechromatographed on Fractogel EMD SO3-650 M under analogous conditions, and desalted on Sephadex G-25. Further purification of toxins was performed by reversed-phase HPLC.
We used the toxicity test in crab to follow the toxic activity of chromatographic fractions during the purification procedures. The toxicity of the different fractions was evaluated as paralysing activity after injection on shore crabs (Uca thayeri and Carcinus maenas).
Mass spectrometry analysis
The masses and sequences were obtained using a Finnigan LCQ-Duo ion trap mass spectrometer, equipped with an electrospray ion source. The analysis was performed as recently reviewed (Dongre et al., 1997).
Right ventricular strips
Small right ventricular strips were dissected out from adult rat hearts fixed to a bath chamber and continuously superfused (10 ml min−1) with normal Tyrode solution (NaCl, 140; KCl, 2.5; CaCl2, 2; MgCl2, 0.5; hydroxyethylpiperazine-N′-2-ethanesulfonic acid (HEPES), 10 and glucose, 5 (pH=7.4, gassed with O2)) at 35°C and field stimulated with 2 ms pulses at a frequency of 1.25 Hz (cycle length, 800 ms). Standard fine-tipped microelectrodes (tip resistance, 15 to 20 MΩ) were used to record action potentials (Alvarez et al., 1981). Action potential duration was measured at 0 (D0) and −60 (D−60) mV.
Isolation of adult ventricular cardiomyocytes
Single rat ventricular cells were dispersed by a collagenase/trypsin enzymatic method similar to that previously described (Galan et al., 1998). In brief, rat hearts were cannulated through the aorta and perfused with a physiological solution for 5 min (in mM): NaCl 112.0, KCl 2.5, CaCl2 1.8, MgCl2 0.6, HEPES 10.0, glucose 5.0, Na2-pyruvate 5.0, and pH adjusted to 7.4. Thereafter, the hearts were perfused with a low-Ca2+ (CaCl2 30 μM) solution for 5 min. The hearts were then perfused for 30 min with the same solution containing collagenase (Boehringer Mannheim) and trypsin (Merck) at 1.5 mg ml−1 and 0.4 mg ml−1 respectively and supplemented with minimum essential medium (1 μl ml−1 MEM; Sigma), creatine (5 mM) and taurine (20 mM). At the end of this period, the right ventricle was cut off and gently shaken in the same solution without enzymes. Isolated myocytes were kept in this physiological solution (Ca2+=1 mM) at room temperature (21 – 23°C) and used within 6 – 8 h.
Patch-clamp recordings
The ‘whole-cell' variant of the patch-clamp method was used (Hamill et al., 1981). Recordings were made using an RK-300 (Biologic, Claix, France) patch-clamp amplifier. Patch pipettes were made from borosilicate glass and had tip resistances between 1.0 and 1.2 MΩ. The pipette ‘intracellular' solution contained (mM): CsCl 100, TEA-Cl 20, MgCl2 4.0, Na2-GTP 0.5, Na2-ATP 3, ethyleneglycol-bis-(-aminoethyl ether) N,N,N′,N′-tetraacetic acid (EGTA) 10, HEPES 10, the pH was adjusted to 7.2 with CsOH. For recording the Na+ current (INa), Ca2+ and K+ currents were blocked by CoCl2 (extracellular, 3 mM), tetraethylammonium chloride (TEA-Cl) and CsCl (intracellular and extracellular; see below), respectively. A cell aliquot was put in a Petri dish containing the control solution (mM): NaCl 112, KCl 2.5, CaCl2 1.8, MgCl2 0.6, HEPES 10, glucose 5.0, Na2-pyruvate 5.0; pH was adjusted to 7.4 with NaOH. Experiments were performed at room temperature (22±2°C).
After achieving the whole-cell patch-clamp configuration, each cell was exposed to different extracellular solutions by positioning it at the extremity of one of six capillaries (inner diameter of each capillary was 250 μm). Such a system allowed rapid changes of solution (<2 s). Currents were routinely evoked by 50 ms voltage-clamp pulses to −40 mV from a holding potential of −100 mV at a frequency of 0.25 Hz (cycle length 4 s). The current amplitude was estimated as the difference between the peak inward current and the current level at the end of a 50 ms pulse. The composition of the extracellular solution in these experiments was (in mM): NaCl 5, TEA-Cl 110, CaCl2 1.8, MgCl2 2, glucose 10, HEPES 10, pH was adjusted to 7.4 at 21°C. Pulse generation and data acquisition were done, using computer facilities and ACQUIS1 (CNRS License, France) software. Currents were filtered at 3 KHz and digitized at 20 KHz with a LabMaster DMA (model TM 125, Scientific Solutions, U.S.A.).
Current to voltage (I – V) relationships and availability curves were constructed using a standard double pulse voltage protocol. From a holding potential of −100 mV, a 50 ms test pulse to −40 mV was preceded by 50 ms prepulses to various membrane potentials. The time interval between pulses was 5 ms. I – V relationships were constructed from currents elicited by prepulse potentials. Normalization of current amplitudes at a given test pulse by the maximal current recorded as a function of prepulse potential gave the availability curve. The experimental points were fitted to a Boltzmann function:
where Vp is the prepulse potential, V1/2 is the potential for half-availability and s a slope factor.
The inactivation time course of currents was described by fitting the current traces between the inward peak and the end of the pulse using the fitting procedures of ACQUIS1 software. Recovery from inactivation was studied by a two pulse protocol. From a holding potential of −100 mV, a pair of 50 ms pulses to −40 mV was applied; the interval between the two pulses was varied from 5 to 650 ms. Recovery from inactivation (or reactivation) was obtained by the ratio of current measured at the second pulse to the current measured at the first pulse as a function of the time interval. Experimentally obtained means of the availability curves were fitted to a Boltzmann function to obtain the potential for half inactivation and slope factors.
Values are expressed as means±s.e.means.
Expression in Xenopus oocytes
For the expression in Xenopus oocytes, the hH1 gene was subcloned into pSP64T (Gellens et al., 1992). For in vitro transcription, hH1/pSP64T has been first linearized by XbaI. Then, capped cRNAs were synthetized from the linearized plasmid using the large-scale SP6 mMESSAGE-mMACHINE transcription kit (Ambion, U.S.A.). The in vitro synthesis of cRNA encoding hH1 and isolation of Xenopus oocytes was as previously described (Liman et al., 1992). Oocytes were injected with 50 nl of hH1 cRNA at a concentration of 1 ng nl−1 using a Drummond microinjector (U.S.A.). The solution used for incubating the oocytes contained (in mM): NaCl 96, KCl 2, CaCl2 1.8, MgCl2 2 and HEPES 5 (pH 7.4), supplemented with 50 mg l−1 gentamicin sulphate.
Electrophysiological recordings in Xenopus oocytes
Whole-cell currents from oocytes were recorded from 1 to 3 days after injection using the two-electrode voltage clamp technique. Voltage and current electrodes were filled with 3 M KCl. Resistances of both electrodes were kept as low as possible (ca. 0.1 – 0.2 MΩ). During the experiments, the external NaCl concentration has been reduced in order to facilitate the clamp. Bath solution composition was (in mM): NaCl 40, KCl 2, CaCl2 1.8, MgCl2 2 and Tris-HCl 60 (pH 7.4). Experiments were performed using a GeneClamp 500 amplifier (Axon instruments, U.S.A.) controlled by a pClamp data acquisition system (Axon instruments, U.S.A.). Using a four-pole low-pass Bessel filter, currents were filtered at 5 kHz and sampled at 10 kHz. Digital leak subtraction of the current records was carried out using a P/4 protocol.
Current to voltage (I – V) relationships and steady-state activation curves were constructed using a standard single pulse voltage protocol. From a fixed holding potential of −90 mV, 25 ms test pulses to membrane potentials varying from −70 to +45 mV by 5 mV increments were performed. I – V relationships were constructed from peak currents elicited at different potentials. Normalization of conductance (g/gmax) as a function of the test pulse voltage gave the steady-state activation curve. The experimental points were fitted to a Boltzmann function:
where Vp is the pulse potential, V1/2act is the potential for half-activation and s a slope factor. Steady-state inactivation curves were constructed using a standard double pulse voltage protocol. From a fixed holding potential of −90 mV, a 50 ms test pulse to −20 mV was preceded by 50 ms prepulses to membrane potentials varying from −120 to 30 mV.
Normalization of the current amplitude at the test pulse by the maximal current recorded (I/Imax) as a function of prepulse potential gave the steady-state inactivation curve. The experimental points were fitted to a Boltzmann function:
where Vp is the prepulse potential, V1/2inact is the potential for half-inactivation and s a slope factor. The permeability ratio of Na+ as compared to K+ (PNa/PK) has been calculated using the Goldman-Hodgkin-Katz (GHK) equation:
where Erev is the reversal potential determined from the I – V relationship, PNa and PK are the permeabilities of the channel for Na+ and K+, respectively. The external concentrations of Na+, [Na]o, and K+, [K]o, were 40 and 2 mM, respectively. The internal concentrations of Na+, [Na]i, and K+, [K]i, have been estimated to be 6 and 92 mM, respectively (Dascal, 1987).
The inactivation time course of Na+ currents was described by fitting the current traces between the inward peak and the end of the pulse using the fitting procedures of pClamp6 software (Axon instruments, U.S.A.).
Data are presented as mean±s.e.mean. All experiments were performed at room temperature (20±2°C).
Results
Toxin identification
After gel filtration chromatography on Sephadex G-50, only fraction three eluting between 70 – 80% of one whole column volume was found to be toxic in the crab bioassay. This fraction was submitted to a cation exchange chromatography on Fractogel EMD SO3-650M. The fractions eluting in the gradient at ammonium acetate concentrations between 0.1 and 0.2 M were further chromatographed under the same conditions to give the toxins (BgIII and BgII respectively) that were earlier reported by Loret et al. (1994).
For BgII toxin, a direct sequencing by Edman degradation of reduced and alkylated toxin provided an unequivocal identification of the 21 first amino acids. Then, following a digestion of this toxin with Arg-C endopeptidase a HPLC separation gave three additional fragments with molecular masses of 1401.6, 1059.4 and 1259.4, respectively. The sequences were obtained by ionization, using MS/MS mass spectrometry, and correspond to sequences from position 15 to 27, 28 to 36 and 37 to 48. The entire sequence corresponds exactly to that earlier reported by Loret et al., 1994 (Figure 1).
Figure 1.

Amino acid sequence determination of toxins. BgII: Direct sequencing by Edman degradation of reduced and alkylated toxin provided unequivocal identification of the 21 first amino acids, shown by underlining (-d->). Three additional fragments were obtained by HPLC separation of toxin BgII digested with Arg-C endopeptidase, which gave peptides with molecular masses of 1401.6, 1059.4 and 1259.4, respectively. The sequences were obtained by ionization, using MSIMS mass spectrometry, and correspond to sequence from positions 15 to 27, 28 to 36 and 37 to 48. The entire sequence corresponds exactly to that reported earlier (Loret et al., 1994). BgIII: Direct sequencing of reduced and alkylated toxin provided the 29 first amino acid residues, as indicated by the underlining (-d->). Three additional overlapping peptides were obtained by Arg-C endopeptidase digestion conducted as mentioned above for BgII, in which the only difference was found in the peptide corresponding to positions 15 to 27 (molecular mass 1402.6) where an aspartic acid substitutes an asparagine in position 16. The MSIMS spectrometry analysis confirmed the sequences shown, which also corresponds exactly to those earlier reported (Loret et al., 1994). The numbers on top of the sequences indicate the positions of the amino acids in the sequences.
For BgIII toxin, a direct sequencing by Edman degradation of reduced and alkylated toxin provided an unequivocal identification of the 29 first amino acids. Then, overlapping peptides were obtained by Arg-C endopeptidase digestion conducted as mentioned above for BgII, in which the only difference was found in the peptide corresponding to positions 15 to 27 (molecular mass 1402.6) where an aspartic acid substitutes an asparagine in position 16. The MS/MS spectrometry analysis confirmed the sequences shown, which also corresponds exactly to those earlier reported by Loret et al., 1994 (Figure 1).
BgII and BgIII consist in two peptides of 48 amino acids which each contain six cysteine residues forming three disulphide bridges. The measured masses for BgII and BgIII, 5072.7 Da and 5073.1 Da, respectively, are almost identical to the theoretical masses determined according to the amino acid sequences, 5071.7 Da and 5072.7 Da for BgII and BgIII, respectively. In Figure 2, amino acid sequences of BgII and BgIII are shown and compared with other sea anemone Na+ channel toxins: ATXII from Anemonia sulcata (Wunderer et al., 1976), ApA and ApB from Anthopleura xanthogrammica (Norton, 1981; Reimer et al., 1985; Tanaka et al., 1977) and ShI from Stichodactyla helianthus (Kem et al., 1989; Wilcox et al., 1993). The amino acid sequence of Bgll possesses a higher similarity with type 1 sea anemone toxins (72.9%, 64.5% and 70.8% identity with ApA, ApB and ATX II, respectively) than with the toxins of type 2 (42.6% identity with ShI).
Effects of Bunodosoma granulifera toxins on action potentials of rat ventricular strip
Superfusion of rat ventricular strips with 10 μM BgII or BgIII toxins induced a fast increase in action potential duration (Figure 3). Within 1 min, action potential duration at 0 mV (D0) was increased to 160 and 130% of control in the presence of BgII and BgIII, respectively. Action potential duration at −60 mV (D−60) was increased to 230 and 185% in the presence of BgII and BgIII, respectively. During this initial period, no major changes were observed in action potential amplitude or resting membrane potential. At longer superfusion times (5 min), action potential duration was further increased and the resting potential decreased. Interestingly, these changes were more marked with BgII which increased D0 to 560% of control and decreased resting potential from −92 to −45 mV. Under the action of BgIII, D0 was only further increased to 216% of control while the resting potential was decreased from −93 to −87 mV.
Figure 3.
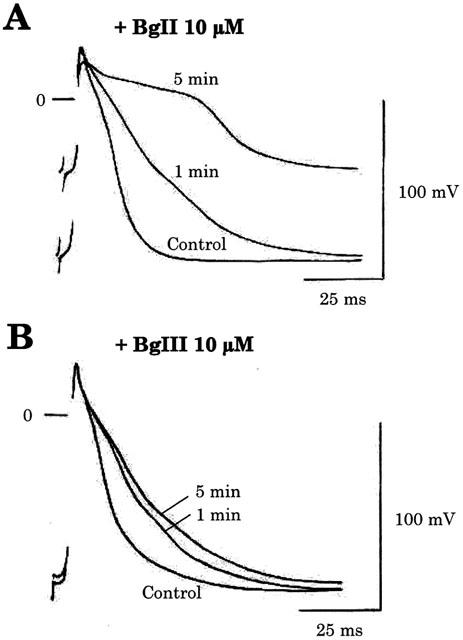
Effects of Bunodosoma granulifera toxins on the resting and action potential characteristics of rat ventricular strips. Both BgII (A) and BgIII (B) were applied at a concentration of 10 μM. Within 1 min, both toxins markedly increased the action potential duration. After a longer time (5 min) the resting potential was decreased. Note that the increase in action potential duration and the decrease in resting potential were more marked with BgII.
The increase of the action potential duration induced by BgII and BgIII is dose-dependent. The effect of BgII and BgIII has been studied at concentrations ranging from 0.01 to 10 μM and revealed that BgII is more potent than BgIII. The toxin concentration producing the half maximum effect (EC50) on the action potential duration at 0 mV determined by a sigmoidal fit is 60±4 nM for BgII (n=3) and 663±380 nM for BgIII (n=3).
Effects of BgII and BgIII on Na+ current of rat ventricular cardiomyocytes
To study the effect of Bunodosoma granulifera toxins on the Na+ current (INa), rat ventricular myocytes were superfused with an extracellular solution containing 5 mM NaCl so as to decrease INa amplitude and to obtain a better voltage-clamp control. Myocytes were held at −100 mV and routinely depolarized to −40 mV with 50 ms voltage-clamp pulses to monitor INa. Membrane capacity was determined in voltage-clamped cells (Galan et al., 1998) and the mean value obtained from 27 cells was 124.5±18.3 pF. Ionic current amplitudes were normalized to membrane capacity and are expressed as current density (pA/pF). Under these experimental conditions INa at −40 mV had a density of −7.4±0.9 pA/pF (n=27).
Both BgII and BgIII exerted concentration-dependent effects on the INa amplitude and the inactivation time course. At low concentrations (10−9, 10−8 M), no significant effects were seen. At higher concentrations (10−7 to 10−5 M), both toxins increased the peak INa density at −40 mV and delayed its inactivation time course. Table 1 summarizes the obtained results.
Table 1.
Effects of BgII and BgIII on INa of rat ventricular cardiomyocytes (values are expressed as per cent increase over control value)
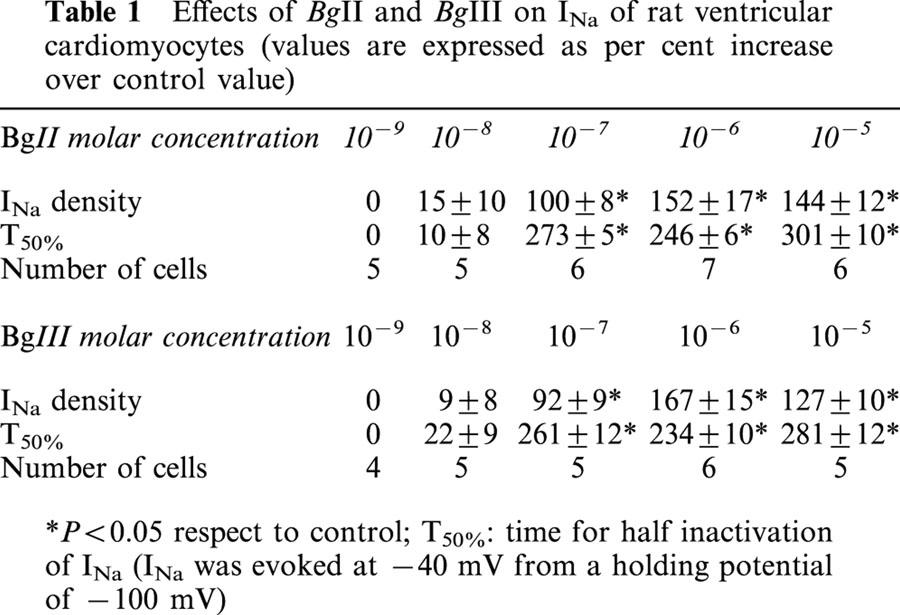
In the present experimental conditions (i.e. 5 mM external Na+ concentration), the inactivation time course of INa could be described by a double exponential function in about 50% of the cells. In the other half, a best fit was obtained by a single exponential function. In addition, in most cells, the inactivation time course in the presence of toxin was best described by a single exponential. For these reasons, we chose to measure the half inactivation time of INa (T50%) as an indication of the slowing of the inactivation time course induced by BgII and BgIII (Table 1).
The effects of both BgII and BgIII on INa were rapidly achieved (Figure 4A, B). Steady-state effects were commonly reached in 10 – 15 s and were stable throughout the 4 – 5 min period of exposure to the toxins. Washout of toxin effect was complete for both BgII and BgIII and took approximately 30 s (Figure 4A,B). The EC50 of the toxins on INa determined by a sigmoidal fit is 58±8 nM for BgII (n=5) and 78±48 nM for BgIII (n=5). Moreover, these actions of BgII and BgIII on INa were voltage-dependent, the effects being more marked at positive potentials. Normalized current to voltage relationships were established under control condition and after superfusion of the myocytes with 0.1 and 10 μM BgII or BgIII (Figure 4C,D). Under non-normalized conditions, 10 μM BgII and BgIII increased INa at −40 mV to 144.3±12.0% and 127.4±10.2%, of controls, respectively. At +60 mV, the large outward INa was increased to 306.2±15.2% and 304.6±16.5% of controls, respectively.
Figure 4.
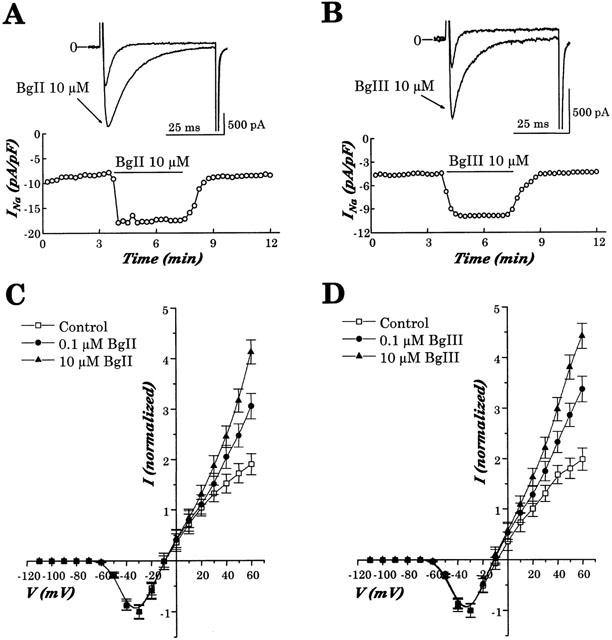
Effect of BgII and BgIII toxins on Na+ currents in rat ventricular cardiomyocytes. (A,B) Time course of the effects of 10 μM BgII or 10 μM BgIII on INa of a rat ventricular cardiomyocyte. Points represent INa density values (pA/pF) at −40 mV. The holding potential was −100 mV. The thick horizontal line indicates the period during which the cell was superfused with the toxin. The inset on the top shows current traces recorded in control condition and during the action of toxins. Note in these cells, the marked increase in current amplitude and the slowing of inactivation. (C,D) Averaged normalized current-voltage relationship of INa established in six cells under control conditions and in the presence of 0.1 and 10 μM BgII (A) or 0.1 10 μM BgIII (B).
The actions of BgII and BgIII on INa were further characterized at a concentration of 10 μM by establishing availability and recovery from inactivation (reactivation) curves (see Methods). Under control condition, best fittings of the experimental means to a Boltzmann function gave a half inactivation potential and slope factor of −80±2 mV and 6.6±0.3 mV, respectively. After application of 10 μM BgII (n=6) and BgIII (n=5) they were −83±3 mV and 7.8±0.4 mV and −77±3 mV and 6.2±0.4 mV, respectively. Except for the change in the slope factor in the presence of BgII (P<0.05), these values were not significantly different from the ones measured in control. However, it should be emphasized that both BgII or BgIII increased INa availability at potentials between −60 and −40 mV with respect to control. For example, at −40 mV availability increased from 0 to 0.13±0.01 and to 0.10±0.01 with BgII and BgIII respectively (data not shown).
Recovery from inactivation (reactivation) was established in control condition and after superfusion of the cells with 10 μM BgII and BgIII. In control condition, the time to reach the half reactivation level of INa was 35.2±6.0 ms. Neither BgII (n=4) nor BgIII (n=4) exerted significant effects on INa recovery from inactivation; times for half reactivation were 32.5±5.3 ms and 31.8±7.3 ms, respectively.
Effect of BgII and BgIII toxins on cloned hH1 Na+ channel expressed in Xenopus oocytes
Families of Na+ current recorded using the two-electrode voltage-clamp technique on Xenopus oocytes expressing hH1 channels are shown in Figure 5. Currents traces were evoked by step depolarizations of 25 ms ranging from −70 to +45 mV, using intervals of 5 mV, from a holding potential of −90 mV. While in control conditions (Figure 5A), Na+ currents inactivated rapidly, the inactivation kinetics strongly slowed down at the different voltages in the presence of 1 μM BgII (Figure 5B) or 60 μM BgIII (Figure 5C) toxin. The slowing of the inactivation process induced by 1 μM of BgII was stronger than the one induced by 60 μM of BgIII. While almost all the channels closed after 25 ms in control conditions, a bigger proportion of channels remained open and led to a persistent current in the presence of the toxins. Another interesting observation is that, although in control conditions only inward Na+ currents can be seen in the voltage range of −70 to +45 mV, clear outward Na+ currents appeared at the most positive voltages, in the presence of 1 μM BgII or 60 μM BgIII toxin.
Figure 5.
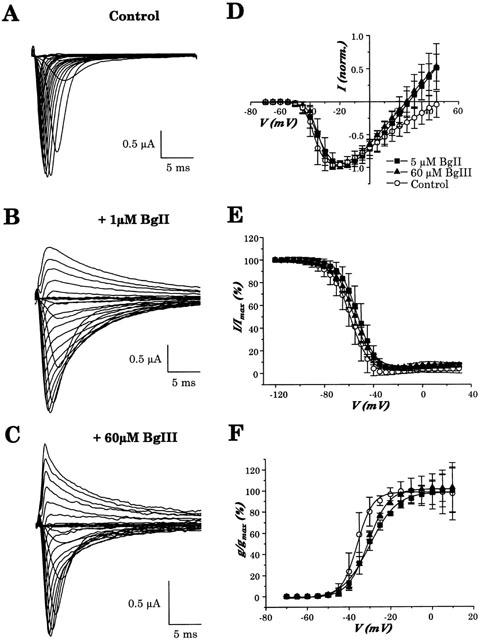
Effect of BgII and BgIII toxins on the cloned hH1 channel expressed in Xenopus oocytes. (A – C) Representative family of current traces evoked in an oocyte expressing hH1 channel by depolarizations ranging between −70 to+45 mV, using 5 mV increments, from a holding potential of −90 mV, in control conditions (A), in the presence of 1 μM BgII toxin (B) and in the presence of 60 μM BgIII toxin (C). Note that the slowing of inactivation is more marked with BgII than with BgIII and also the emergence of outward currents in the presence of both toxins. (D) Averaged normalized current-voltage relationship of hH1 in control conditions, in the presence of 5 μM of BgII toxin or 60 μM of BgIII toxin. (E) Averaged normalized steady-state inactivation in control condition, in the presence of 5 μM of BgII toxin or 60 μM of BgIII toxin. (F) Average normalized steady-state activation in the absence of toxins, in the presence of 5 μM of BgII toxin or 60 μM of BgIII toxin. In (D – F), data are mean±s.e.mean of n=20, n=6 and n=4 experiments, respectively.
Current to voltage (I-V) relationships are displayed in the Figure 5D, in the absence and presence of 5 μM and 60 μM of BgII and BgIII, respectively. The I-V curves reveal that BgII and BgIII induced a depolarizing shift in the threshold of activation of the hH1 channel. This shift is confirmed by the voltage dependence of steady-state activation shown in Figure 5F. In control conditions, the half activation voltage (V1/2act) and the slope factor measured by a Boltzmann fit of the activation curve are −37.0±0.2 mV and 3.8±0.2 mV (n=20), respectively. In the presence of 5 μM BgII, V1/2act is −30.0±0.4 mV and the slope factor is 6.5±0.4 mV (n=6). In the presence of 60 μM BgIII, V1/2act is −30.8±0.3 mV and the slope factor is 5.0±0.3 mV (n=4). Thus, BgII and BgIII induce a relatively similar shift of +7.0 mV and +6.2 mV of the V1/2act values, respectively but BgII is more potent than BgIII. Moreover, as can be anticipated from the emergence of outward currents upon toxin application in Figure 5A – C, the reversal potential (Erev) changes in the presence of toxins (Figure 5D). In control conditions, Erev is 44.4±7.4 mV (n=20) while Erev measured in the presence of 5 μM BgII and 60 μM BgIII is 26.3±6.4 mV (n=6) and 27.0±7.4 mV (n=4), respectively. This will be further discussed below.
Figure 5E shows the voltage dependence of steady-state sodium channel inactivation in the absence and presence of 5 and 60 μM of BgII and BgIII, respectively. Measurements of steady-state inactivation were made following a standard two-pulse protocol. A 50 ms conditioning pulse ranging from −120 to 30 mV was followed by a 50 ms test pulse at −20 mV. In control conditions, a prepulse of −120 mV forces all the channels in the resting state and make these fully available for activation during the test pulse. In contrast to this, a prepulse of −30 mV moves all the channels in the inactivated state and thus non-available for activation during the test pulse. The voltage at which half of the channels are inactivated (V1/2inact) and the slope factor determined by a Boltzmann fit of the inactivation curve are −58.9±0.4 and 7.4±0.3 mV, respectively. BgII and BgIII induce a depolarizing shift in the steady-state inactivation of the hH1 channel. In the presence of 5 μM BgII, V1/2inact and the slope factor are −53.2±0.3 mV and 7.1±0.2 mV (n=6), respectively. In the presence of 60 μM BgIII, V1/2inact and the slope factor are −56.1±0.3 mV and 7.3±0.3 mV (n=4), respectively. Thus, saturating concentrations of BgII and BgIII cause a shift of+5.7 mV and +3.8 mV, respectively, in the voltage at which half the hH1 Na+ channels are inactivated.
The inactivation time course of hH1 increased with the concentration of toxin (Figure 6). Figure 6A,B display the effect of different concentrations of BgII and BgIII on Na+ currents carried by hH1 and evoked by a step depolarization to 0 mV for 25 ms from a holding potential of −90 mV. As can be seen on these different current traces, the slowing of the inactivation induced by BgII and BgIII is clearly concentration-dependent, with an effect more pronounced for BgII than BgIII. The inactivation process can be fitted satisfactorily by a single exponential. The relationship between BgII and BgIII concentration and the obtained τ-values are displayed in Figure 6C,D, respectively. It can be seen that τ increased as a function of the concentration of the toxins. In control condition, τ was 1.0±0.1 ms (n=17). In the presence of 1 μM BgII or 100 μM BgIII, τ increased to 5.8±0.6 (n=5) and 4.6±0.4 ms (n=3), respectively. The increase of τ is thus approximately 25% more important in the presence of BgII than in the presence of BgIII. The concentration of the toxin producing the half maximum effect on the inactivation time constant (EC50) determined by a sigmoidal fit is 382±140 nM for BgII and 7.8±1.2 μM for BgIII. From this follows that BgII is approximately 20 times more potent than BgIII.
Figure 6.
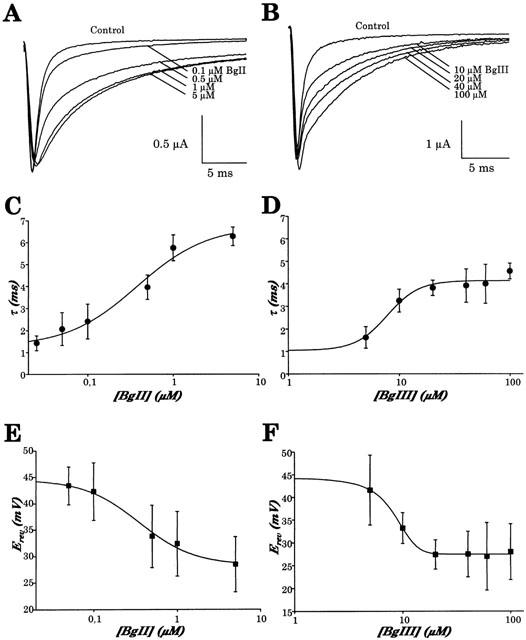
Concentration dependence of the slowing of inactivation and of the modification of the reversal potential induced by BgII and BgIII toxins on hH1 Na+ channels. (A) Current traces were evoked by a step depolarization to 0 mV lasting 25 ms from a holding potential of −90 mV, in the absence (control) and in the presence of increasing concentrations of BgII toxin (as indicated), using the same oocyte. (B) Same protocol as in (A) but in the absence (control) and in the presence of increasing concentrations of BgIII toxin (as indicated), in the same oocyte. (C) Averaged time constant of inactivation (τ) plotted vs concentration of BgII toxin. Time constants of inactivation were calculated from a first order exponential fit of current traces evoked by a step depolarization to 0 mV from a holding potential of −90 mV. The EC50 value determined by a sigmoidal fit is 382±140 nM. Data are the mean±s.e.mean of at least three experiments. (D) Averaged time constant inactivation (τ) plotted vs concentration of BgIII toxin. Time constants of inactivation were calculated as in (C). The EC50 value as determined by a sigmoidal fit of τ is 7.8±1.2 μM. (E) Averaged reversal potential plotted vs concentration of BgII toxin. The EC50 value determined by a sigmoidal fit is 340±70 nM. (F) Averaged reversal potential plotted vs concentration of BgIII toxin. The EC50 value determined by a sigmoidal fit is 8.4±0.3 μM. Data are the mean±s.e.mean of at least three experiments at each concentration.
As can be seen in the current-voltage relationships in Figure 5D, BgII and BgIII induce a shift of the reversal potential. In control conditions, the reversal potential of Na+ currents generated by hH1 channels is 44.4±7.4 mV (n=20). In the presence of 5 μM BgII and 100 μM BgIII, Erev is 28.5±5.2 mV and 28.0±6.1 mV, respectively (n=4 and 3). The maximal shifts of Erev as induced by BgII and BgIII are very similar, −15.9 and −16.4 mV, respectively. A statistical analysis shows that the differences of Erev in the absence or in the presence of toxins are significant (Student's t-test, P<0.05). The permeability ratio of Na+ over K+ (PNa/PK) determined from the measured Erev using the GHK equation (see Methods) decreases from 84 in control condition to 12.3 and 12.7 in the presence of 5 μM BgII and 100 μM BgIII, respectively. This represents a 6.8 and 6.6 fold decrease in the ionic selectivity for Na+ over K+ of the hH1 channel in the presence of the toxins. Similarly, the modification of the reversal potential induced by BgII and BgIII observed in a different external concentration of Na+ (96 mM) resulted in a 7 and 6.7 fold decrease in the ionic selectivity for Na+ over K+ in the presence of 5 μM BgII and 100 μM BgIII, respectively (data not shown). In the same experimental conditions (40 mM external Na+), the reversal potential of hH1 is not modified in the presence of 1 μM ATX II (data not shown).
Figure 6E,F display the concentration dependence of the reversal potential of hH1 channels as a function of the concentration of BgII or BgIII toxins. The EC50 determined by a sigmoidal fit is 340±70 nM for BgII and 8.4±0.3 μM for BgIII. These EC50 values are very comparable with the ones obtained by fitting the toxin concentration dependence of the time constant of inactivation. In both cases, i.e. Erev and time constant of inactivation measurements, it is clear that BgII is more potent than BgIII.
Discussion
In rat ventricular strips, BgII and BgIII significantly prolong the action potentials and induce a decrease of the resting potential in a concentration-dependent way. The half maximum effect (EC50) on the action potential duration at 0 mV is 60 nM for BgII and 663 nM for BgIII. In rat cardiac myocytes, patch-clamp studies point out that BgII and BgIII, at concentrations ranging from 0.1 to 10 μM, increased Na+ current density (with an effect more pronounced at positive voltages) and delayed its inactivation time course (Figure 4). The electrophysiological effect of BgII and BgIII relates to the mechanisms of action of other Na+ channels toxins from sea anemone, for example the well known ATX II from Anemonia sulcata (Alsen et al., 1981; Chahine et al., 1996; Ravens, 1976; Ulbricht & Schmidtmayer, 1981), ApA and ApB from Anthopleura xanthogrammica (Benzinger et al., 1997; Kudo & Shibata, 1980; Shibata et al., 1976), or the recently isolated APE 1.1, APE 1.2, APE 2.1, APE 2.2 and APE 5.3 from Anthopleura elegentissima (Bruhn et al., 2001). Similarly to BgII and BgIII, these toxins inhibit the inactivation process of Na+ channels which leads to cardiotoxic (arrhythmia) and neurotoxic (repetitive firing) effects. In comparison, the EC50 values of the Na+ influx increase induced by ApA, ApB and ATXII in rat cardiac tissues are 3, 2 and 15 nM, respectively (Schweitz et al., 1981).
Although in this study, we focused on the cardiotoxicity of these toxins, Loret et al. (1994) have shown in their experiments that BgII and BgIII are also potent neurotoxins. When injected intracerebroventricularly to mice, BgII and BgIII present LD50 values of 0.4 and 21 μg kg−1, respectively. Moreover, binding competition assays of these toxins with Aahll, a scorpion toxin also acting on site 3 of Na+ channels, resulted in a Kd of 9 nM for BgII and 72 nM for BgIII. Likewise, recent patch-clamp experiments in cultured neurons have shown that BgII is able to interact with neuronal Na+ channels (E. Salceda, personal communication).
In order to deepen our comprehension on the molecular mechanisms of action of BgII and BgIII toxins, we performed voltage-clamp studies of the cloned cardiac Na+ channel hH1 expressed in Xenopus oocytes. Our experiments show that the main effect induced by BgII and BgIII is a marked and concentration-dependent slowing of the inactivation process of Na+ current associated with a decrease of the ionic selectivity of the channel. Interestingly, in comparison to the data obtained in cardiomyocytes, no clear effects on the current amplitude were observed. Our results indicate that BgII is about 20 times more potent than BgIII. The concentrations for which the half maximal effect on the inactivation process are observed (EC50) are 0.38 and 7.8 μM for BgII and BgIII, respectively. In comparison, the EC50 of ATX II on cloned hH1 channels expressed in mammalian cells is 11 nM (Chahine et al., 1996). BgII, and even more BgIII, thus appear to be less powerful inhibitors of the Na+ channel inactivation process than ATX II. Nevertheless, this conclusion has to be considered, at least in part, by the fact that the pharmacological characteristics of the heterologous expression system used (i.e. mammalian cells vs Xenopus oocytes) may be different.
The difference in efficiency between BgII and BgIII could be surprising regarding the almost identical amino acid sequences of these two toxins. However, our findings are consistent with the results of a previous study by Loret et al. (1994) who have shown that the toxicity of BgII injected intracerebroventricularly in mice is more than 50 times superior than the toxicity of BgIII. In the same study, the higher toxicity of BgII is correlated with a higher competition binding with the scorpion α-toxin AaH II to rat brain synaptosomes. All together, these data strongly suggest that the asparagine in position 16, which is a very conservative residue among type 1 toxins, is an important residue for the toxicity of these toxins.
In parallel to the action on the inactivation process of Na+ currents, our study also reveals that BgII and BgIII induce several other modifications of the electrophysiological properties of hH1 channels. Firstly, the steady-state activation and inactivation parameters are shifted to more positive values by the toxins. In comparison, the steady-state activation of hH1 expressed in mammalian cells is not affected by ATX II while the steady-state inactivation is shifted to a more positive potential by about 3 mV (Chahine et al., 1996).
The most intriguing observation, in the presence of both toxins, is the shift of the reversal potential of Na+ current generated by hH1. The measured shifts correspond to a 7 fold decrease of the ionic selectivity for Na+ over K+. In comparison, in myocytes, no modifications of the Erev are observed upon addition of BgII and BgIII. In control conditions and in the presence of toxins, Erev remains ∼−10 mV. In these experiments, the internal concentration of Na+ is higher than the external one (7 mM and 5 mM, respectively) and there is no K+ in solution since it has been replaced by TEA+ and Cs+. One explanation could be that the Erev in the presence of the toxin is not shifted, because, even if the selectivity of the channel is modified, K+ is not present and, except Na+, no other monovalent cations are able to pass through the channel. The EC50 values for the observed changes in Erev are 0.34 and 8.4 μM for BgII and BgIII respectively. Here again the potency of BgII is higher than the potency of BgIII, confirming the importance of the asparagine in position 16 for the binding and the toxicity of these toxins. Interestingly, the efficacy of BgII and BgIII on the change in reversal potential is very similar (a shift of approximately −16 mV in both cases), suggesting that the mechanism by which BgII and BgIII affect the ionic selectivity of hH1 is different from the one affecting the inactivation time course or steady-state activation. To the best of our knowledge, the modification of the ionic selectivity of Na+ channel has never been observed with any other sea anemone toxins. Nonetheless, several other toxins have been reported to affect the ionic selectivity of Na+ channels. For example, two alcaloid toxins (batrachotoxin isolated from the skin of the Colombian arrow poison frog of the genera Dendrobates and Phyllobates, as well as aconitine from the plant Aconitinum napellus) are able to decrease the ionic selectivity of Na+ channels (Denac et al., 2000; Hille, 1992). It is thought that these toxins act by a widening of the selectivity filter. The receptor site of these lipid-soluble toxins is localized within the plasma membrane (receptor site 2) while BgII and BgIII bind to a site localized in the extracellular loop between segments S3 and S4, in the fourth domain (D4) (receptor site 3) (Loret et al., 1994). It should be mentioned that another toxin which binds to site 3, the α-toxin AaH II from the scorpion Androctonus australis Hector, has also been reported to modify the reversal potential of Na+ currents (Benoit & Dubois, 1987). On the one hand, it remains difficult to explain how toxin binding to an external site not localized within the pore could decrease the ionic selectivity of the channel. On the other hand, given the negatively charged residues in the outer mouth of rat sodium channels controlling the ion flux and selectivity (Chiamvimonvat et al., 1996), it could be hypothesized that the apparent decrease in ionic selectivity of the channel could result from a modification of the surface charges of the external part of the channel due to the binding of the toxin. In this view, the toxin can be considered as a pre-filter interfering with the access of ions to the pore.
In the present study, we have characterized and compared the mode of action of two toxins isolated from the sea anemone Bunodosoma granulifera, BgII and BgIII, on action potentials of rat ventricular strips, on Na+ currents in rat cardiomyocytes and on the cloned cardiac Na+ channel hH1 expressed in Xenopus oocytes. This work is the first electrophysiological characterization of these toxins and reveals that BgII and BgIII interact with Na+ channels in cardiac cells, pointing out that these toxins, generally known as neurotoxins, are also cardiotoxic. From a mechanical point of view, our work discloses that, beside the ‘classical' effects of sea anemone Na+ channels toxins (slowing of inactivation kinetics, shift of steady-state activation and inactivation parameters), BgII and BgIII also induce a decrease of the ionic selectivity of Na+ channels.
Acknowledgments
We are grateful to Evelyne Dubois and Chris Ulens for fruitful discussions. The hH1 clone was kindly provided by R.G. Kallen. This work was supported by a K.U. Leuven post-doctoral fellowship to C. Goudet and a grant Z-005 from Consejo Nacional de Ciencia y Tecnologia (CONACyT), Mexican Government to L.D. Possani.
Abbreviations
- ApA and ApB
Anthopleura xanthogrammica toxins A and B
- ATX I and ATX II
Anemonia sulcata toxins I and II, BgII and BgIII, Bunodosoma granulifera toxins II and III
- HEPES
2-[4-(2-hydroxyethyl)-1-piperazinyl] ethanesulphonic acid
- Sh I
Stychodactyla helianthus toxin I
- Tris
Tris(hydroxymethyl)aminomethane
- TTX
Tetrodotoxin
- VGSC
Voltage-Gated Sodium Channel
References
- ALSEN C., HARRIS J.B., TESSERAUX I. Mechanical and electrophysiological effects of sea anemone (Anemonia sulcata) toxins on rat innervated and denervated skeletal muscle. Br. J. Pharmacol. 1981;74:61–71. doi: 10.1111/j.1476-5381.1981.tb09955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ALVAREZ J., DORTICO S.F., MORLANS J. Changes in electrical and mechanical activities of rabbit papillary muscle during hypoxic perfusion. J. Physiol. (Paris) 1981;77:807–812. [PubMed] [Google Scholar]
- ANEIROS A., GARCIA I., MARTINEZ J.R., HARVEY A.L., ANDERSON A.J., MARSHALL D.L., ENGSTROM A., HELLMAN U., KARLSSON E. A potassium channel toxin from the secretion of the sea anemone Bunodosoma granulifera. Isolation, amino acid sequence and biological activity. Biochim. Biophys. Acta. 1993;1157:86–92. doi: 10.1016/0304-4165(93)90082-j. [DOI] [PubMed] [Google Scholar]
- BALSER J.R. Structure and function of the cardiac sodium channels. Cardiovasc. Res. 1999;42:327–338. doi: 10.1016/s0008-6363(99)00031-0. [DOI] [PubMed] [Google Scholar]
- BENOIT E., DUBOIS J.M. Properties of maintained sodium current induced by a toxin from Androctonus scorpion in frog node of Ranvier. J. Physiol. (Lond.) 1987;383:93–114. doi: 10.1113/jphysiol.1987.sp016398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BENZINGER G.R., DRUM C.L., CHEN L.Q., KALLEN R.G., HANCK D.A., HANCK D. Differences in the binding sites of two site-3 sodium channel toxins. Pflugers Arch. 1997;434:742–749. doi: 10.1007/s004240050460. [DOI] [PubMed] [Google Scholar]
- BRUHN T., SCHALLER C., SCHULZE C., SANCHEZ-RODRIGUEZ J., DANNMEIER C., RAVENS U., HEUBACH J.F., ECKHARDT K., SCHMIDTMAYER J., SCHMIDT H., ANEIROS A., WACHTER E., BERESS L. Isolation and characterisation of five neurotoxic and cardiotoxic polypeptides from the sea anemone Anthopleura elegantissima. Toxicon. 2001;39:693–702. doi: 10.1016/s0041-0101(00)00199-9. [DOI] [PubMed] [Google Scholar]
- CATTERALL W.A. Structure and function of voltage-gated ion channels. Annu. Rev. Biochem. 1995;64:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- CATTERALL W.A. From ionic currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- CESTELE S., CATTERALL W.A. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie. 2000;82:883–892. doi: 10.1016/s0300-9084(00)01174-3. [DOI] [PubMed] [Google Scholar]
- CHAHINE M., PLANTE E., KALLEN R.G. Sea anemone toxin (ATX II) modulation of heart and skeletal muscle sodium channel alpha-subunits expressed in tsA201 cells. J. Memb. Biol. 1996;152:39–48. doi: 10.1007/s002329900083. [DOI] [PubMed] [Google Scholar]
- CHIAMVIMONVAT N., PEREZ-GARCIA M.T., TOMASELLI G.F., MARBAN E. Control of ion flux and selectivity by negatively charged residues in the outer mouth of rat sodium channels. J. Physiol. (Lond.) 1996;491:51–59. doi: 10.1113/jphysiol.1996.sp021195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DASCAL N. The use of Xenopus oocytes for the study of ion channels. CRC Crit. Rev. Biochem. 1987;22:317–387. doi: 10.3109/10409238709086960. [DOI] [PubMed] [Google Scholar]
- DENAC H., MEVISSEN M., SCHOLTYSIK G. Structure, function and pharmacology of voltage-gated sodium channels. Naunyn Schmiedebergs Arch. Pharmacol. 2000;362:453–479. doi: 10.1007/s002100000319. [DOI] [PubMed] [Google Scholar]
- DONGRE A.R., ENG J.K., YATES J.R., 3RD Emerging tandem-mass-spectrometry techniques for the rapid identification of proteins. Trends Biotechnol. 1997;15:418–425. doi: 10.1016/S0167-7799(97)01110-4. [DOI] [PubMed] [Google Scholar]
- GALAN L., TALAVERA K., VASSORT G., ALVAREZ J.L. Characteristics of Ca2+ channel blockade by oxodipine and elgodipine in rat cardiomyocytes. Eur. J. Pharmacol. 1998;357:93–105. doi: 10.1016/s0014-2999(98)00543-3. [DOI] [PubMed] [Google Scholar]
- GELLENS M.E., GEORGE A.L., JR, CHEN L.Q., CHAHINE M., HORN R., BARCHI R.L., KALLEN R.G. Primary structure and functional expression of the human cardiac tetrodotoxin-insensitive voltage-dependent sodium channel. Proc. Natl. Acad. Sci. U.S.A. 1992;89:554–558. doi: 10.1073/pnas.89.2.554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOLDIN A.L., BARCHI R.L., CALDWELL J.H., HOFMANN F., HOWE J.R., HUNTER J.C., KALLEN R.G., MANDEL G., MEISLER M.H., NETTER Y.B., NODA M., TAMKUN M.M., WAXMAN S.G., WOOD J.N., CATTERALL W.A. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- GORDON D., SAVARIN P., GUREVITZ M., ZINN-JUSTIN S. Functional anatomy of scorpion toxins affecting sodium channels. J. Toxicol-Toxin Rev. 1998;17:131–159. [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- HILLE B. Sinauer Associates Inc., Sunderland, Massachusetts, USA; 1992. Ionic channels of excitable membranes. [Google Scholar]
- KEM W.R., PARTEN B., PENNINGTON M.W., PRICE D.A., DUNN B.M. Isolation, characterization, and amino acid sequence of a polypeptide neurotoxin occurring in the sea anemone Stichodactyla helianthus. Biochemistry. 1989;28:3483–3489. doi: 10.1021/bi00434a050. [DOI] [PubMed] [Google Scholar]
- KUDO Y., SHIBATA S. The potent excitatory effect of a novel polypeptide, anthopleurin-B, isolated from a sea anemone (Anthopleura xanthogrammica) on the frog spinal cord. J. Pharmacol. Exp. Ther. 1980;214:443–448. [PubMed] [Google Scholar]
- LAZDUNSKI M., FRELIN C., BARHANIN J., LOMBET A., MEIRI H., PAURON D., ROMEY G., SCHMID A., SCHWEITZ H., VIGNE P., VIJVERBERG H.P.M. Polypeptide toxins as tools to study voltage-sensitive Na+ channels. Ann. N. Y. Acad. Sci. 1986;479:204–220. doi: 10.1111/j.1749-6632.1986.tb15571.x. [DOI] [PubMed] [Google Scholar]
- LIMAN E.R., TYTGAT J., HESS P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- LORET E.P., DEL VALLE R.M., MANSUELLE P., SAMPIERI F., ROCHAT H. Positively charged amino acid residues located similarly in sea anemone and scorpion toxins. J. Biol. Chem. 1994;269:16785–16788. [PubMed] [Google Scholar]
- MÉNEZ A. Functional architectures of animal toxins: a clue to drug design. Toxicon. 1998;36:1557–1572. doi: 10.1016/s0041-0101(98)00148-2. [DOI] [PubMed] [Google Scholar]
- NICHOLSON G.M., WALSH R., LITTLE M.J., TYLER M.I. Characterisation of the effects of robustoxin, the lethal neurotoxin from the Sydney funnel-web spider Atrax robustus, on sodium channel activation and inactivation. Pflügers. Arch. 1998;436:117–126. doi: 10.1007/s004240050612. [DOI] [PubMed] [Google Scholar]
- NICHOLSON G.M., WILLOW M., HOWDEN M.E., NARAHASHI T. Modification of sodium channel gating and kinetics by versutoxin from the Australian funnel-web spider Hadronyche versuta. Pflügers. Arch. 1994;428:400–409. doi: 10.1007/BF00724524. [DOI] [PubMed] [Google Scholar]
- NORTON R.S. Structure and structure-function relationships of sea anemone proteins that interact with the sodium channel. Toxicon. 1991;29:1051–1084. doi: 10.1016/0041-0101(91)90205-6. [DOI] [PubMed] [Google Scholar]
- NORTON R.S. Structure and function of peptide and protein toxins from marine organisms. J. Toxicol-Toxin Rev. 1998;17:99–130. [Google Scholar]
- NORTON T.R. Cardiotonic polypeptides from Anthopleura xanthogrammica (Brandt) and A. elegantissima (Brandt) Fed. Proc. 1981;40:21–25. [PubMed] [Google Scholar]
- POSSANI L.D., BECERRIL B., DELEPIERRE M., TYTGAT J. Scorpion toxins specific for Na+-channels. Eur. J. Biochem. 1999;264:287–300. doi: 10.1046/j.1432-1327.1999.00625.x. [DOI] [PubMed] [Google Scholar]
- RAVENS U. Electromechanical studies of an Anemonia sulcata toxin in mammalian cardiac muscle. Naunyn Schmiedebergs Arch. Pharmacol. 1976;296:73–78. doi: 10.1007/BF00498842. [DOI] [PubMed] [Google Scholar]
- REIMER N.S., YASUNOBU C.L., YASUNOBU K.T., NORTON T.R. Amino acid sequence of the Anthopleura xanthogrammica heart stimulant, anthopleurin-B. J. Biol. Chem. 1985;260:8690–8693. [PubMed] [Google Scholar]
- RENAUD J.F., FOSSET M., SCHWEITZ H., LAZDUNSKI M. The interaction of polypeptide neurotoxins with tetrodotoxin-resistant Na+ channels in mammalian cardiac cells. Correlation with inotropic and arrhythmic effects. Eur. J. Pharmacol. 1986;120:161–170. doi: 10.1016/0014-2999(86)90536-4. [DOI] [PubMed] [Google Scholar]
- ROGERS J.C., QU Y., TANADA T.N., SCHEUER T., CATTERALL W.A. Molecular determinants of high affinity binding of alpha-scorpion toxin and sea anemone toxin in the S3-S4 extracellular loop in domain IV of the Na+ channel alpha subunit. J. Biol. Chem. 1996;271:15950–15962. doi: 10.1074/jbc.271.27.15950. [DOI] [PubMed] [Google Scholar]
- SALINAS E.M., CEBADA J., VALDES A., GARATEIX A., ANEIROS A., ALVAREZ J.L. Effects of a toxin from the mucus of the Caribbean sea anemone (Bunodosoma granulifera) on the ionic currents of single ventricular mammalian cardiomyocytes. Toxicon. 1997;35:1699–1709. doi: 10.1016/s0041-0101(97)00010-x. [DOI] [PubMed] [Google Scholar]
- SCHWEITZ H., VINCENT J.P., BARHANIN J., FRELIN C., LINDEN G., HUGUES M., LAZDUNSKI M. Purification and pharmacological properties of eight sea anemone toxins from Anemonia sulcata, Anthopleura xanthogrammica, Stoichactis giganteus, and Actinodendron plumosum. Biochemistry. 1981;20:5245–5252. doi: 10.1021/bi00521a023. [DOI] [PubMed] [Google Scholar]
- SHIBATA S., NORTON T.R., IZUMI T., MATSUO T., KATSUKI S. A polypeptide (AP-A) from sea anemone (Anthopleura xanthogrammica) with potent positive inotropic action. J. Pharmacol. Exp. Ther. 1976;199:298–309. [PubMed] [Google Scholar]
- TANAKA M., HAINU M., YASUNOBU K.T., NORTON T.R. Amino acid sequence of the Anthopleura xanthogrammica heart stimulant, anthopleurin A. Biochemistry. 1977;16:204–208. doi: 10.1021/bi00621a007. [DOI] [PubMed] [Google Scholar]
- ULBRICHT W., SCHMIDTMAYER J. Modification of sodium channels in myelinated nerve by Anemonia sulcata toxin II. J. Physiol. (Paris) 1981;77:1103–1111. [PubMed] [Google Scholar]
- WILCOX G.R., FOGH R.H., NORTON R.S. Refined structure in solution of the sea anemone neurotoxin ShI. J. Biol. Chem. 1993;268:24707–24719. [PubMed] [Google Scholar]
- WUNDERER G., EULITZ M. Amino-acid sequence of toxin I from Anemonia sulcata. Eur. J. Biochem. 1978;89:11–17. doi: 10.1111/j.1432-1033.1978.tb20890.x. [DOI] [PubMed] [Google Scholar]
- WUNDERER G., FRITZ H., WACHTER E., MACHLEIDT W. Amino-acid sequence of a coelenterate toxin: toxin II from Anemonia sulcata. Eur. J. Biochem. 1976;68:193–198. doi: 10.1111/j.1432-1033.1976.tb10778.x. [DOI] [PubMed] [Google Scholar]