

Abstract
In this study we investigated the effects of the putative cardioselective sulphonylurea derivative HMR 1098 on ATP-sensitive potassium (KATP) channels from cardiac ventricular myocytes, the INS-1 β-cell line and from recombinant KATP channels composed of SUR2A/Kir6.2, SUR1/Kir6.2, SUR1/Kir6.1 an Kir6.2,ΔC26. Recombinant channels were expressed in tsA201 or COS-1 cells.
The effects of HMR 1098 on single channel and whole-cell currents were recorded using the patch-clamp technique.
At the single channel level, using excised inside-out membrane patches, HMR 1098 inhibited KATP channels from ventricular cells and INS-1 cells with IC50s of 0.88 and 720 μM respectively. Similar results to those in cardiac cells were obtained using recombinant SUR2A/Kir6.2 KATP channels. HMR 1098 inhibition of SUR2A/Kir6.2 KATP channels was unaffected by the presence of internal ADP.
In whole-cell recordings, HMR 1098 inhibited SUR2A/Kir6.2 and SUR1/Kir6.2 currents with IC50s of 2.1 and 860 μM respectively. HMR 1098 was without effect on currents either from the Kir6.2,ΔC26 truncation mutant or from Kir2.1.
Our results demonstrate that HMR 1098 is a selective inhibitor of cardiac KATP channels, showing a 400–800-fold selectivity over β-cell KATP channels. The non-aromatic substitutions in the sulphonylurea moiety greatly increase the cardioselectivity of this compound while reducing the overall blocking potency of this sulphonylurea derivative.
Keywords: KATP channels, patch-clamp, sulphonylureas, heart, pancreas, β-cell
Introduction
Sulphonylurea drugs are used extensively in the treatment of diabetes. The principal target of these drugs is the pancreatic β-cell ATP-sensitive potassium (KATP) channel. Sulphonylurea-mediated inhibition of β-cell KATP channels leads to membrane depolarization and subsequent influx of calcium through activation of voltage-dependent calcium channels, leading to insulin granule exocytosis. Sulphonylureas such as glibenclamide display a clinical selectivity for β-cell KATP channels over plasma-membrane KATP (KATP) channels found in the heart and vasculature, although the question of whether undesired cardiovascular side effects occur is still under debate (Garrat et al., 1999; Smits & Thien, 1995; Leibowitz & Cerasi, 1996). Recent advances in sulphonylurea chemistry have lead to the synthesis of novel derivatives that display tissue-specific selectivity. For example, the benzoic acid derivative mitiglinide is ∼1000-fold more selective for β-cell KATP channels (Reimann et al., 2001), while the sulphonylurea HMR 1098 seems to display selectivity for the cardiac versus β-cell plasma-membrane KATP channel (Liu et al., 2001; Gögelein et al., 1999; 1998). It has been proposed that HMR 1098 possesses antiarrhythmic actions via a direct inhibitory effect on KATP channels in the myocardium (Wirth et al., 2000). However, the apparent cardioselectivity of the sulphonylurea HMR 1098 has not been well-characterized at the single-channel and recombinant levels. Given the potential clinical importance of new sulphonylurea compounds such as HMR 1098 in the management of arrhythmias, and the possibility of undesired effects on glucose homeostasis, we set out to characterize the pharmacology of HMR 1098 with respect to inhibition of cardiac and β-cell KATP channels.
Recent molecular studies have revealed that KATP channels consist of two distinct subunits, a sulphonylurea receptor (SUR) and a pore-forming potassium channel Kir6.2 in a 4 : 4 stoichiometry (Inagaki et al., 1995; Seino, 1999). In β-cells, the SUR isoform is SUR1, while in heart and skeletal muscle it is SUR2A (Aguilar-Bryan & Bryan, 1999; Inagaki et al., 1996). It is known that these distinct SUR isoforms bestow the unique pharmacological characteristics upon the KATP channel complex (Aguilar-Bryan & Bryan, 1999; Inagaki et al., 1996). In this study we used the patch clamp technique to record single-channel and whole-cell KATP channel currents from: (1) native KATP channels in cardiac myocytes and the insulin secreting INS-1 β-cell line; and (2) expressed recombinant KATP channels of differing isoform compositions, SUR1/Kir6.2, SUR2A/Kir6.2 and SUR1/Kir6.1. Although wild-type KATP channels require both subunits present to form functional channels, the removal of the last 20 to 40 amino acids from the Kir6.2 subunit carboxy terminus (for example, Kir6.2,ΔC26) enables functional channel activity to be recorded in the absence of the sulphonylurea receptor (Tucker et al., 1997; Light et al., 2000). As such, this truncation mutant provides a useful tool in this study and the effects of HMR 1098 on Kir6.2,ΔC26 and the structurally-related strong inward rectifier Kir2.1 currents were determined. All subunit constructs were heterologously expressed in tsA201 or COS-1 cells.
Our results demonstrate that HMR 1098 is a very selective inhibitor of cardiac plasma-membrane KATP channels and that this selectivity is conferred by the SUR2A isoform found in cardiac tissue rather than the SUR1 isoform expressed in β-cells or the associated pore-forming Kir6.2 subunit.
Methods
Cell isolation and culture
Right ventricular myocytes from rat were enzymatically isolated using standard protocols described previously (Light et al., 1998). The insulin secreting β-cell line INS-1 was maintained in culture with RPMI medium supplemented with 10 mM glucose, 2 mM L-glutamine, 10% foetal calf serum and 0.1% penicillin/streptomycin. tsA201 cells (a SV40-transformed variant of the HEK293 human embryonic kidney cell line) and COS-1 cells were maintained in Dulbecco's modified Eagle's medium supplemented with 10 mM glucose, 2 mM L-glutamine, 10% foetal calf serum and 0.1% penicillin/streptomycin. INS-1, tsA201 and COS-1 cells were kept at 37°C with 10% CO2. The KATP channel subunit clones Kir6.1 and Kir6.2 from mouse were generously provided by Dr S Seino (Inagaki et al., 1995). The truncated Kir6.2,ΔC26 construct was made by the introduction of a stop codon at the appropriate position (Light et al., 2000). The SUR2A from rabbit heart was cloned in our laboratories (GENBANK accession No. AF087468). The strong inward rectifier Kir2.1 clone from mouse was generously provided by Dr L.-Y. Jan (Kubo et al., 1993). tsA201 and COS-1 cells were plated at 30–40% confluency on 35 mm culture dishes 4 h prior to transfection. Clones were inserted into the mammalian expression vector pCDNA3, and transfected into tsA201 cells using the calcium phosphate precipitation technique, or into COS-1 cells using Lipofectamine reagent as per manufacturers instructions (Life Technologies). Transfected cells were identified using fluorescence optics in combination with co-expression of the green fluorescent protein plasmid (pGreenLantern, Life Technologies). Recordings were made from cells 48–72 h after transfection.
Patch-clamp experiments
The pipette solution used for all patch recordings contained the following (in mM): KCl 140; HEPES 10; MgCl2 1.4; EGTA 1; glucose 10. The pH of the solution was adjusted to 7.4 with KOH. This solution was also used in the recording chamber to superfuse the cells/patches for experiments using symmetrical [K+]. Patch pipettes were pulled using borosilicate glass (G85150T, Warner Instrument Corp.) to yield pipettes with a resistance of 2–6 Mω when filled with pipette solution.
Single-channel recording
Standard patch-clamp techniques were used to record single-channel currents in the inside-out patch configuration. Single-channel currents were recorded at fixed holding potentials, amplified (Axopatch 200B, Axon Instruments), digitized and acquired using pClamp 8.0 software (Axon Instruments). Data were sampled at 1000 Hz and filtered at 400 Hz except where otherwise stated.
Whole-cell recordings
Once a Gω seal was formed, the patch was ruptured and cells were allowed to dialyze with the pipette solution containing 10 μM ATP. All whole cell recordings were made under symmetrical K+ conditions (140 mM) at a holding potential of 0 mV using 200 ms long voltage steps from holding potentials of −100 mV applied every 10 s. Data were acquired and analysed using the same hardware/software mentioned above. After waiting several minutes for the stabilization of whole-cell current, the amplitude of potassium current was estimated as the current blocked by 2 mM barium chloride. Experiments were started after this initial period. All patch clamp experiments were performed at room temperature (20–22°C).
Cells or membrane patches were directly exposed to test solutions via a multi-input perfusion pipette (time to change solution at the tip of the recording pipette was less than 2 s).
Analysis and statistics
For experiments on single KATP channels from ventricular myocytes and INS-1 cells, open probability was expressed as relative NPo, the product of N, the number of channels in the patch, and Po, the mean open probability. NPo was calculated by dividing the mean patch current (over a 10–30 s test period) by the mean unitary current amplitude. Mean unitary current amplitude was calculated from all-points histograms using pClamp 8.0 software (Axon Instruments). Single-channel data were fitted to the Hill equation:
where relative NPo=NPo(test[HMR 1098])/NPo(control) and n is the Hill coefficient.
Recombinant single-channel and all whole-cell current data were fitted to the Hill equation Irel=1/{1+([HMR 1098]/IC50)n}, where n is the Hill coefficient and Irel is the current relative to the maximal current observed in the absence of HMR 1098 i.e. I([HMR 1098])/I(control). Statistical significance was evaluated by Student's paired t-test. Differences with values of probability P<0.05 were considered to be significant. All values in the text are given as mean±s.e.mean.
Experimental compounds
MgATP or K2 ADP (Sigma, St. Louis, MO, U.S.A.) was added as required from a 10 mM stock, which was prepared immediately before use. HMR 1098 (sodium salt of HMR 1883) was dissolved as stock solutions of various concentrations in distilled water and stored at 4°C.
Results
The effects of HMR 1098 on native KATP channels
Figure 1A shows the inhibitory effect of HMR 1098 on cardiac KATP channels recorded from ventricular membrane patches. In the presence of 10 μM ATP, application of 5 μM HMR 1098 to the internal face of the membrane caused a marked inhibition in KATP channel activity that was fully reversible upon washout. Analysis of current amplitude histograms revealed HMR 1098 does not affect the single-channel amplitude. Before addition of HMR 1098, the mean unitary current amplitude was 3.46±0.045 pA (n=3 patches, Figure 1Ai) and did not change significantly in the presence of 5 μM HMR 1098 (3.49±0.055 pA, P>0.05, Figure 1Aii). The use of 0.1, 0.5, 1, 5, 10, 50 and 1000 μM HMR 1098 applied to inside-out membrane patches inhibited ventricular KATP channels in a dose-dependent manner and fitting of this data set yielded an HMR 1098 inhibition curve (Figure 1C) with an IC50 of 0.88 μM (n=3–9 patches at each concentration). Using inside-out membrane patches from the insulin secreting β-cell line INS-1, much higher concentrations of HMR 1098 were required to inhibit β-cell KATP channels. Plotting of an HMR 1098 inhibition curve yielded an IC50 of 720 μM (n=6 patches), a value ∼800-fold greater than that observed with ventricular KATP channels (see Table 1).
Figure 1.
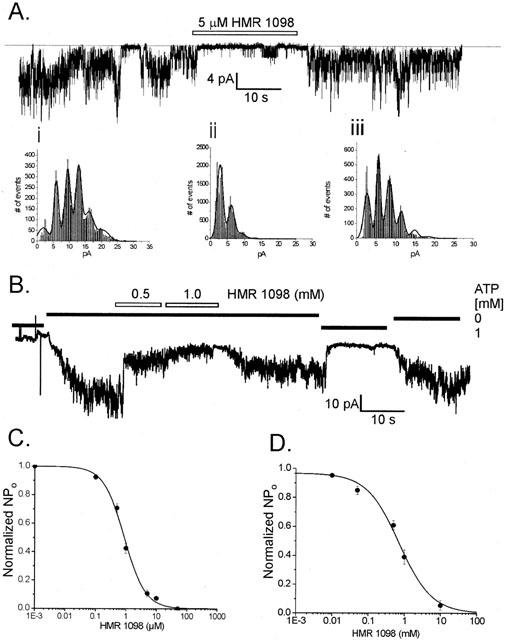
(A) Representative recording of an inside-out ventricular membrane patch containing at least five cardiac KATP channels showing the response to application of 5 μM HMR 1098. All points amplitude histograms constructed from the single channel data in (Ai) before addition of HMR 1098, (ii) in the presence of 5 μM HMR 1098 and (iii) washout of HMR 1098. (B) Typical recording of an inside-out INS-1 β-cell membrane patch containing at least 10 KATP channels in response to application of 0.5 and 1.0 mM HMR 1098. (C,D) HMR 1098 dose-response curves obtained from pooled data recordings of KATP channels from rat ventricular myocytes (C) and INS-1 β-cells (D). The IC50 estimate was obtained by grouping data from 4–9 patches at each HMR 1098 concentration and fitting to the Hill equation. ATP-sensitivity was assessed prior to HMR 1098 application (in the presence of 10 μM internal ATP). The patches were excised and held at −50 mV in symmetrical K+ (140 mM) and exposed to HMR 1098 concentrations of 1 nM, 0.1, 0.5, 1, 5, 10, 50, 100 and 1000 μM.
Table 1.
Comparison of HMR 1098 block on KATP channels using inside-out membrane patches recorded in the absence of ATP and ADP (no symbol) or presence of ATP and ADP (†) (see Results)

The effects of HMR 1098 on recombinant KATP channels
In order to compare the results from native cardiac KATP channels with recombinant cardiac KATP channel isoforms under similar conditions, inside-out patch experiments were performed. The cardiac KATP channel is encoded by a hetero-octamer consisting of the SUR2A isoform and the pore-forming Kir6.2 subunit (Inagaki et al., 1995; Seino, 1999). Co-expression of the SUR2A and Kir6.2 clones in COS-1 cells enabled the excision of inside-out membrane patches containing recombinant cardiac KATP channels. HMR 1098 inhibited macroscopic SUR2A/Kir6.2 currents in the absence of ATP (Figure 2A,B) with an IC50 of 1.02 μM (Figure 2C) and a Hill coefficient of 1.04.
Figure 2.

The effects of HMR 1098 on recombinant SUR2A/Kir6.2 KATP channels. (A,B) Representative inside-out membrane patch recordings of SUR2A/Kir6.2 multi-channel macroscopic currents. HMR 1098 inhibits current in a dose-dependent manner. The effect of HMR 1098 was tested in the absence of internal ATP. (C) HMR 1098 dose-response relationship obtained from 7–9 patches at each concentration. The data set was fitted with the Hill equation (see Methods). (D) Inside-out patch recording showing the inhibitory effect of HMR 1098 in the presence of internal 0.1 mM ATP and 0.1 mM ADP. (E) HMR 1098 dose-response relationship in the presence of ATP and ADP (n=7–8 patches at each concentration). All currents were recorded at a holding potential of −60 mV under symmetrical (140 mM K+) conditions.
It has previously been found that the selectivity of pharmacological modulators such as sulphonylureas and diazoxide for KATP channels is altered in response to changes in intracellular nucleotide levels (Gribble et al., 1998; D'hahan et al., 2000). For example, the physiological selectivity of glibenclamide for SUR1 over SUR2A is thought to result from the reduction in affinity for SUR2A in the presence of ADP (Gribble et al., 1998; 1997). Therefore, experiments were performed in the presence of 0.1 mM MgATP and 0.1 mM ADP to test whether the potency of HMR 1098 block is affected by intracellular nucleotides. Figure 2D shows a typical macroscopic SUR2A/Kir6.2 current recording in response to HMR 1098 in the continued presence of 0.1 mM MgATP and ADP. Fitting of the grouped data from 7–8 patches at each concentration yielded an IC50 of 0.82 μM and a Hill coefficient of 0.93 for HMR 1098 inhibition (Figure 2E). These values were very similar to those observed in both native ventricular and recombinant SUR2A/Kir6.2 KATP channels (see Table 1).
In the above inside-out patch experiments, HMR 1098 was applied directly to the internal face of membrane patches. However, HMR 1098 is much less lipophilic than antidiabetic sulphonylureas such as glibenclamide and the pharmacological route of entry of such drugs is from the outside of the cell. Therefore, whole-cell experiments were performed when HMR 1098 was applied to the external surface of the cell membrane. In these experiments, tsA201 cells were transiently transfected with different combinations of the subunit clones. HMR 1098 inhibited SUR2A/Kir6.2 current in a dose dependent manner (Figure 3Ai). Currents were measured at 0.1, 0.5, 1, 10 and 1000 μM HMR 1098. Fitting of the HMR 1098 dose-response relationship determined the IC50 and Hill coefficient to be 2.08 μM and 0.68 respectively (Figure 3Aii). This IC50 value was 2.36 fold higher than that observed for SUR2A/Kir6.2 and the native cardiac KATP channel recorded using the inside-out patch configuration.
Figure 3.
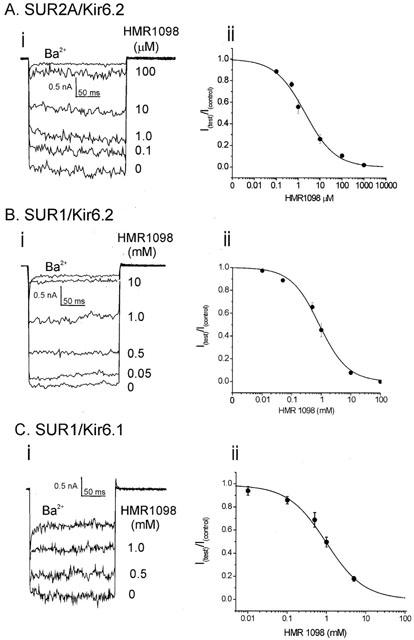
The effects of HMR 1098 on recombinant KATP channel whole-cell currents. (A, B and C) are (i) representative whole-cell currents traces and (ii) grouped data fits from tsA201 cells expressing SUR2A/Kir6.2, SUR1/Kir6.2 and SUR1/Kir6.1 KATP channels respectively (data pooled from 4–8 cells for each group). Whole-cell currents were recorded under symmetrical K+ (140 mM) conditions and were elicited by stepping to −100 mV for 200 ms from a holding potential of 0 mV. The amount of potassium selective inward current is indicated by the external application of 2 mM barium chloride (Ba2+). Grouped data were fitted to the Hill equation (see Methods) and parameters are summarized in Table 1.
The effect of HMR 1098 was also studied on the β-cell KATP isoform, SUR1/Kir6.2. In contrast to SUR2A/Kir6.2, inhibition of SUR1/Kir6.2 current required much higher concentrations of HMR 1098 for inhibition (Figure 3Bi). SUR1/Kir6.2 currents were measured at HMR concentrations of 0.01, 0.05, 0.5, 1, 10 and 100 mM. Fitting of the HMR 1098 dose-response relationship determined the IC50 and Hill coefficient to be 860 μM and 0.83 respectively (Figure 3Bii). The recombinant SUR1/Kir6.2 is ∼400 fold less sensitive to HMR 1098 than the SUR2A/Kir6.2 cardiac KATP channel subunit combination in whole-cell recordings.
In addition to KATP channels being found in the plasma-membrane, KATP channels are also expressed in the mitochondrial inner-membrane (mitoKATP channels). Although the molecular makeup of mitoKATP channels is unknown, it has recently been suggested that these channels possess a very similar pharmacological profile to the recombinant SUR1/Kir6.1 (Liu et al., 2001). Therefore, the effects of this sulphonylurea were tested using tsA201 cells transiently transfected with the SUR1 and Kir6.1 clones. HMR 1098 inhibited whole-cell SUR1/Kir6.1 currents in a dose-dependent manner (Figure 3Ci). Fitting of the grouped data from five cells yielded an IC50 of 980 μM with a Hill coefficient of 0.90 (Figure 3Cii). These values were very similar to those observed with the SUR1/Kir6.2 subunit combination.
The principal high-affinity binding site of sulphonylurea compounds is the sulphonylurea subunit of the KATP channel complex (Gribble et al., 1998). However recent studies have suggested that sulphonylureas may also bind to and inhibit the pore-forming Kir6.2 subunit with a lower affinity (Song & Ashcroft, 2001; Reimann et al., 2001; Gribble et al., 1998). We therefore decided to test the effects of HMR 1098 on the Kir6.2 subunit in the absence of the SUR subunit using the Kir6.2,ΔC26 truncation mutant. Whole cell-currents were measured using HMR 1098 concentrations of 10, 50, 500 and 1000 μM. Unlike other sulphonylureas, HMR 1098 did not inhibit the Kir6.2,ΔC26 current at any of the concentrations tested (Figure 4A,C).
Figure 4.
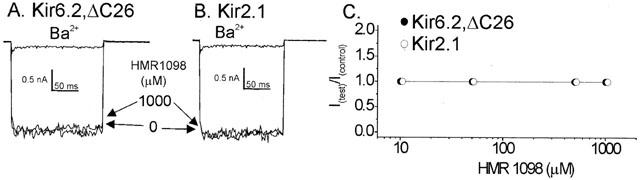
The lack of effect of HMR 1098 on Kir6.2,ΔC26 (A) and Kir2.1 (B) whole-cell currents measured from transfected tsA201 cells. (C) Pooled data from 4–7 cells for each group. Whole-cell currents were recorded under symmetrical K+ (140 mM) conditions at were elicited by stepping to −100 mV for 200 ms from a holding potential of 0 mV. The amount of potassium selective inward current is indicated by the external application of 2 mM barium chloride (Ba2+).
In order to test for non-selective effects of HMR 1098 on other inward rectifier potassium channels, the strong inward rectifier Kir2.1 was studied in tsA201 cells. Whole cell-currents were measured in the presence of 10, 50, 500 and 1000 μM HMR 1098. Kir2.1 current was not inhibited by HMR 1098 at any concentration (Figure 4B,C).
Discussion
Results from this study demonstrate that the sulphonylurea HMR 1098 is a selective inhibitor of cardiac plasma-membrane KATP channels. In inside-out patches, the IC50s were 0.88 and 1.02 μM for native ventricular and recombinant SUR2A/Kir6.2 KATP channels. In whole-cell recordings, HMR 1098 blocked SUR2A/Kir6.2 currents with an IC50 of 2.08 μM and this value is consistent with the value of 1.5 μM found previously for SUR2A/Kir6.2 channels (Liu et al., 2001).
In contrast to the observed IC50s of ∼1–2 μM for HMR 1098 on cardiac KATP channels, HMR 1098 inhibited β-cell KATP channels with IC50s of 720 and 860 μM for INS-1 β-cell and SUR1/Kir6.2 KATP channels respectively (Table 1). It has previously been reported, using inside-out patches, that sulphonylurea such as glibenclamide and glimepiride inhibit cardiac and β-cell KATP channels with similar potency – IC50s between 3–30 nM (Gribble et al., 1998; Song & Ashcroft, 2001). Our results show that while HMR 1098 inhibits cardiac KATP channels in the low micromolar range, this compound is 400–800 fold more potent for the cardiac KATP channel than for the β-cell isoform (Table 1). However, a recent study reported an HMR 1098 IC50 of ∼10 μM for SUR1/Kir6.2 and a lowered potency of HMR 1098 block with the SUR2A/Kir6.1 combination compared to SUR2A/Kir6.2 (Liu et al., 2001). These apparent discrepancies between their study and ours may be due to differences in recording techniques. In the study by Liu et al. (2001) SUR1/Kir6.2 , SUR2A/Kir6.2(or 6.1) currents were activated by pinacidil, a KATP channel opener, whereas in our study we activated KATP channels by either removal of ATP from the inside of the patch or by dialyzing low ATP in whole-cell experiments. The interactions between KATP channel openers and inhibitors remains to be tested experimentally. At the mechanistic level, single-channel analysis demonstrates that HMR 1098, like other sulphonylurea, inhibits potassium flow through KATP channels by reducing the open probability of KATP channels rather than any effects on the unitary current amplitude through individual channels (Figure 1A).
Comparison of sulphonylurea structures indicate that non-aromatic substitutions within the sulphonylurea moiety may confer the distinct cardioselectivity of HMR 1098 (Figure 5). It seems likely that the benzamido group confers binding to the SUR family, whereas modifications in the sulphonylurea group, while reducing overall affinity for SURs in general, also bestows a pharmacological selectivity between the SUR2A and SUR1 isoforms. Removal of the sulphonylurea group alone is likely not sufficient to afford isoform selectivity as meglitinide, a sulphonylurea truncated glibenclamide derivative (Figure 5), is equipotent in blocking both cardiac and β-cell KATP channels (Gribble et al., 1998).
Figure 5.

Chemical structures of the sulphonylureas HMR 1098, glibenclamide, glimepiride and the benzoic acid derivative meglitinide.
It is known that the effects of glibenclamide on β-cell KATP channels are essentially irreversible (Gribble et al., 1998). In contrast, the effects of even millimolar HMR 1098 were reversible in this present study (see Figure 1A,B). This may be explained by a combination of the much higher aqueous solubility (reduced lipophilicity) of HMR 1098 and the lower affinity of HMR 1098 for the SUR subunit compared to other sulphonylureas. Moreover, the 2 fold increase in IC50 for HMR 1098 on SUR2A/Kir6.2 currents observed in whole-cell recordings compared to inside-out patches may reflect the requirement for the compound to pass through the cell-membrane when applied externally in whole-cell experiments.
The absence of HMR 1098 inhibition of current using the Kir6.2,ΔC26 truncation mutant strongly suggests that this compound normally binds to a high affinity site on the sulphonylurea receptor. The non-aromatic substitutions in the sulphonylurea group likely account for the loss of a low affinity site on Kir6.2, as the aromatic sulphonylureas glibenclamide and glimepiride also possess low affinity sites on Kir6.2 (Gribble et al., 1998; Song & Ashcroft, 2001). Best fit of the HMR 1098 dose-dependent inhibition data to a single binding site yielded Hill coefficients ranging between 0.68–1.24, suggesting that there is one binding site on the SUR subunit for HMR 1098. These findings, in combination with the IC50 data, indicate that the marked preference of HMR 1098 for the cardiac KATP channel isoform can be attributed to a much higher binding affinity for the SUR2A subunit than for the SUR1 subunit. The lack of HMR 1098 effect on Kir2.1 current also suggests that there are no general effects of HMR 1098 on other members of the inward rectifier family of potassium channels.
Of importance is the low affinity of HMR 1098 for the SUR1/Kir6.1 isoform combination. There is growing evidence that sulphonylurea blockade of the mitochondrial isoform of the KATP channel in the heart (mitoKATP) prevents the cardioprotective effects of ischemic preconditioning (for reviews see Gross & Fryer, 1999; Gross, 2000). Although, to date the molecular identity of the mitoKATP channels is unknown, a recent study suggests that SUR1/Kir6.1 channels possess an almost identical pharmacological profile to native mitoKATP channels (Liu et al., 2001). Data from our study also confirm that HMR 1098 is a weak inhibitor SUR1/Kir6.1 channels with an IC50 of 980 μM. Exploitation of the anti-diabetic and potentially beneficial antiarrhythmic effects of KATP channel inhibitors may be complicated by undesired effects during and post ischemia (Garrat et al., 1999), perhaps via blockade of mitoKATP channels. Evidence from our study and others (Liu et al., 2001) suggests the antiarrhythmic activity of HMR 1098, is likely to occur in the absence of mitoKATP channel blockade via selective inhibition of the SUR2A/Kir6.2 isoform.
HMR 1098 has been recently shown to have higher IC50s for the putative smooth muscle KATP channel isoforms SUR2B/Kir6.1 and SUR2B/Kir6.2 compared to SUR2A/Kir6.2 (Liu et al., 2001). These findings suggest that HMR 1098 also inhibits cardiac KATP channels more effectively than smooth muscle KATP channels. SUR2A and SUR2B are splice variants of the last 42 amino acids in the carboxy terminus (Inagaki et al., 1996). These findings, suggest the carboxy terminus is significant in the binding of HMR 1098. Indeed, this has been elegantly demonstrated by Ashfield et al. (1999). Using a chimeric approach, they show the importance of the carboxy-terminus in conferring isoform selectivity to the sulphonylureas. However, the similar IC50s observed with the benzoic acid derivative, mitiglinide, on SUR2A and SUR2B suggest that the carboxy-terminus is not important in the binding of all KATP channel inhibitors (Reimann et al., 2001).
The efficacy of sulphonylurea inhibition has been shown to be dependent on the internal nucleotide concentration. For example, glibenclamide block of cardiac KATP channels is reduced in the presence of nucleotides and this may account for the selectivity of this compound for β-cell KATP channels under physiological conditions (Gribble et al., 1998). In our study, HMR 1098 inhibition of SUR2A/Kir6.2 KATP channels was equally effective in the absence or presence of MgATP and ADP. Indeed, it has been reported that the HMR 1098 analogue HMR 1883 has an IC50 of 0.6 μM in intact cardiac tissue (Gögelein et al., 1998) where physiological nucleotide levels are present. At the structural level, the nucleotide sensitivity of sulphonylureas may be conferred by compounds containing aromatic sulphonylurea groups (Figure 5).
In summary, our data demonstrate that HMR 1098 is a selective blocker of the cardiac plasma-membrane KATP channel. Although the potency of KATP channel block by HMR 1098 is not as high as some sulphonylureas, its cardioselectivity is a unique characteristic of this derivative. In the clinical setting, effective plasma concentrations in the 10–20 μM range will likely cause >90% inhibition of cardiac KATP channels with little or no effect on the β-cell KATP channel and hence insulin secretion. HMR 1098 inhibits KATP channel activity exclusively via interactions with the sulphonylurea receptor, binding to the SUR2A isoform with much greater affinity than the β-cell SUR1 isoform.
Acknowledgments
Funding for this study was provided by the Canadian Institutes of Health Research (CIHR, to P.E. Light and R.J. French) and the Canadian Diabetes Association (to P.E. Light) and by core funds from a CIHR Group Grant (P.I. Dr W.R. Giles). R.J. French received salary support as an Alberta Heritage Foundation for Medical Research (AHFMR) Senior Scientist and CIHR Distinguished Scientist. P.E. Light is an AHFMR Scholar and CIHR New Investigator. J.E. Manning Fox is an AHFMR Postdoctoral Fellow. Samples of HMR 1098 were generously provided by Dr Heinz Gögelein, Aventis Pharma, Deutschland. We thank Robert Winkfein for assistance in cloning the rabbit SUR2A gene.
Abbreviations
- ADP
adenosine diphosphate
- ATP
adenosine triphosphate
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulphonic acid
- KATP
ATP-sensitive potassium channels
- RPMI 1640
Roswell Park Memorial Institute 1640 medium
- SUR
sulphonylurea receptor
References
- AGUILAR-BRYAN L., BRYAN J. Molecular biology of ATP-sensitive potassium channels. Endocrine Reviews. 1999;20:101–135. doi: 10.1210/edrv.20.2.0361. [DOI] [PubMed] [Google Scholar]
- ASHFIELD R., GRIBBLE F.M., ASHCROFT S.J., ASHCROFT F.M. Identification of the high-affinity tolbutamide site on the SUR1 subunit of the K(ATP) channel. Diabetes. 1999;48:1341–1347. doi: 10.2337/diabetes.48.6.1341. [DOI] [PubMed] [Google Scholar]
- D'HAHAN N., MOREAU C., PROST A.-L., JACQUET H., ALEXANDER S.A., ALEKSEEV A.E., TERZIC A., VIVAUDOU M. Pharmacological plasticity of cardiac ATP-sensitive potassium channels toward diazoxide revealed by ADP. PNAS U.S.A. 2000;96:12162–12167. doi: 10.1073/pnas.96.21.12162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARRAT K.N., BRADY P.A., HASSINGER N.L., GRILL D.E., TERZIC A., HOMES D.R., JR Sulfonylurea drugs increase early mortality in patients with diabetes mellitus after direct angioplasty for acute myocardial infarction. J. Am. Coll. Cardiol. 1999;33:119–124. doi: 10.1016/s0735-1097(98)00557-9. [DOI] [PubMed] [Google Scholar]
- GÖGELEIN H., HARTUNG J., ENGLERT H.C. Molecular basis, pharmacology and physiological role of cardiac K(ATP) channels. Cell Physiol. Biochem. 1999;9:227–241. doi: 10.1159/000016319. [DOI] [PubMed] [Google Scholar]
- GÖGELEIN H., HARTUNG J., ENGLERT H.C., SCHOLKENS B.A. HMR 1883, a novel cardioselective inhibitor of the ATP-sensitive potassium channel. Part I: effects on cardiomyocytes, coronary flow and pancreatic beta-cells. J. Pharmacol. Exp. Ther. 1998;286:1453–1464. [PubMed] [Google Scholar]
- GRIBBLE F.M., TUCKER S.J., ASHCROFT F.M. The interaction of nucleotides with the tolbutamide block of cloned ATP-sensitive K+ channel currents expressed in Xenopus oocytes: a reinterpretation. J. Physiol. 1997;504:35–45. doi: 10.1111/j.1469-7793.1997.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIBBLE F.M., TUCKER S.J., SEINO S., ASHCROFT F.M. Tissue specificity of sulfonylureas: studies on cloned cardiac and beta-cell K(ATP) channels. Diabetes. 1998;47:1412–1418. doi: 10.2337/diabetes.47.9.1412. [DOI] [PubMed] [Google Scholar]
- GROSS G.J. The role of mitochondrial KATP channels in cardioprotection. Basic Res. Cardiol. 2000;95:280–284. doi: 10.1007/s003950050004. [DOI] [PubMed] [Google Scholar]
- GROSS G.J., FRYER R.M. Plasma-membrane versus mitochondrial ATP-sensitive K+ channels and myocardial preconditioning. Circ. Res. 1999;84:973–979. doi: 10.1161/01.res.84.9.973. [DOI] [PubMed] [Google Scholar]
- INAGAKI N., GONOI T, , CLEMENT J.P., IV, NAMBA N., INAZAWA J., GONZALEZ G., AGUILAR BRYAN L., SEINO S., BRYAN J. Reconstitution of IKATP: An inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- INAGAKI N., GONOI T., CLEMENT J.P., IV, WANG C.-Z., AGUILAR-BRYAN L., BRYAN J., SEINO S. A family of sulfonylurea receptors determines the pharmacological properties of ATP sensitive K+ channels. Neuron. 1996;16:1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- KUBO Y., BALDWIN T.J., JAN Y.N., JAN L.Y. Primary structure and functional expression of a mouse inward rectifier potassium channel. Nature. 1993;362:127–133. doi: 10.1038/362127a0. [DOI] [PubMed] [Google Scholar]
- LEIBOWITZ G., CERASI E. Sulphonylurea treatment of NIDDM patients with cardiovascular disease: a mixed blessing. Diabetologia. 1996;39:503–514. doi: 10.1007/BF00403296. [DOI] [PubMed] [Google Scholar]
- LIGHT P.E., BLADEN C., WINKFEIN R., WALSH M.P., FRENCH R.J. Molecular basis of protein kinase C-induced activation of ATP-sensitive potassium channels. PNAS U.S.A. 2000;97:9058–9063. doi: 10.1073/pnas.160068997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIGHT P.E., SHIMONI Y., HARBISON S., GILES W.R., FRENCH R.J. Hypothyroidism decreases the ATP-sensitivity of KATP channels from rat heart. J. Memb. Biol. 1998;162:217–233. doi: 10.1007/s002329900359. [DOI] [PubMed] [Google Scholar]
- LIU Y., REN G., O'ROURKE B., MARBAN E., SEHARASEYON J. Pharmacological comparison of native mitochondrial K(ATP) channels with molecularly defined surface K(ATP) channels. Mol. Pharmacol. 2001;59:225–230. [PubMed] [Google Scholar]
- REIMANN F., PROKS P., ASHCROFT F.M. Effects of mitiglinide (S 21403) on Kir6.2/SUR1, Kir6.2/SUR2A and Kir6.2/SUR2B types of ATP-sensitive potassium channel. Br. J. Pharmacol. 2001;132:1542–1548. doi: 10.1038/sj.bjp.0703962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SEINO S. ATP-sensitive potassium channels: a model of heteromultimeric potassium channel/receptor assemblies. Annu. Rev. Physiol. 1999;61:337–362. doi: 10.1146/annurev.physiol.61.1.337. [DOI] [PubMed] [Google Scholar]
- SMITS P., THIEN T. Cardiovascular effects of sulphonylurea derivatives. Implications for the treatment of NIDDM. Diabetologia. 1995;38:116–121. doi: 10.1007/BF02369361. [DOI] [PubMed] [Google Scholar]
- SONG D.K., ASHCROFT F.M. Glimepiride block of cloned beta-cell, cardiac and smooth muscle K(ATP) channels. Br. J. Pharmacol. 2001;133:193–199. doi: 10.1038/sj.bjp.0704062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TUCKER S.J., GRIBBLE F.M., ZHAO C., TRAPP S., ASHCROFT F.M. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- WIRTH K.J., UHDE J., ROSENSTEIN B., ENGLERT H.C., GÖGELEIN H., SCHOLKENS B.A., BUSCH A.E. K(ATP) channel blocker HMR 1883 reduces monophasic action potential shortening during coronary ischemia in anesthetised pigs. Naunyn schmiedebergs Arch. Pharmacol. 2000;361:155–160. doi: 10.1007/s002109900166. [DOI] [PubMed] [Google Scholar]