

Abstract
The human HERG gene encodes the cardiac repolarizing K+ current IKr and is genetically inactivated in inherited long QT syndrome 2 (LQTS2). The antihistamine terfenadine blocks HERG channels, and can cause QT prolongation and torsades de pointes, whereas its carboxylate fexofenadine lacks HERG blocking activity.
In the present study the ability of fexofenadine to block the K897T HERG channel variant was investigated. The underlying single nucleotide polymorphism (SNP) A2960C was identified in a patient reported to develop fexofenadine-associated LQTS.
K897T HERG channels produced wild-type-like currents in Xenopus oocytes. Even at a concentration of 100 μM, fexofenadine did not inhibit wild-type or K897T HERG channels. Coexpression of wild-type and K897T HERG with the ß-subunit MiRP1, slightly changed current kinetics but did not change sensitivity to terfenadine and fexofenadine.
Western blot analysis and immunostaining of transiently transfected COS-7 cells demonstrated that overall expression level, glycosylation pattern and subcellular localization of K897T HERG is indistinguishable from wild-type HERG protein, and not altered in the presence of 1 μM fexofenadine.
We provide the first functional characterization of the K897T HERG variant. We demonstrated that K897T HERG is similar to wild-type HERG, and is insensitive to fexofenadine. Although the polymorphism changes PKA and PKC phosphorylation sites, regulation of K897T HERG by these kinases is not altered.
Our results strongly indicate that QT lengthening and cardiac arrhythmia in the reported case of drug-induced LQT are not due to the K897T exchange or to an inhibitory effect of fexofenadine on cardiac IKr currents.
Keywords: Fexofenadine, HERG, K+ channel, polymorphism, arrhythmia
Introduction
The delayed rectifier potassium current plays a critical role in the repolarization of the cardiac cellular membrane potential. It is composed of a rapid component, IKr, and a slow component, IKs. The pore-forming α-subunits encoded by the HERG gene associate with MiRP1 subunits, the protein products of the KCNE2 gene, to form the rapid component. The channel underlying the IKs current is composed of the α-subunits encoded by KCNQ1 and the accessory MinK subunits encoded by the KCNE1 gene. The functional importance of these genes is underscored by the finding that mutations in all four genes are responsible for the long QT syndrome (LQTS), an inherited form of cardiac arrhythmia. Almost 90% of identified LQTS mutations occur in the KCNQ1 and HERG genes (Tristani-Firouzi et al., 2001), predisposing affected individuals to torsades de pointes-type ventricular arrhythmias that finally can cause sudden cardiac death.
LQTS also can be acquired, usually as a result of pharmacological therapy. Acquired LQTS is a common side-effect of antiarrhythmic, antiallergic, antibiotic, antipsychotic, and gastrointestinal prokinetic drugs (Dhein, 2000; Escande, 2001; Lacerda et al., 2001). All known drugs with QT-prolonging activity preferentially block IKr. This has been demonstrated by their effects on either native IKr or heterologously expressed HERG currents. It remains unknown why all these structurally unrelated compounds block HERG channels, and only recently first insights in the structural basis of drug binding were gained (Mitcheson et al., 2000). However, IKr blocking ability constitutes a serious problem in the development of new and safe drugs.
HERG-blocking activity of a given drug might not be determined solely by its chemical structure, but may also be influenced by sequence variations within the HERG and MiRP1 subunits. Induced mutation in the S6 domain of HERG has been shown to decrease its sensitivity to known HERG blockers (Lees-Miller et al., 2000). In addition, naturally occurring mutations in MiRP1 conferred a higher sensitivity to antibiotic drugs (Abbott et al., 1999; Sesti et al., 2000), suggesting that allelic variants of the HERG gene might also strongly contribute to drug sensitivity.
The non-sedating antihistamine terfenadine blocks IKr and HERG channels (Salata et al., 1995; Roy et al., 1996; Süssbrich et al., 1996). Elevated plasma levels of terfenadine, resulting from overdosing, hepatic disease, or coadministration with drugs that inhibit drug metabolism, can result in QT prolongation and ventricular arrhythmia (Honig et al., 1992; 1993; Escande, 2001). In contrast, terfenadine carboxylate (fexofenadine), the major metabolite of terfenadine, does not block HERG channels in vitro (Roy et al., 1996), and clinical trials did not reveal any effect of fexofenadine on QTc time (Pratt et al., 1999; Craig-Mcfeely et al., 2001).
Recently, Pinto et al. (1999) described a single patient presenting with a prolonged QT interval at baseline. The patient had a ventricular tachycardia, which progressed to ventricular fibrillation during medication with fexofenadine; the authors concluded that in contrast to previous findings, fexofenadine may increase QT time in susceptible patients.
In the present study, we tested the possibility that the patient carries a mutation in the HERG gene which might confer novel fexofenadine sensitivity to the IKr current, resulting in the observed QT lengthening under fexofenadine medication.
Methods
Study subject
A detailed description of the patient was given in the report of Pinto et al. (1999). Briefly, the patient was a 67-year-old male of normal posture and without a history of significant cardiac disease. He was known to have mild hypertension and mild left ventricular hypertrophy, and used fexofenadine successfully against an unexplained but severe general itching problem. The possible presence of additional risk factors for QTc prolongation and ventricular dysrhythmia in the patient was discussed previously (Giraud, 1999; Dhar et al., 2000).
PCR analysis of the genomic DNA
The QIAamp Blood Kit (Qiagen) was used to purify the total DNA from the patient's blood. Primers to amplify exons of the HERG, KCNQ1, and KCNE1 genes, and the respective PCR conditions have been described previously in detail (Splawski et al., 1998). The primer pair used to amplify the single human MiRP1 exon was: forward primer: 5′-CCG AGG ATC CGT TTT CCT AAC CTT GTT CGC C-3′; reverse primer: 5′-GCG CAA GCT TGG TGC CTT TCT CCC TTA TC-3′. The MiRP1 coding sequence was subcloned into pSGEM oocyte expression vector (Villmann et al., 1997) as BamHI/HindIII fragment. Amplification conditions were: 94°C for 3 min followed by 10 cycles at 94°C for 15 s, 55°C for 20 s, and 72°C for 1 min, and 25 cycles at 94°C for 15 s, 60°C for 20 s, and 72°C for 1 min, and a 5-min extension at 72°C. PCR products were purified by agarose gel electrophoresis and the QIAquick Gel Extraction Kit (Qiagen), and sequenced with an ABI PRISM 310 sequencer (Perkin Elmer).
Construction of the K897T HERG mutant and in vitro transcription of cRNA
Site-directed mutagenesis was performed by PCR using Pfu-Polymerase (Promega). The construct was verified by DNA sequencing. cRNAs for injection into oocytes were prepared with the mMESSAGE mMACHINE in vitro transcription kit (Ambion).
Electrophysiology
Handling and injection of Xenopus oocytes have been described previously (Lerche et al., 2000). Briefly, oocytes were stored in recording solution ND96, containing (in mM): NaCl 96, KCl 2, CaCl2 1.8, MgCl2 1, HEPES 5, pH 7.4, with additional NaPyruvate (275 mg l−1), theophylline (90 mg l−1), and gentamycin (50 mg l−1) at 18°C. The two-electrode voltage clamp (TEVC) configuration was used to record ionic currents from Xenopus oocytes. Experiments were performed at room temperature with a Turbo Tec 10CD (NPI) amplifier, an ITC-16 interface combined with Pulse software (Heka) and Origin version 5.0 (Microcal software) for data acquisition on Pentium II PC. In several sets of experiments, oocytes were individually injected with 10 ng of cRNA encoding for wild-type HERG and HERG K897T, or coinjected with 10 ng of wild-type HERG or HERG K897T plus 5 ng of hMiRP1. Macroscopic currents were recorded 2–4 days after injection. The microelectrodes were filled with 3 M KCl solution and had resistances between 0.5 and 1 MΩ. The oocytes were continuously perfused with ND96 during measurements. Terfenadine and fexofenadine were synthesized in house, and were added from a 100 mM stock solution in DMSO to the recording solution.
Protocols
Holding potential in all cases was −80 mV. Data were sampled at 4 kHz, and filtered at 500 Hz. (1) Deactivation, current amplitude, concentration-inhibition curves (IC50): prepulse for 1 s to 40 mV followed by a test pulse for 1 s to −80 mV; interpulse interval was 3 s. Current amplitude was measured at the beginning of the test pulse at the maximum of the current trace. For the dose-response curves, the oocytes were perfused for 5 min by each test solution (5 concentrations from 0.03 to 3 μM terfenadine). (2) Inactivation (Smith et al., 1996): a preceding pulse for 15 s to −15 mV followed by a prepulse for 40 ms to −100 mV, and a test pulse for 100 ms to −15 mV. (3) Activation: prepulse for 1 s from −80 to 40 mV in 10 mV increments followed by a test pulse for 1.5 s to −80 mV; interpulse interval was 3 s. All fitting procedures were based on the simplex algorithm.
Statistical analysis
Data are presented as mean±s.e.m. Statistical significance determined by Student's t-test (P<0.05). Significant results are indicated by asterisks.
Antibodies
The anti-HERG polyclonal antibody, used in Western blots and immunostaining, was generated in rabbits against a GST fusion protein containing the last 112 amino acids of HERG (residues 1048–1159). HERG-antiserum was purified on an affinity column consisting of a short C-terminal peptide corresponding to either HERG residues 1102–1121 (TLTLDSLSQVSQFMACEELP), or the entire fusion protein. The epitope recognized by the antibody is located far away from the K897T mutation; both the wild-type and K897T HERG proteins are recognized by the antibody.
Western blot analysis and immunostaining
For Western blotting, HEK 293 cells were transfected with Lipofectamine/Plus (Life Technologies). Forty-eight hours after transfection, cells were solubilized for 1 h at 4°C in lysis buffer containing (in mM): NaCl 150, EDTA 1, Triton X-100 1%, Tris-HCl 50 (pH 7.5), and a protease inhibitor mix (Complete, Roche Biochemicals).
Protein concentrations were determined by the BCA method (Pierce). For Western blotting, proteins were separated on 8% SDS polyacrylamide gels, and transferred to polyvinylidene difluoride membranes. Membranes were blocked overnight with 5% nonfat dry milk in phosphate-buffered saline (PBS) plus 0.1% Tween, and immunoblotted with polyclonal rabbit anti-HERG antibody (1 : 200 dilution; 1 h at RT) followed by horseradish peroxidase-conjugated secondary antibody (1 : 3000; 1 h at RT; Amersham Pharmacia Biotech). Western blots were developed with the ECL-Plus detection system (Amersham Pharmacia).
Immunocytochemistry and confocal laser scanning microscopy
COS cells were grown in Nunc cover glass chamber slides. Transfection was performed using FuGENE 6 Transfection Reagent (Roche Molecular Biochemicals), followed by an overnight cultivation. Then cells were washed once with PBS and fixed in 3.7% paraformaldehyde in PBS with 0.2% Triton X-100 for 10 min, followed by blocking in PBS with 1% BSA for an additional 10 min. The cells then were sequentially incubated with anti-HERG antibody, diluted 1 : 200 in PBS with 0.2% Triton X-100, for 45 min and with FITC-labelled goat anti-rabbit antibody (Chemicon), diluted 1 : 200 in PBS with 0.2% Triton X-100, for 45 min. Both incubation steps were followed by three washings with PBS. Stained specimens were mounted in 0.1 M Tris/HCl, pH 8.5, 25% glycerol, 10% Mowiol 4–88 (Hoechst), and 2% DABCO (Sigma). Cells were observed using a Zeiss LSM-410 laser scanning confocal microscope. Images were processed with Photoshop 5.5 (Adobe Systems).
SSCP analysis
A SSCP system was established for the detection of mutations and screening of controls. After PCR amplification of exon 11 (forward primer: 5′-GGC AGT ACG GAG TTA GAG GGT-3′; reverse primer: 5′-CAC CCC GCC TTC CAG CTC C-3′; PCR conditions were 94°C for 4 min followed by 10 cycles at 94°C for 30 s, 66°C for 30 s, and 68°C for 40 s, and 26 cycles at 94°C for 30 s, 64°C for 30 s, and 68°C for 30 s, and a 4-min extension at 68°C; Expand Long Template PCR system, Roche), SSCP analysis was performed using 10% non-denaturing polyacrylamide gels (CleanGels, Pharmacia) on the Multiphor II electrophoresis system (Pharmacia) at 4°C. Bands were visualized after separation for 5 h at 250 V by standard silver-staining protocols.
Results
Identification of the HERG K897T sequence variation
Genomic DNA was isolated from the peripheral blood of a patient reported to have fexofenadine-induced arrhythmia (Pinto et al., 1999). Intron-flanking primers were used to amplify exonic sequences of the HERG, KCNQ1, KCNE1, and KCNE2 genes. PCR fragments were then sequenced, and compared to published sequences. Three sequence differences were identified: in the HERG gene, we detected (1) a silent T1956C exchange in exon 8 (Larsen et al., 1999); and (2) in exon 11 an A2690C exchange, leading to an exchange of a threonine for a lysine (K897T) within the C-terminus of the HERG protein (Figure 1B) (Scherer et al., 2000). (3) The patient also carried the common MinK polymorphism A112G that results in a S38G exchange in the MinK protein (Lai et al., 1994). The patient was heterozygous for the three alleles. The novel A2690C (K897T) HERG variation was verified in independent sequencing reactions, and by sequencing of subcloned PCR fragments (Figure 1A). We found cDNA clones encoding both the wild-type and the variant sequence as expected for the heterozygous patient. The exchanged lysine is highly conserved in HERG proteins from different species, as well as in the related ether-à-go-go (eag) K+ channels (Figure 1C), and is located C-terminally of the cyclic nucleotide binding domain. The K897T amino acid substitution creates a novel consensus motif for PKA phosphorylation, and destroys a putative PKC phosphorylation site.
Figure 1.
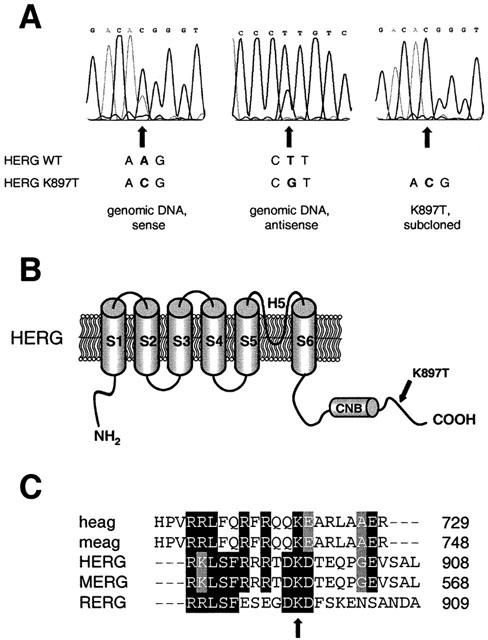
Identification of the A2690C single nucleotide polymorphism resulting in the K897T exchange in the patient's HERG protein. (A) Electropherograms from sequencing analyses of genomic DNA (left and middle panel) indicate heterogeneity for the position 897 in the HERG protein. An A–C substitution is observed at position 2960 (K897T) in the coding sequence. Right panel shows the variant sequence of a subcloned PCR fragment. (B) Topological model of the HERG protein, showing the location of the mutation K897T within the intracellular C-terminus, 41 amino acids downstream of the putative cyclic nucleotide binding domain (CNB). (C) Protein sequence alignment of members of the eag potassium channel family (human and mouse eag channels, and human, rat and mouse ERG channels), showing conservation of the lysine at codon 897 (HERG numbering, indicated by arrow). GCG software (Wisconsin Package, Version 10.1) was used.
SSCP analysis of the K897T HERG variation
The abundance within the Caucasian population of the novel A2690C nucleotide exchange in the HERG gene was investigated in genomic DNA isolated from blood of 47 healthy individuals. Exon 11 and its flanking intronic sequences were amplified by polymerase chain reaction, and the fragments were analysed by SSCP analysis. Fragments presenting aberrant conformers encoded by the A2690C mutation were detected within the genomic DNA of the patient. However, the sequence variation could not be detected within the 94 control chromosomes (data not shown).
Protein processing studied by Western blot analysis and immunostaining
Many LQT2 mutants show defective processing and intracellular trafficking (Ficker et al., 2000). Figure 2A shows a Western blot analysis of wild-type and K897T HERG proteins. Both the wild-type HERG and the K897T mutation expressed two protein bands, an upper band representing a fully glycosylated mature HERG protein found in the plasma membrane, and a lower band representing a core-glycosylated immature protein. Presence of 1 μM fexofenadine in the culture medium of transfected HEK 293 cells did not alter the processing and trafficking of wild-type and K897T HERG protein. Culturing the cells at 37°C or 26°C had no influence on protein maturation; however at lower temperatures, higher HERG protein levels were observed. We studied the subcellular localization of the HERG protein by immunostaining of transiently transfected COS-7 cells. As shown in Figure 2B, in cells expressing either wild-type or K897T HERG protein, staining was visible throughout the cells. This corresponding subcellular distribution pattern for both proteins is consistent with the results of Western blot experiments, and shows that the K897T protein is properly processed and trafficked to the plasma membrane.
Figure 2.
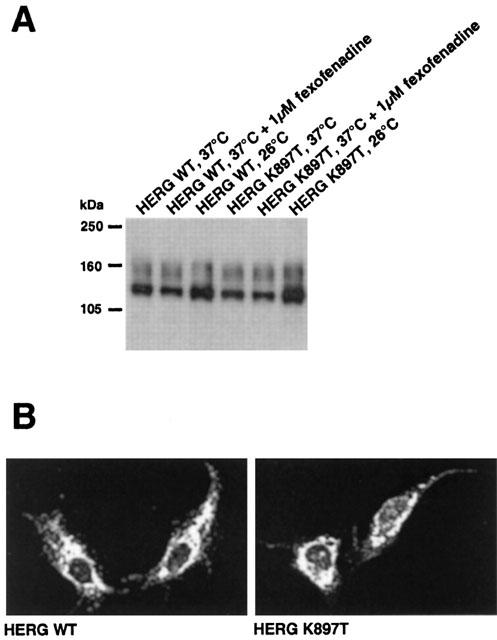
Expression of wild-type and K897T HERG protein in mammalian cells. (A) Western blot analysis of wild-type and K897T HERG channels. After transfection HEK 293 cells were grown for 2 days at 37 or 26°C. Fexofenadine was added for 24 h to cells grown at 37°C. HERG K897T was present in two forms. A 135 kDa band represents a core-glycosylated form of the protein. A band of about 155 kDa represents the mature fully glycosylated form of the protein delivered to the plasma membrane. (B) Immunostaining of COS cells expressing either wild-type or K897T HERG protein. Both proteins were expressed at equal levels and displayed a similar subcellular distribution pattern.
Electrophysiological properties of K897T HERG channels
We used site-directed mutagenesis to generate the K897T HERG variant, and expressed it in Xenopus oocytes to investigate possible effects on channel function using standard two-microelectrode voltage clamp techniques. First we compared the amplitudes and kinetics of wild-type and K897T HERG channels, either expressed alone or together with MiRP1.
HERG current was activated by 1 s test pulses from −80 to +40 mV. The magnitude of HERG current increased progressively with test pulses up to −30 mV, and then gradually decreased at more positive test potentials (Figure 3A,B). Deactivation of current (tail current) was assessed after returning the membrane to the holding potential of −80 mV. The amplitude of the tail currents progressively increased after depolarization, and saturated at +10 mV. Macroscopic current traces obtained with this protocol were the same for wild-type and K897T HERG channels (Figure 3A,B; Table 1).
Figure 3.
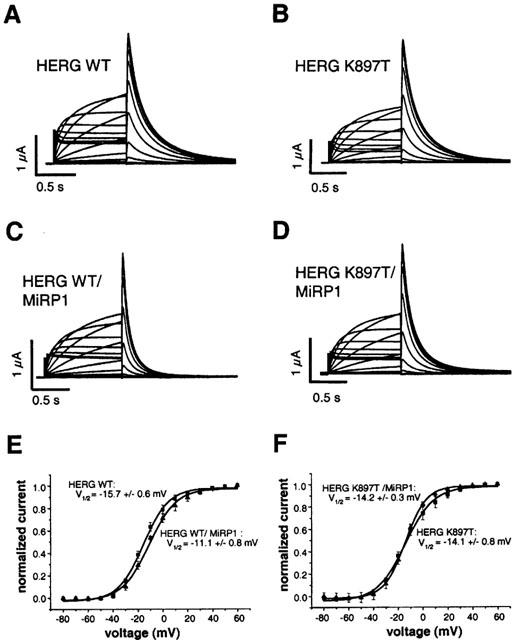
Two-electrode voltage clamp records of wild-type and K897T HERG and the respective IKr currents expressed in Xenopus oocytes. (A–D), Representative current traces from oocytes expressing the indicated proteins using protocol (3). In (C) and (D) deactivation kinetics are faster as compared to homomeric expression of the HERG subunit in (A) and (B) respectively. (E) Current voltage relationships from (A) and (C) showing the influence of MiRP1 on activation of wild-type HERG channels. (F) Current voltage relationships from (B) and (D) showing the influence of MiRP1 on activation of HERG K897T channels. Tail current amplitude was measured at the maximum of the current trace, squares represent values from homomeric HERG expression, triangles represent values from coexpression with the MiRP1 subunit; numbers of experiments as indicated in Table 1.
Table 1.
Kinetic analysis of wild-type and K897T HERG and the respective IKr currents expressed in Xenopus oocytes
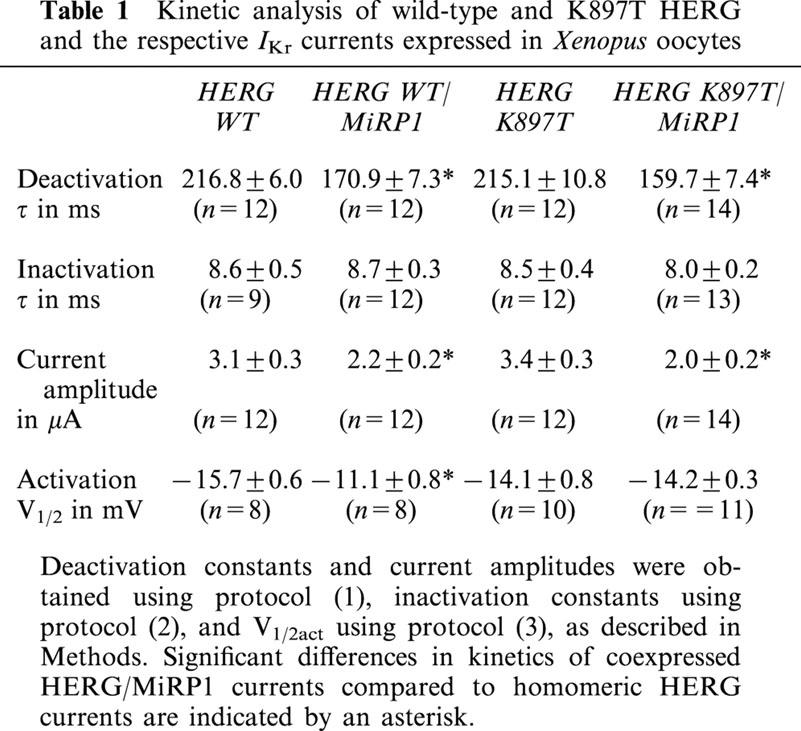
Coexpression of MiRP1 reduced HERG current amplitudes, with similar magnitudes of reduction found for wild-type and K897T currents (from 3.1±0.3 to 2.2±0.2 μA, and from 3.4±0.3 to 2.0±0.2 μA, respectively) (Table 1). Deactivation time constants of tail currents were 216.8±6.0 ms for homomeric wild-type, and 215.1±10.8 ms for homomeric K897T channels (Table 1). Coexpression of MiRP1 accelerated deactivation of both wild-type and K897T HERG channels (Figure 3C,D), with deactivation constants of 170.9±7.3 and 159.7±7.4 ms, respectively (Table 1). To separate the inactivation from the deactivation process, protocol (2) was used. With a prepulse to −15 mV all channels were inactivated. Channels were then released from inactivation by a short pulse to −100 mV, without enabling deactivation (Smith et al., 1996). Inactivation kinetics were determined during a final pulse to −15 mV. Wild-type and K897T HERG channel kinetics did not differ. Additionally and in agreement with previous findings, coexpressed MiRP1 had no effect on inactivation kinetics of wild-type channels (Abbott et al., 1999), and this was also the case for K897T channels (Table 1).
The voltage dependence of channel activation was assessed by plotting the relative amplitude of tail currents as a function of test potential (Figure 3E,F). Homomeric HERG currents reached half maximal activation (V1/2) at a potential of −15.7±0.6 mV, and heteromeric HERG/MiRP1 currents at a more depolarized potential of −11.1±0.8 mV. This difference in half maximal activation reached statistical significance (n=8; P<0.05). The values for K897T and K897T/MiRP1 channels were −14.1±0.8 and −14.2±0.3 mV, and were not statistically different from those of wild-type homomeric HERG currents. Therefore, current amplitude and current kinetics of activation, deactivation, and inactivation of the K897T HERG channel were very similar to the wild-type HERG channel. Effects of MiRP1 on wild-type HERG currents were found as described previously (Abbott et al., 1999), and were similar between wild-type and K897T HERG, with the exception of a slight shift in V1/2 of activation induced by the coexpressed MiRP1 that was missing in K897T HERG/MiRP1 channels.
PMA and forskolin shift activation of wild-type and K897T HERG channels
Phorbol ester (PMA) causes a shift in the activation curve of HERG channels (Kiehn et al., 1998). The substitution of lysine for a threonine at position 897 destroys the putative PKC phosphorylation site at position 895 (TDK897T), and introduces a novel consensus PKA phosphorylation site (RTDT897). To investigate whether these changes affect the PMA-induced shift in HERG channel activation, we determined the half maximal activation voltage of wild-type and K897T HERG channels in the presence of PMA. Perfusion of oocytes for 30 min with 100 nM PMA shifted the average V1/2 of activation in wild-type channels from −10.5±1.1 to 19.6±2.0 mV (n=5; Figure 4A). Similarly, V1/2 of activation in K897T HERG channels was shifted from −8.8±0.9 to 17.6±0.9 mV (n=5; Figure 4A). Thus, the shift in channel activation by the phorbol ester was similar with wild-type and K897T HERG channels (30.1 vs. 26.4 mV). To compare the sensitivity of wild-type and K897T HERG channels to forskolin, oocytes were exposed to 400 μM forskolin for 60 min. Maximum outward current amplitude of the K897T HERG channels was reduced by 51.7±5.5% (n=5), and the voltage of half maximal activation was shifted by +9.8±3.0 mV (n=5). Similarly, wild-type channels showed a reduction of maximum outward current by 56.3±7.1% (n=5), and the V1/2 of activation was shifted by +8.1±0.9 mV (n=5). Thus, introduction of an additional PKA phosphorylation site in the K897T HERG channels did not alter the sensitivity to PKA (Figure 4B). In addition, current amplitude and V1/2 of activation of wild-type and K897T HERG channels were not affected when fexofenadine (200 μM) was present during forskolin treatment of HERG expressing oocytes (n=5) (Figure 4B).
Figure 4.
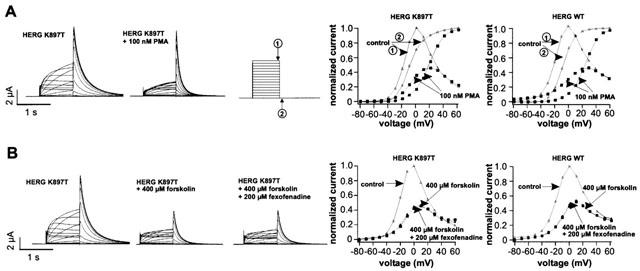
Effects of protein kinase stimulation by PMA or forskolin on K897T HERG activation. (A) Current traces of K897T HERG channels before and after 30 min perfusion of oocytes with 100 nM PMA. Normalized I-V-relationships were determined either at the end of the test pulse (arrow 1) or at the tail current at −80 mV (arrow 2). The shift of the activation curve was similar for both HERG channels. (B) Control measurements of K897T HERG and the effect of 400 μM forskolin after 60 min in the same oocyte. I-V-relationships were measured at the end of the test pulse. A similar reduction in current amplitudes and shift in the voltage of half-maximal activation was observed for K897T and WT HERG channels by PKA stimulation. Addition of 200 μM fexofenadine to the bath solution following stimulation of PKA had no effect on HERG currents.
Effects of fexofenadine on wild-type and K897T HERG channels
We next investigated the possible effects of fexofenadine on both wild-type and K897T HERG channels, either expressed alone or in combination with MiRP1. Current amplitudes, activation, deactivation, and inactivation kinetics of HERG and HERG/MiRP1 currents were measured in both the absence and presence of 100 μM fexofenadine. Figure 5 shows representative current traces of homomeric and heteromeric expressed K897T HERG currents. Again, fexofenadine had no significant effect on current amplitudes and kinetics of wild-type (not shown) and K897T HERG channels.
Figure 5.
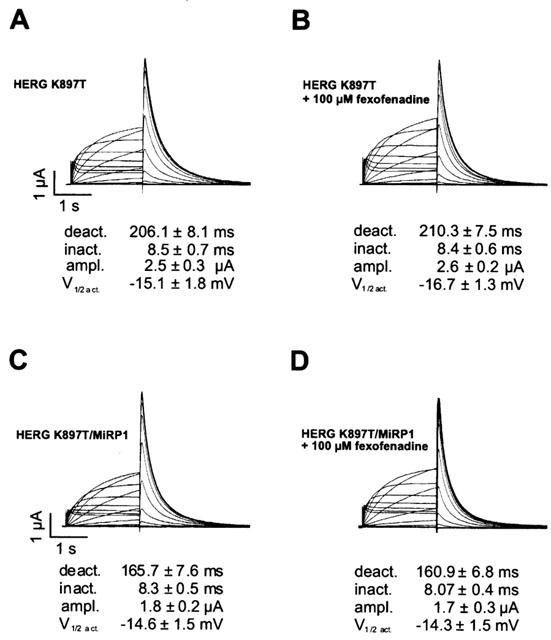
Effect of fexofenadine on HERG current properties. Representative currents recorded in oocytes expressing K897T HERG alone or together with MiRP1. The voltage protocols were as described in Methods. Currents were recorded before (A) and (C) or after addition of 100 μM fexofenadine to the bath for 5 min (B) and (D). Values for kinetic parameters given below are means obtained from eight oocytes. None of the analysed parameters differed significantly from the controls, when fexofenadine was present.
MiRP1 does not change inhibition of HERG currents by terfenadine
It has been described that terfenadine efficiently blocks HERG channels (Roy et al., 1996). In our experiments, terfenadine inhibited homomeric HERG channels with a half maximal concentration of 0.35 μM, very similar to the reported affinity. It remains unknown whether coexpression with MiRP1 changes its affinity to terfenadine (or its metabolite fexofenadine). We therefore determined IC50 values of homomeric HERG and heteromeric HERG/MiRP1 (IKr) currents with terfenadine. As shown in Table 2, MiRP1 did not change the inhibitory activity of terfenadine on wild-type HERG channels. In addition, K897T exchange did not alter terfenadine sensitivity to expressed homomeric and heteromeric channels.
Table 2.
Concentration values at half maximal inhibition (IC50) of terfenadine on wild-type HERG, wild-type HERG/MiRP1, K897T HERG, and K897T HERG/MiRP1

Discussion
Ventricular arrhythmias are rare side effects of a number of commonly prescribed drugs, like the non-sedating antihistamines terfenadine and astemizole. Blockade of the HERG K+ channel, which produces the cardiac repolarizing IKr current, has been recognized as the mechanism underlying the cardiotoxicity of these compounds (Salata et al., 1995; Taglialatela et al., 1999). Consequently, terfenadine and astemizole have been withdrawn from the market, and replaced by structurally-related noncardiotoxic H1 receptor antagonists. However, even after introduction of these third-generation antihistamines, the discussion is still ongoing if they can lead to arrhythmias in isolated cases.
Terfenadine blocks HERG at a concentration slightly above therapeutic concentrations (Roy et al., 1996). It is metabolized in the liver by a P450 cytochrome enzyme, partly to its carboxylate, fexofenadine, which still blocks histamine receptors but in contrast to the parent drug terfenadine, does not inhibit the rapid delayed rectifier current IKr in the heart (Roy et al., 1996). Therefore, accumulation of terfenadine, due to an impaired cytochrome system, is the likely explanation for its cardiotoxicity. Supporting the hypothesis that both intrinsic HERG channel blocking activity and plasma accumulation are necessary to trigger arrhythmia, mizolastine, a second-generation antihistamine, was recently shown to potently block HERG channels in vitro (Taglialatela et al., 2000). No cases of mizolastine-induced LQTS have been reported. Mizolastine is limitedly metabolised via the P450 cytochrome system (Delauche-Cavallier et al., 1999). As fexofenadine neither blocks HERG channels in vitro, nor is it metabolized in the liver, the first case of QT lengthening after intake of fexofenadine was surprising (Pinto et al., 1999).
The present study aimed to investigate the possible sensitivity to fexofenadine of this patient. We systematically screened K+ channel genes associated with LQTS, for the presence of mutations, since mutations in the genes encoding HERG, MiRP1, MinK and KCNQ1 subunits are responsible for more than 90% of inherited LQTS cases (Vincent, 1998). By DNA sequence analyses of amplified exonic sequences, we identified a single heterozygous nucleotide substitution in the HERG gene, which resulted in the exchange of a threonine for a lysine within the C-terminal part of the HERG protein (K897T). The mutated position is highly conserved in HERG proteins of different species and in related eag channels, and lies within a region that is critical for HERG assembly and expression (Kupershmidt et al., 1998). The identical sequence variation was recently identified in Japanese patients with LQTS, but is rarely found in the normal Japanese population (Iwasa et al., 2000), suggesting that K897T represents a polymorphism rather than a disease-causing mutation. We did not detect this sequence variation in 94 control chromosomes, suggesting a similar low frequency of this polymorphism in the Caucasian population. In contrast to our finding, Laitinen et al. (2000) identified this SNP in the Finnish population at a much higher frequency. Surprisingly, their data suggested a possible functional relevance for the QT interval in female LQT1 patients. In healthy controls of both gender, the presence of the SNP was not associated with an altered QTc. However, in females with a particular LQT1 mutation, its presence was associated with a shorter QTc, but only if both alleles were affected. Since our patient is male, heterozygous for the SNP, and also had no LQT1 mutation, accordingly no influence of the SNP on QT time should be expected. Further studies are needed to determine the exact frequency of K897T in the Caucasian population.
To analyse the putative functional effects of K897T, we first tested the hypothesis that the identified amino acid exchange affects channel activity, and might be responsible for the prolonged QT interval in the patient, even after withdrawal of fexofenadine medication (Pinto et al., 1999). However, when expressed in Xenopus oocytes, the K897T HERG alone produced currents with wild-type-like voltage dependence and current amplitude. Corresponding IKr currents, produced by coinjection of either wild-type or K897T HERG with MiRP1, were also very similar. We did not identify significant differences in activation, deactivation, or inactivation kinetics of the variant compared to the wild-type HERG and IKr current, which could explain the observed QT lengthening in the patient at baseline.
PMA reduced HERG currents by shifting channel activation to more positive voltages (Kiehn et al., 1998). Interestingly, this effect is caused by PKA-mediated phosphorylation of the HERG channel protein (Thomas et al., 1999). Since the K897T exchange introduced a novel PKA site, an additional effect on channel activation was possible. However, the voltage of half-maximal activations of wild-type and K897T HERG channels were identical at baseline, and differences in the half-maximal activation voltage resulting from PMA or forskolin treatment were similar for K897T HERG and wild-type HERG currents, suggesting that the K897T exchange does not interfere with HERG current regulation by PKA.
Several HERG mutations in the C-terminal tail result in impaired plasma membrane trafficking due to misprocessing of the mutant proteins (January et al., 2000). We investigated this possibility by biochemical and immunocytochemical methods in transiently transfected mammalian cells. Expression levels of wild-type and K897T variant channels were identical in cells grown at 37°C. The variant protein is also correctly processed as was evident from the presence of the fully glycosylated form of the HERG channel protein.
As reported in a previous study, a rare polymorphism in the MiRP1 potassium channel subunit can be associated with a drug-induced LQTS, while showing no phenotype at baseline (Sesti et al., 2000). Similar to this observation, the K897T sequence variation may only affect channel activity in the presence of fexofenadine.
However, fexofenadine did not block K897T or wild-type HERG channels either formed by the α-subunits alone or by the α-subunits together with the MiRP1 β-subunits. Even at a concentration of 100 μM, which far exceeds the concentrations achieved in plasma of treated patients (about 100 nM; Roy et al., 1996), no inhibition of HERG currents was observed. In contrast, both wild-type mutant and K897T HERG channels were effectively blocked by terfenadine, with identical IC50 values. MiRP1 did not significantly alter the affinity of HERG proteins to terfenadine. Current kinetics also were not significantly affected by the presence of fexofenadine. Additionally, fexofenadine did not influence the expression levels and maturation of both wild-type and variant proteins.
In conclusion, we have demonstrated that the K897T HERG variant produces currents with very similar kinetic properties, and PKC and PKA regulation behaviours, to the wild-type HERG channel. Our functional analyses experimentally confirm the results of the population study of Laitinen et al. (2000) that the K897T polymorphism has no effect on the QT interval, and in line with this suggest that it does not cause the reported LQTS susceptibility in the investigated patient.
We confirmed that fexofenadine does not block wild-type HERG currents, and as a new result showed that it neither affects K897T HERG currents, nor interferes with processing of the HERG proteins. Therefore, our results strongly indicate that QT prolongation and cardiac arrhythmias in the reported patient are not due to an effect of fexofenadine on cardiac IKr currents. Since the HERG channel is the predominant target of drugs with known QT-prolonging activity, and since fexofenadine was proven safe in preclinical and clinical testing for cardiac events, the possibility of a drug-induced LQT in this patient seems also highly unlikely.
We cannot explain the occurrence of arrhythmia in this single case. The presence of left ventricular hypertrophy and possibly of other risk factors for arrhythmia, like old age, hypertension, and cessation of antihypertensive therapy (Pinto et al., 1999; Giraud, 1999; Dhar et al., 2000), offer alternative explanations for the cardiac events, independently of fexofenadine administration. We have not found a disease causing mutation in LQTS associated genes. Therefore, mutations in yet unidentified genes may also be responsible for the arrhythmia susceptibility of the patient.
Acknowledgments
We thank the patient for his consent to this study, Dr Y.M. Pinto for providing the blood sample, and Pearly Lee for critically reading the manuscript.
Abbreviations
- HERG
human ether-à-go-go related gene
- IKr
rapid component of the delayed rectifier K+ current
- MinK
minimal K+ channel
- MiRP
MinK-related protein
- PKA
PKC, protein kinases A and C
- PMA
phorbol 12-myristate 13-acetate
- QTc
QT interval of the electrocardiogram, corrected for the heart rate
- SSCP
single-strand conformation polymorphism
References
- ABBOTT G.W., SESTI F., SPLAWSKI I., BUCK M.E., LEHMANN M.H., TIMOTHY K.W., KEATING M.T., GOLDSTEIN S.A.N. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- CRAIG-McFEELY P.M., ACHARYA N.V., SHAKIR S.A.W. Evaluation of the safety of fexofenadine from experience gained in general practice use in England in 1997. Eur. J. Clin. Pharmacol. 2001;57:313–320. doi: 10.1007/s002280100292. [DOI] [PubMed] [Google Scholar]
- DELAUCHE-CAVALLIER M.C., CHAUFOUR S., GUERALT E., LACROUX A., MURRIETA M., WAJMAN A. QT interval monitoring during clinical studies with mizolastine, a new H1 antihistamine. Clin. Exp. Allergy. 1999;29:206–211. doi: 10.1046/j.1365-2222.1999.0290s3206.x. [DOI] [PubMed] [Google Scholar]
- DHAR S., HAZRA P.K., MALAKAR S., MISTRI G. Fexofenadine-induced QT prolongation: a myth or fact. Br. J. Dermatol. 2000;142:1260–1261. doi: 10.1046/j.1365-2133.2000.03576.x. [DOI] [PubMed] [Google Scholar]
- DHEIN S. Arzneimittel-induzierte QT-Verlängerung und Torsade de Pointes-Arrhythmien. Dtsch. Med. Wschr. 2000;125:703–708. doi: 10.1055/s-2007-1024441. [DOI] [PubMed] [Google Scholar]
- ESCANDE D. Pharmacogenetics of cardiac K+ channels. Eur. J. Pharmacol. 2001;410:281–287. doi: 10.1016/s0014-2999(00)00821-9. [DOI] [PubMed] [Google Scholar]
- FICKER E., THOMAS D., VISWANATHAN P.C., DENNIS A.T., PRIORI S.G., NAPOLITANO C., MEMMI M., WIBLE B.A., KAUFMAN E.S., IYENGAR S., SCHWARTZ P.J., RUDY Y., BROWN A.M. Novel characteristics of a misprocessed mutant HERG channel linked to hereditary long QT syndrome. Am. J. Physiol. 2000;279:H1748–H1756. doi: 10.1152/ajpheart.2000.279.4.H1748. [DOI] [PubMed] [Google Scholar]
- GIRAUD T. QT lengthening and arrhythmias associated with fexofenadine. The Lancet. 1999;353:2072–2073. doi: 10.1016/S0140-6736(05)77890-9. [DOI] [PubMed] [Google Scholar]
- HONIG P.K., WOOSLEY R.L., ZAMANI K., CONNER D.P., CANTILENA L.R. Changes in the pharmacokinetics and electrocardiographic pharmacodynamics of terfenadine with concomitant administration of erythromycin. Clin. Pharmacol. Ther. 1992;52:231–238. doi: 10.1038/clpt.1992.135. [DOI] [PubMed] [Google Scholar]
- HONIG P.K., WORTHAM D.C., ZAMANI K., CONNER D.P., MULLIN J.C., CANTILENA L.R. Terfenadine-ketoconazole interaction. JAMA. 1993;269:1513–1518. [PubMed] [Google Scholar]
- IWASA H., ITOH T., NAGAI R., NAKAMURA Y., TANAKA T. Twenty single nucleotide polymorphisms (SNPs) and their allelic frequencies in four genes that are responsible for familial long QT syndrome in the Japanese population. J. Hum. Genet. 2000;45:182–183. doi: 10.1007/s100380050207. [DOI] [PubMed] [Google Scholar]
- JANUARY C.T., GONG Q., ZHOU Z. Long QT syndrome: Cellular basis and arrhythmia mechanism in LQT2. J. Cardiovasc. Electrophysiol. 2000;11:1413–1418. doi: 10.1046/j.1540-8167.2000.01413.x. [DOI] [PubMed] [Google Scholar]
- KIEHN J., KARLE C., THOMAS D., YAO X., BRACHMANN J., KÜBLER W. HERG potassium channel activation is shifted by phorbol esters via protein kinase A-dependent pathways. J. Biol. Chem. 1998;273:25285–25291. doi: 10.1074/jbc.273.39.25285. [DOI] [PubMed] [Google Scholar]
- KUPERSHMIDT S., SNYDERS D.J., RAES A., RODEN D.M. A K+ channel splice variant common in human heart lacks a C-terminal domain required for expression of rapidly activating delayed rectifier current. J. Biol. Chem. 1998;273:27231–27235. doi: 10.1074/jbc.273.42.27231. [DOI] [PubMed] [Google Scholar]
- LACERDA A.E., KRAMER J., SHEN K.-Z., THOMAS D., BROWN A.M. Comparison of block among cloned cardiac potassium channels by non-antiarrhythmic drugs. Eur. Heart J. Supplements. 2001;3:K23–K30. [Google Scholar]
- LAI L.P., DENG C.L., MOSS A.J., KASS R.S., LIANG C.S. Polymorphism of the gene encoding a human minimal potassium ion channel (minK) Gene. 1994;151:339–340. doi: 10.1016/0378-1119(94)90685-8. [DOI] [PubMed] [Google Scholar]
- LAITINEN P., FODSTAD H., PIIPPO K., SWAN H., TOIVONEN L., VIITASALO M., KAPRIO J., KONTULA K. Survey of the coding region of the HERG gene in long QT syndrome reveals six novel mutations and an amino acid polymorphism with possible phenotypic effects. Human Mutation. 2000;15:580–581. doi: 10.1002/1098-1004(200006)15:6<580::AID-HUMU16>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- LARSEN L.A., CHRISTIANSEN M., VUUST J., ANDERSEN P.S. High-throughput single-strand conformation polymorphism analysis by automated capillary electrophoresis: robust multiplex analysis and pattern-based identification of allelic variants. Hum. Mutat. 1999;13:318–327. doi: 10.1002/(SICI)1098-1004(1999)13:4<318::AID-HUMU9>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- LEES-MILLER J.P., DUAN Y., TENG G.Q., DUFF H.J. Molecular determinant of high-affinity dofetilide binding to HERG1 expressed in Xenopus oocytes: involvement of S6 sites. Mol. Pharmacol. 2000;57:367–374. [PubMed] [Google Scholar]
- LERCHE C., SCHERER C.R., SEEBOHM G., DERST C., WEI A.D., BUSCH A.E., STEINMEYER K. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J. Biol. Chem. 2000;29:22395–22400. doi: 10.1074/jbc.M002378200. [DOI] [PubMed] [Google Scholar]
- MITCHESON J.S., CHEN J., LIN M., CULBERSON C., SANGUINETTI M.C. Proc. Natl. Acad. Sci. U.S.A. 2000. pp. 12329–12333. [DOI] [PMC free article] [PubMed]
- PINTO Y.M., VAN GELDER I.C., HEERINGA M., CRIJNS H.J.G.M. QT lengthening and life-threatening arrhythmias associated with fexofenadine. The Lancet. 1999;353:980. doi: 10.1016/s0140-6736(99)01009-0. [DOI] [PubMed] [Google Scholar]
- PRATT C.M., MASON J., RUSSELL T., REYNOLDS R., AHLBRANDT R. Cardiovascular safety of fexofenadine HCl. Am. J. of Cardiol. 1999;83:1451–1454. doi: 10.1016/s0002-9149(99)00124-1. [DOI] [PubMed] [Google Scholar]
- ROY M.-L., DUMAINE R., BROWN A.M. HERG, a primary human ventricular target of the nonsedating antihistamine terfenadine. Circulation. 1996;94:817–823. doi: 10.1161/01.cir.94.4.817. [DOI] [PubMed] [Google Scholar]
- SALATA J.J., JURKIEWICZ N.K., WALLACE A.A., STUPIENSKI R.F., GUINOSSO P.J., LYNCH J.J. Cardiac electrophysiological actions of the H1-receptor antagonists astemizole and terfenadine compared with chlorpheniramine and pyrilamine. Circ. Res. 1995;76:110–119. doi: 10.1161/01.res.76.1.110. [DOI] [PubMed] [Google Scholar]
- SCHERER C.R., LERCHE C., PINTO Y.M., BUSCH A.E., STEINMEYER K. The antihistamine fexofenadine does not affect IKr currents in a patient reported to have fexofenadine-induced cardiac arrhythmia. Biophys. J. 2000;78:A–342. [Google Scholar]
- SESTI F., ABBOTT G.W., WEI J., MURRAY K.T., SAKSENA S., SCHWARTZ P.J., PRIORI S., RODEN D.M., GEORGE A.L., JR, GOLDSTEIN S.A.N. A common polymorphism associated with antibiotic-induced cardiac arrhythmia. Proc. Natl. Acad. Sci. U.S.A. 2000;97:10613–10618. doi: 10.1073/pnas.180223197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH P., BAUKROWITZ T., YELLEN G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- SPLAWSKI I., SHEN J., TIMOTHY K.W., VINCENT G.M., LEHMANN M.H., KEATING M.T. Genomic structure of three long QT syndrome genes: KvLQT1, HERG, and KCNE1. Genomics. 1998;51:86–97. doi: 10.1006/geno.1998.5361. [DOI] [PubMed] [Google Scholar]
- SÜSSBRICH H., WALDEGGER S., LANG F., BUSCH A.E. Blockade of HERG channels expressed in Xenopus oocytes by the histamine receptor antagonists terfenadine and astemizole. FEBS Lett. 1996;385:77–80. doi: 10.1016/0014-5793(96)00355-9. [DOI] [PubMed] [Google Scholar]
- TAGLIALATELA M., CASTALDO P., PANNACCIONE A., GIORGIO G., GENOVESE A., MARONE G., ANNUNZIATO L. Cardiac ion channels and antihistamines: possible mechanisms of cardiotoxicity. Clin. Exp. Allergy. 1999;29 Suppl 3:182–189. doi: 10.1046/j.1365-2222.1999.0290s3182.x. [DOI] [PubMed] [Google Scholar]
- TAGLIALATELA M., PANNACCIONE A., CASTALDO P., GIORGIO G., ANNUNZIATO L. Inhibition of HERG K+ channels by the novel second-generation antihistamine mizolastine. Br. J. Pharmacol. 2000;131:1081–1088. doi: 10.1038/sj.bjp.0703654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMAS D., ZHANG W., KARLE C.A., KATHÖFER S., SCHÖLS W., KÜBLER W., KIEHN J. Deletion of protein kinase A phosphorylation sites in the HERG potassium channel inhibits activation shift by protein kinase A. J. Biol. Chem. 1999;274:27457–27462. doi: 10.1074/jbc.274.39.27457. [DOI] [PubMed] [Google Scholar]
- TRISTANI-FIROUZI M., CHEN J., MITCHESON J.S., SANGUINETTI M.C. Molecular Biology of K+ channels and their role in cardiac arrhythmias. Am. J. Med. 2001;110:50–59. doi: 10.1016/s0002-9343(00)00623-9. [DOI] [PubMed] [Google Scholar]
- VILLMANN C., BULL L., HOLLMANN M. Kainate binding proteins possess functional ion channel domains. J. Neurosci. 1997;17:7634–7643. doi: 10.1523/JNEUROSCI.17-20-07634.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VINCENT G.M. The molecular genetics of the long QT syndrome: genes causing fainting and sudden death. Annu. Rev. Med. 1998;49:263–274. doi: 10.1146/annurev.med.49.1.263. [DOI] [PubMed] [Google Scholar]