

Abstract
In the presence of genotoxic stress poly(ADP-ribose) polymerase-1 (PARP-1) leads to NAD+ and ATP depletion, participating in the pathogenesis of several disorders including inflammation. Accordingly, chemical inhibitors of PARP-1 are efficacious anti-inflammatories, albeit the underlying molecular mechanisms are still under debate.
This study investigated the effect of the PARP-1 inhibitors 6(5H)-phenanthridinone and benzamide as well as that of benzoic acid, an inactive analogue of benzamide, on development of experimental allergic encephalomyelitis (EAE) in rats. Both 6(5H)-phenanthridinone and benzamide attenuated development of EAE, reducing clinical score, neuroimmune infiltration and expression of inflammatory mediators such as inducible nitric oxide synthase, interleukin-1β and -2, cyclooxygenase-2, tumour necrosis factor-α and interferon-γ in the spinal cord of myelin-immunized rats. Importantly, no evidence of NAD+ and ATP depletion as well as poly(ADP-ribose) formation was detected in the spinal cord.
By contrast, a robust formation of poly(ADP-ribose) occurred in B- and T-cell areas in lymph nodes of myelin-immunized rats and was suppressed by the treatment with 6(5H)-phenanthridinone and benzamide. In cultures of activated rat lymphocytes, 6(5H)-phenanthridinone and benzamide reduced the DNA-binding activity of NF-κB and AP-1 and transcription of pro-inflammatory cytokines such as interleukin-2, interferon-γ and tumour necrosis factor-α.
Notably, benzoic acid did not reproduce the in vivo and in vitro effects of its parent compound.
These findings indicate that PARP-1 promotes transcriptional activation in lymphocytes and inhibitors of its enzymatic activity are useful for the treatment of autoimmune disorders of the central nervous system.
Keywords: PARP, NF-κB, AP-1, multiple sclerosis, autoimmunity
Introduction
Poly(ADP-ribose) polymerase-1 (PARP-1) is a nuclear enzyme activated by DNA damage. After binding to single- or double-DNA strand breaks, PARP-1 transforms β-nicotinamide adenine dinucleotide (NAD+) into long branched polymers of ADP-ribose that are targeted to chromatin-interacting proteins. By so doing, PARP-1 modulates nuclear functions and assists DNA repair (D'amours et al., 1999; Shall & de Murcia, 2000). However, once hyper-activated by massive genotoxic stress, PARP-1 causes NAD+ and ATP depletion, eventually leading to cell death by energy failure (Berger, 1985; Ha & Snyder, 1999; Herceg & Wang, 2001). This cascade of events, known as the ‘suicide pathway' (Gaal et al., 1987), is causative in necrosis (Ha & Snyder, 1999; Moroni et al., 2001) with a still controversial role in apoptosis (Leist et al., 1997; Scovassi & Poirier, 1999; Smulson et al., 2000; Chiarugi, 2002).
A large body of evidence demonstrates that PARP-1 is activated during the inflammatory response, contributing to tissue damage. Accordingly, pharmacological inhibition of PARP-1 is of therapeutic efficacy in models of inflammation (Szabo & Dawson, 1998; Szabo, 1998) and PARP−/− mice are protected from endotoxic shock (Oliver et al., 1999; Kuhnle et al., 1999). However, the mechanisms underlying these anti-inflammatory effects are still under debate (Szabo, 1998).
The unexpected identification of multiple PARPs with different cellular localization (Smith, 2001; Chiarugi, 2002) suggests that poly(ADP-ribose) formation has pleiotropic effects under both physiological and pathological conditions. Furthermore, a growing body of evidence demonstrates that, in addition to its role in DNA repair, PARP-1 also regulates transcriptional activation (D'amours et al., 1999; Butler & Ordahl, 1999; Vyas et al., 2001; Akiyama et al., 2001; Nirodi et al., 2001) and modulates the activity of transcription factors such as activator-protein-2 (AP-2) (Kannan et al., 1999), p53 (Wang et al., 1998), nuclear factor-κB (NF-κB) (Hassa & Hottinger, 1999; Oliver et al., 1999; Chang & Alvarez-Gonzalez, 2001) and octamer-binding protein-1 (Oct-1) (Nie et al., 1998; Ha et al., 2002) (for reviews see Ziegler & Oei, 2001; Chiarugi, 2002). The key role of poly(ADP-ribose) in transcription is well exemplified by the profound alteration in gene expression revealed by the oligonucleotide microarray analysis in PARP-1−/− cells (Simbulan-Rosenthal et al., 2000). Thus, the involvement of PARP-1 in the inflammatory response may be more complex than previously expected, and the therapeutic effects of PARP-1 inhibitors mediated by several mechanisms in addition to the prevention of NAD+ and ATP shortage (Chiarugi, 2002).
To further investigate the mechanisms underlying the anti-inflammatory effects of inhibitors of poly(ADP-ribose) formation, the present study analysed the effects of 6(5H)-phenanthridinone (PHE) and benzamide (BZD) (two enzymatic inhibitors of PARP-1) and of benzoic acid (BA, an inactive analogue of BZD) (Banasik et al., 1992), on the development of experimental allergic encephalomyelitis (EAE) in rats, an experimental model of multiple sclerosis. The working hypothesis was that PARP-1 inhibitors might protect from EAE through two major mechanisms. First, they could directly prevent cellular energy depletion and subsequent demyelination and axonal injury in glia and neurones exposed to DNA-damaging reactive radicals formed during EAE (Smith et al., 1999). Second, considering the role of poly(ADP-ribose) in transcription, PARP-1 inhibitors might modulate gene expression in immune cells, thereby influencing the autoimmune attack on the central nervous system (CNS).
Methods
Materials
Myelin basic protein, complete Freund's adjuvant (CFA), PHE, BZD, BA, ATP, NAD+, alcohol dehydrogenase, bicine, bovine serum albumin (BSA), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium, phenazine ethosulfate, 12-myristate 13-acetate (PMA), ionomycin and REDTaq DNA polymerase were from Sigma (St. Louis, MO, U.S.A.). Mycobacterium tuberculosis H37 Ra was from DIFCO Laboratories (Detroit, MI, U.S.A.). ATP contents were measured using the ATP Determination Kit from Molecular Probes (Eugene, OR, U.S.A.).
Induction of EAE, drug treatment and evaluation of neurological score
EAE has been induced in rats as previously reported (Chiarugi et al., 2001a) in accordance with standard approved protocols. At day 1 post immunization (p.i.), animals (five per group) were intraperitoneally injected every 12 h with 30/60 mg kg−1 PHE, 100/200 mg kg−1 BZD or 100/200 mg kg−1 BA dissolved in DMSO. Myelin-immunized vehicle-treated animals received the same amount of DMSO. Each drug was evaluated in a separate experiment. The neurological deficit was daily monitored from day 1 to 20 p.i. according to the following scoring system: 0, no impairment in muscle tone and motility; 1, tail paralysis; 2, hind limbs weakness; 3, hind limbs paralysis and incontinence; 4, forelimb weakness and/or loss of righting reflex; 5, tongue paralysis or death.
Tissue preparation
Four groups (five animals each) of myelin-immunized rats were treated every 12 h from day 1 p.i. with vehicle (DMSO), 60 mg kg−1 PHE, 200 mg kg−1 BZD or 200 mg kg−1 BA. Five sham-immunized rats were used as controls. At day 14 p.i., animals were anaesthetized, transcardially perfused with saline isotonic solution and the spinal cord as well as the brain and inguinal lymph nodes collected, snap frozen and stored at −80°C. Organs were cut with cryostat in 14 μm sections, mounted on slides, air dried and stored at −80°C. Portions of lumbar spinal cord were collected in eppendorf tubes for RT–PCR, Western blotting and measurements of NAD+ and ATP contents.
Immunohistochemistry
For immunohistochemistry, sections on slides were fixed in cold ethanol for 10 min. Spinal cord sections were incubated for 1 h with phosphate buffered saline pH 7.4 (PBS) containing 10% horse serum (HS) and then for 6 h at room temperature (RT) with a fluorescein-conjugated anti-rat class II major histocompatibility complex (MHC II) antibody (Pharmingen, San Diego, CA, U.S.A.) diluted 1 : 100 in PBS/2% HS. After washing with PBS (three times/10 min each), fluorescence was examined with a Nikon Labophot-2 inverted microscope using a fluorescein filter (excitation 460–490, emission >515 nm). Later on, sections were incubated overnight at 4°C with a biotin-conjugated anti rat ED1 antibody (Serotec, Oxford, U.K.) or monoclonal anti rat Pan-T antibody (Pharmingen, San Diego, CA, U.S.A.) diluted 1 : 100 in PBS/2% HS. After washes with PBS, sections were incubated 1 h at RT with streptavidin-conjugated Cy3 (for ED1 visualization) or Cy3-conjugated anti-mouse antibody (for PanT visualization) diluted 1 : 300 in PBS/2% HS. Fluorescence was visualized using a rhodamine filter (Ex 520, Em >580). Spinal cord sections were subsequently subjected to haemotoxylin-eosin staining for histological evaluation. For immunohistochemical analysis of poly(ADP-ribose), sections were incubated with the anti poly(ADP-ribose) antibody 10H (Alexis, Zurich, CH) diluted 1 : 100 in PBS containing 0.3% Triton X-100 and 2% HS for 6 h at RT. Sections were then incubated 1 h with a Cy3-conjugated anti-mouse antibody. Specificity of immune-detection was established by the omission of the primary antibody.
In vitro lymphocyte activation
Lymphocytes from lymph nodes of healthy Lewis rats were isolated by teasing through stainless steel wire mesh in PBS. Macrophage/dendritic cells were eliminated by plastic adherence. Lymphocytes were seeded in 24-well plates and cultured in AIM V (Gibco, Rockville, MD, U.S.A.) for 24 h and then stimulated with 50 ng ml−1 PMA and 2 μM ionomycin. PHE, BZD and BA were dissolved in dimethyl formamide. Cells were harvested in eppendorf tubes, pelleted and stored at −80°C.
Western blotting
For Western blotting, spinal cord sections or lymphocytes were processed as previously described (Chiarugi et al., 2001a). Twenty μg of protein/lane were subjected to SDS–PAGE in 10% acrylamide mini-gels and then blotted to nitrocellulose membranes (Hybond-ECL, Amersham, U.K.). Membranes were blocked with PBS containing 0.1% Tween-20 and 5% skimmed milk (TBPS/5% milk) and then incubated 1 h with the primary antibody (1 : 1000) in TBPS/5% milk. For phospho c-Jun NH(2)-terminal kinase (JNK) determination, 5% BSA was used instead of milk. Membranes were then washed with TBPS and incubated 1 h in TBPS/5% milk containing a peroxidase-conjugated anti rabbit antibody (1 : 2000). After washing in TBPS, ECL (Amersham) was used to visualize the peroxidase-coated bands. Polyclonal anti-inducible NO synthase (iNOS) and monoclonal anti β-actin antibodies were from (Sigma, St. Louis, MO, U.S.A.); anti cyclooxygenase (COX)-2, anti IkBα, anti phospho-JNK and anti interleukin (IL)-1β were polyclonal antibodies from Santa Cruz (Palo Alto, CA, U.S.A.).
Semi-quantitative RT–PCR
One μg of total RNA extracted with Trizol (GIBCO-BRL, Rockville, MD, U.S.A.) from the spinal cord or cultured lymphocytes was reverse transcribed into DNA and subjected to PCR using the following software-designed oligonucleotide primers: IL-2, 5′-GCGCACCCACTTCAAGCCCT-3′ (sense) and 5′-CCACCACAGTTGCTGGCTCA-3′ (antisense); interferon-γ (IFNγ), 5′-TCGAATCGCACCTGATCACTA-3′ (sense) and 5′-GGGTTGTTCACCTCGAACTTG-3′ (antisense); tumour necrosis factor-α (TNFα), 5′-ATGAGCACAGAAAGCATGATC-3′ (sense) and 5′-CAGAGCAATGACTCCAAAGTA-3′ (antisense); GAPDH, 5′-CCCTCAAGATTGTCAGCAATG-3′ (sense) and 5′-GTCCTCAGTGTAGCCCAGGAT-3′ (antisense). The number of PCR cycles (94°C 30 s, 58°C 30 s, 72°C 1 min, 5 min for the last extension) was selected after determining the linear working range for the reaction. RT products from lymphocytes were amplified 26 times for GAPDH, IFNγ and IL-2, and 30 times for TNFα. RT products from the spinal cords were amplified 26 times for GAPDH, 30 times for TNFα and IFNγ and 38 times for IL-2. PCR amplification products were separated on a 1.8% agarose gel.
Quantification of the contents of NAD+ and ATP
NAD+ contents were quantified by means of an enzymatic cycling procedure according to Shah et al. (1995), and those of ATP with the luciferin-luciferase bioluminescence assay according to Chiarugi et al. (2001b). Data were calculated as nmoles of ATP or NAD+ mg−1 protein.
Electrophoresis mobility gel shift assay
The DNA binding activity of NF-κB, nuclear factor of activated T-cells (NFAT) and activator protein-1 (AP-1) was investigated in lymphocytes resuspended in buffer ‘A' containing (in mM): HEPES pH 7.8 10, KCl 10, EDTA 1, EGTA 0.1, PMSF 1, and 4 μg/ml−1 aprotinin and leupeptin. Cells were kept on ice for 15 min, vortexed every 3 min and then centrifuged at 9000×g for 5 min at 4°C. The nuclear pellet was resuspended in 50 μl of buffer ‘B', analogous to ‘A' plus 400 mM NaCl and incubated for 10 min on ice. The mixture was centrifuged at 15,000×g for 10 min at 4°C and the surnatant aliquoted and stored at −80°C. The DNA binding activity was tested by incubating 10 μg of proteins of the nuclear extract in 20 μl of a buffer containing (in mM): Tris pH 7.4 10 (4% glycerol), MgCl2 1, ethylenediamineteraacetic acid (EDTA) 0.5, dithiothreitol 1, NaCl 50, 0.05 mg ml−1 poly(dl-dC), and 10,000 c.p.m. of specific 32P-labelled oligonucleotide for 20 min at RT. The mixture was electrophoresed in 4% non-denaturing polyacrylamide gels that, after drying, were exposed to X-ray film (Amersham). The following double-stranded oligonucleotides were used: 5′-AGTTGAGGGGACTTTCCCAGGC-3′ (NF-κB), 5′-CGCCCAAAGAGGAAAATTTGTTTCATA-3′ (NFAT), 5′-CGCTTGATGAGTCAGCCGGAA-3′ (AP-1).
Statistical analysis
Results are expressed as the mean±s.e.mean for n observations. Evaluation of significant differences among groups was performed using the Student's t-test, Kruskal-Wallis with Dunn's post test, and Mann-Whitney U-test. Difference among groups was considered significant for P values <0.05.
Results
Effects of PHE, BZD and BA on the development of EAE
The effects of PHE, BZD and BA on the development of EAE are summarized in Table 1 and Figure 1. In myelin-immunized rats, the injection of 30 mg kg−1 PHE reduced the peak disease score, whereas it had no effect on day of onset, duration and day of maximal disease. At 60 mg kg−1, PHE also delayed onset and day of maximal disease (Table 1, Figure 1A). Similarly, BZD attenuated development of EAE dose-dependently, delaying onset and day of maximal disease, and reducing duration and peak disease score (Table 1, Figure 1B). Neither PHE nor BZD affected the incidence of EAE. Notably, BA had no effects on the development of EAE (Table 1, Figure 1C).
Table 1.
Effects of PHE, BZD and BA on EAE in the rat
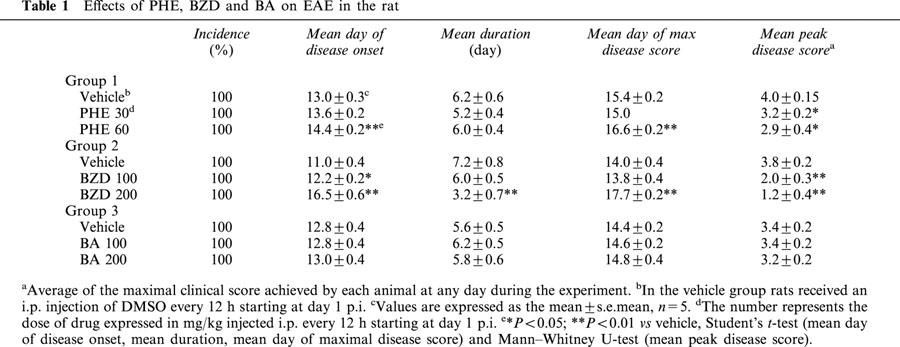
Figure 1.
PHE and BZD but not BA reduce symptoms in rats with EAE. After immunization, animals were treated every 12 h from day 1 p.i. with PHE (A), BZD (B) or BA (C) dissolved in DMSO. Vehicle-treated animals received an equal volume of DMSO. Each drug was evaluated in a separate experiment. Multiple clinical score (MCS) was assessed daily and is shown as mean±s.e.mean of five animals per group. *P<0.05; **P<0.01 vs vehicle, Kruskal-Wallis and Dunn's post test.
Effects of PHE, BZD and BA on neuroinflammatory infiltrates and expression of pro-inflammatory mediators in the spinal cord of rats with EAE
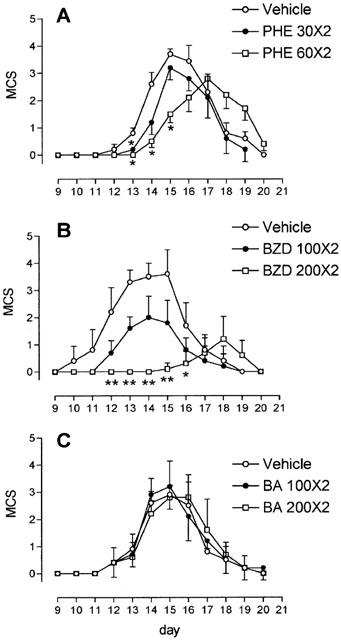
Rats were sacrificed at day 14 p.i. Spinal cords of sham-immunized rats were free of inflammatory infiltrates and MHC II+ cells. Conversely, the spinal cords of myelin-immunized vehicle-treated rats showed massive inflammatory infiltrates of MHC II+ cells in meningeal and perivascular localization. Immune cell infiltration was drastically reduced by the treatment with PHE and BZD but not BA (Figure 2). Similarly, infiltrates of Pan-T+ lymphocytes and ED1+ macrophages were absent in the spinal cord of sham-immunized animals, massive in those of myelin-immunized vehicle- or BA-treated rats and suppressed in those of rats sensitized with myelin and injected with PHE or BZD (data not shown). In the spinal cord of rats with EAE, double labelling immunohistochemistry showed that numerous ED1+ macrophages expressed MHC II, whereas rare Pan-T+ cells were MHC II+, in line with the ability of T lymphocytes to acquire MHC II antigens from MHC II+ cells during EAE (Flugel et al., 2001). A population of MHC II+ cells, probably B lymphocytes, expressed neither ED1 nor PanT (data not shown).
Figure 2.
PHE and BZD but not BA reduce inflammatory infiltrates in the spinal cord of myelin-immunized rats. Inflammatory infiltrates in sections of the lumbar spinal cord of sham- (A, B) and myelin-immunized (C-L) rats at day 14 p.i. are shown with haematoxylin-eosin staining (A, C, E, G, I) or MHC II immunohistochemistry (B, D, F, H, L). Inflammatory infiltrates are not detectable in sham-immunized animals (A, B). Massive meningeal and perivascular inflammatory infiltrates (C) colocalize with MHC II+ cells (D) (arrows) in rats with EAE. Note the striking reduction of infiltrates in rats treated daily with 60 mg kg−1 PHE (E, F, arrows) or 200 mg kg−1 BZD (G, H, arrows). The treatment with 200 mg kg−1 BA does not affect immune infiltration (I, L, arrows). Each section is representative of one group of animals (n=5). Scale bar=550 μm.
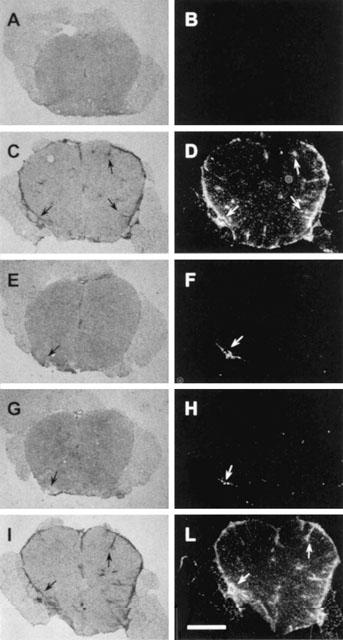
The mRNA levels of IL-2, TNFα and IFNγ were markedly increased in the spinal cord of rats with EAE. The treatment with PHE and BZD but not BA reduced cytokine transcript levels (Figure 3A). iNOS and IL-1β were not detected in the spinal cord of sham-immunized rats, whereas COX-2 was slightly expressed, in line with its constitutive presence in the rat CNS (Quan et al., 1998). Remarkably, PHE or BZD but not BA reduced the expression of the three proteins induced in the spinal cord of myelin-immunized rats (Figure 3B).
Figure 3.
PHE and BZD but not BA reduce the expression of inflammatory mediators in the spinal cord of myelin-immnunized rats. PHE and BZD but not BA reduce the mRNA levels of IL-2, IFNγ and TNFα (A) as well as the expression of iNOS, IL-1β and COX-2 (B) in the spinal cord of animals with EAE at day 14 p.i. Each lane corresponds to one animal, and two representative animals per group (n=5) are shown. GAPDH and β-actin are shown as loading controls.
NAD+ and ATP contents in the spinal cord of rats with EAE: effect of PHE, BZD and BA
The contents of NAD+ and ATP were increased in the lumbar spinal cord of myelin-sensitized vehicle-treated rats at day 14 p.i. compared to sham-immunized animals. NAD+ levels increased about 2 fold from 12.3±4 to 27.5±2.5 nmol mg prot−1, whereas those of ATP augmented from 23.4±2.2 to 37±4.2 nmol mg prot−1. These increments were significantly reduced by the treatment with PHE or BZD but not BA (Table 2). Although various cell types contribute to the spinal cord contents of NAD+ and ATP, these findings, along with the reduction of neuroimmune infiltrates induced by PHE and BZD (Figures 2 and 3), suggest that the nucleotide increase was due to the presence of actively metabolizing immune cells.
Table 2.
Effects of PHE, BZD and BA on the spinal cord contents of NAD+ and ATP in sham- and myelin-immunized rats at day 14 p.i.

Poly(ADP-ribose) formation in the spinal cord and lymph nodes of rats with EAE
Poly(ADP-ribose) formation was evaluated by means of immunohistochemistry in the spinal cord and draining lymph nodes of rats at day 14 p.i. Polymer content was below the detection limit in the spinal cord of sham- and myelin-immunized rats (not shown). Low levels of poly(ADP-ribose) immunoreactivity were detected in the medulla of lymph nodes of sham-immunized rats (Figure 4A). By contrast, numerous poly(ADP-ribose)+ cells were present in medullary cords and paracortex of lymph nodes of rats with EAE (Figure 4B,G,H). Fluorescence was localized in round-shaped structures (Figure 4F), consistent with the nuclear synthesis of poly(ADP-ribose). Some poly(ADP-ribose)+ cells were MHC II+ (data not shown). Because both the anti-poly(ADP-ribose) and anti-PanT antibodies were raised in mouse, it was impossible to test whether PanT+ cells expressed ADP-ribose chains. Notably, polymer formation was reduced in lymph nodes of rats treated with PHE (Figure 4C) or BZD (Figure 4D) but not BA (Figure 4E), demonstrating that administration of PARP-1 inhibitors inhibited poly(ADP-ribose) formation in rats.
Figure 4.
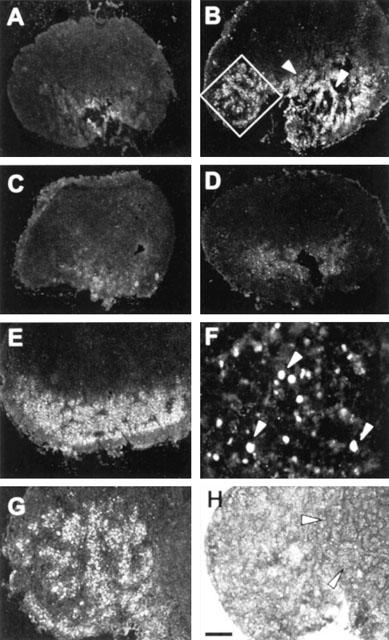
Immunohistochemical evaluation of poly(ADP-ribose) formation in lymph nodes during EAE. (A) Low levels of poly(ADP-ribose) immunoreactivity are present in the medulla of lymph nodes of sham-immunized animals. (B) Numerous poly(ADP-ribose)+ cells are localized in the paracortex (inset) and medullar cords (arrowheads) in lymph nodes of myelin-immunized rats at day 14 p.i. The number of poly(ADP-ribose)+ cells is significantly reduced by the treatment with PHE (C) or BZD (C) but not BA (E). (F) Poly(ADP-ribose)+ nuclei (arrowheads). The inset depicted in (B), is shown at higher magnification with MHC II immuno- (G) and haematoxylin-eosin (H) staining. Note that poly(ADP-ribose)+ cells are localized in the paracortex (G), whereas secondary follicles (H, arrowheads) are not immunoreactive. Each section is representative of one group of animals (n=5). Scale bar=500 μm (A–E), 40 μm (F), 150 μm (G, H).
Effects of PHE, BZD and BA on transcriptional activation and Th1 cytokine expression in lymphocytes
It is well established that EAE is triggered by clonal expansion of autoreactive lymphocytes in draining lymph nodes and subsequent migration into the CNS (Bar-Or et al., 1999). The evidence that the decreased content of poly(ADP-ribose) in lymph nodes correlated with reduced levels of pro-inflammatory cytokines of the Th1 profile in the spinal cord and attenuation of EAE, suggested that PHE and BZD targeted key step(s) of lymphocyte activation. Thus, the effect of the PARP-1 inhibitors on in vitro lymphocytes activation was evaluated.
As shown in Figure 5A, PHE (1–100 μM) or BZD (0.1–10 mM) but not BA (0.1–10 mM) reduced the levels of IL-2 mRNA dose-dependently in lymphocytes activated in vitro for 2.5 h with PMA/ionomycin. The inhibition was maintained over time (Figure 5Ba and b). PHE reduced the levels of IL-2 transcripts even when added 4 h after lymphocyte activation (Figure 5Bc) and transcriptional inhibition was promptly relieved after PHE wash out (Figure 5Bd), indicating that PHE's effects were not due to cytotoxicity. PHE or BZD, but not BA, also reduced the transcript levels of IFNγ and TNFα in activated lymphocytes (Figure 5C).
Figure 5.
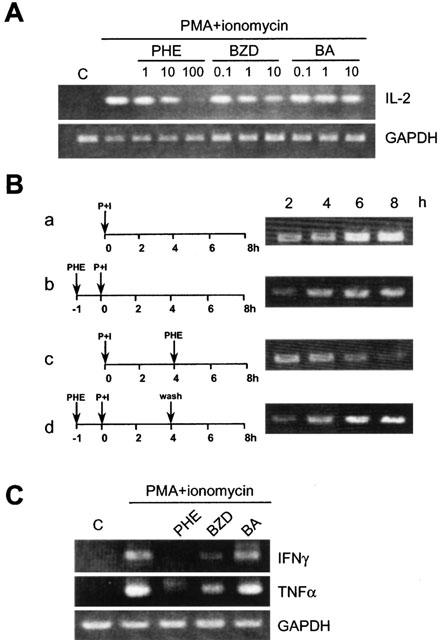
PHE and BZD but not BA reduce the mRNA levels of IL-2, IFNγ and TNFα in rat lymphocytes. Lymphocytes were isolated from inguinal lymph nodes and stimulated in culture with PMA and ionomycin. Levels of IL-2, IFN-γ and TNFα transcripts were evaluated after 2.5 h (A, C) or over time (2–8 h) (B). GAPDH is shown as a loading control. (A) The preincubation (1 h) with PHE or BZD but not BA reduces IL-2 mRNA dose-dependently in activated lymphocytes. (Ba) Over time increase of IL-2 mRNA induced by PMA/ionomycin (P+I). (Bb) The pre-incubation (1 h) with 100 μM PHE reduces the transcript levels of IL-2 at each time point. (Bc) 100 μM PHE decreases the mRNA levels of IL-2 when added 4 h after PMA/ionomycin exposure. (Bd) IL-2 mRNA increases after PHE (100 μM) wash out. (C) The pre-incubation (1 h) with 100 μM PHE or 10 mM BZD but not 10 mM BA reduce the levels of IFNγ and TNFα transcripts induced by PMA/ionomycin. One experiment representative of three (A, C) and two (B) is shown.
The expression of IL-2, IFNγ and TNFα depends on the activation of signalling pathways leading to activation of transcription factors such as NF-κB, AP-1 and NFAT. In lymphocytes exposed 1 h to PMA/ionomycin, 100 μM PHE or 10 mM BZD did not affect both IκBα degradation and JNK phosphorylation (Figure 6A), triggers of NF-κB and AP-1 activation, respectively. These results suggested that PARP-1 inhibitors did not interfere with signalling upstream of transcription factor activation. The exposure to PMA/ionomycin increased the DNA binding activity of NF-κB, AP-1 and NFAT in lymphocytes (Figure 6B–D). The binding activity was reduced by the addition of 50 fold molar excess of specific but not non-specific cold oligoprobes in the binding mixture (not shown), indicating specificity of the retarded bands. Notably, incubation with PHE or BZD selectively reduced the PMA/ionomycin-induced DNA binding activity of NF-κB and AP-1 (Figure 6B and C), having no effects on that of NFAT (Figure 6D).
Figure 6.
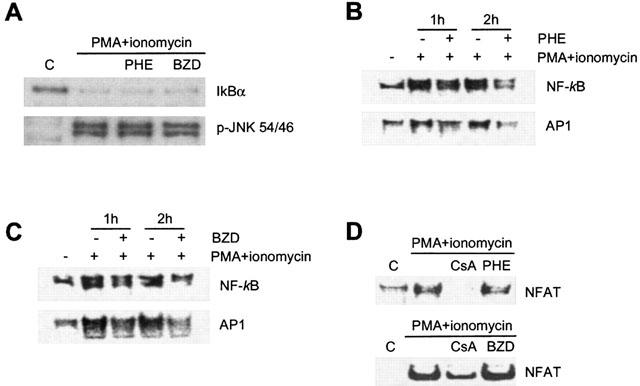
Effects of PHE and BZD on transcriptional activation in rat lymphocytes. Lymphocytes were isolated from lymph nodes, and stimulated in culture with PMA 50 ng ml−1 and 2 μM ionomycin. (A) The pre-incubation (1 h) with 100 μM PHE or 10 mM BZD has no effects on IkBα degradation or JNK phosphorylation induced by 1 h exposure to PMA/ionomycin. (B, C) Identical concentrations of PHE and BZD reduce the DNA binding activity of NF-κB and AP-1 in lymphocytes exposed 1 or 2 h to PMA/ionomycin. (D) Five μM Cyclosporin A (CsA) but not 100 μM PHE or 10 mM BZD inhibits NFAT DNA binding in lymphocytes challenged with PMA/ionomycin for 2 h. One experiment representative of 2 (A, D) and 3 (B, C) is shown.
Discussion
This study reports the novel findings that PARP-1 inhibitors PHE and BZD (i) reduced neuroinflammation and symptoms in rats with EAE; (ii) prevented poly(ADP-ribose) formation in their draining lymph nodes, and (iii) impaired NF-κB and AP-1 activity as well as transcription of pro-inflammatory cytokines in lymphocytes.
It is well established that PARP-1 activation participates in the development of inflammation by inducing cell death by NAD+ and ATP depletion and subsequent immune cell infiltration (Szabo & Dawson, 1998; Szabo, 1998). It is reported here that the contents of NAD+ and ATP were increased in the spinal cord of rats with EAE but, still, PARP-1 inhibitors PHE and BZD exerted anti-inflammatory effects. Therefore, the absence of NAD+ and ATP depletion, together with that of poly(ADP-ribose) formation suggest that, under these experimental conditions, PARP-1 was not hyper-activated in the spinal cord during the acute phase of EAE, in apparent contrast with the results of Scott et al. (2001). The present findings, however, are in agreement with the anti-inflammatory effects of 3-amino BZD in a model of chronic colitis characterized by an increased energy charge in the inflamed tissue (Jijon et al., 2000), and with the induction of the metabolic route of NAD+ synthesis in the CNS of rats with EAE (Chiarugi et al., 2001a). Data are also in line with the recent evidence that PARP−/− mice are protected from ischaemic brain injury through mechanisms apparently independent of energy failure (Goto et al., 2002). Taken as a whole, the present results suggest that PARP-1 participated in the pathogenesis of EAE by mechanism(s) not related to impairment of energy dynamic in the CNS.
It is well established that priming of lymphocytes in draining lymph nodes and ensuing invasion of the CNS with local synthesis of pro-inflammatory cytokines are key events in EAE pathogenesis (Xiao & Link, 1999; Bar-Or et al., 1999). It is reported here the novel finding that poly(ADP-ribose) formation occurred in draining lymph nodes of rats with EAE. DNA damage occurring in proliferating lymphocyte (Greer & Kaplan, 1986) may have been the trigger of poly(ADP-ribose) formation in lymph nodes, consistent with a previous demonstration in activated macrophages (Berton et al., 1991). Notably, inhibition of poly(ADP-ribose) formation in B- and T-cell areas of draining lymph nodes correlated with amelioration of EAE. These results, along with the evidence that PHE and BZD reduced transcription of Th-1 cytokines such as IL-2, IFNγ and TNFα in both cultured lymphocytes and spinal cord of rats with EAE, suggest that PARP-1 inhibitors impaired the autoimmune response by reducing lymphocyte activation. This hypothesis is corroborated by studies demonstrating that PARP-1 expression is increased in proliferating lymphocytes (McNerney et al., 2001) and PARP-1 inhibitors such as BZD analogues and PHE itself impair B- and T-cell activation in vitro and in vivo (Broomhead & Hudson, 1985; McNerney et al., 1987; Exley et al., 1987; Weltin et al., 1995).
It is shown here for the first time that PHE and BZD reduced the DNA-binding activity of NF-κB and AP-1 in lymphocytes. Considering the central role of these two transcription factors in pro-inflammatory cytokine expression by Th1-cells, it is plausible that PHE and BZD reduced Th1 lymphocyte activation in both cultures and myelin-immunized rats by targeting transcriptional activation. This event may have reduced subsequent macrophage/microglia activation, neuroinflammation and neurological symptoms. PARP-1 also regulates the expression of pro-inflammatory molecules such as iNOS (Le Page et al., 1998; Oliver et al., 1999). IL-1β (Ha et al., 2002), CD11a (Ullrich et al., 2001), ICAM1 (Zingarelli et al., 1998; Ha et al., 2002) as well as CXCL1 (Nirodi et al., 2001). It is possible, therefore, that decreased expression of these pro-inflammatory agents may have contributed to the therapeutic effects of PHE and BZD on rats. Results shown in Figure 3B are in line with this assumption.
Although several reports demonstrate a key role of PARP-1 in trans-activation (Ziegler & Oei, 2001; Chiarugi, 2002), the underlying molecular mechanisms are controversial (Hassa et al., 2001; Chang & Alvarez-Gonzalez, 2001; Ha et al., 2002). It has been proposed that ADP-ribose polymer chains regulate the assembly of the transcriptional machinery, thereby modulating RNA synthesis (Vispè et al., 2000) and gene expression (Akiyama et al., 2001; Chang & Alvarez-Gonzalez, 2001; Ullrich et al., 2001). In keeping with this, PARP-1 has been identified as the transcriptional coactivator TFIIC (Slattery et al., 1983), and PARP-1 binding to promoter elements regulates transcription (Butler & Ordahl, 1999; Akiyama et al., 2001; Nirodi et al., 2001; Zhang et al., 2002). The lack of effect of PHE and BZD on the DNA binding activity of NFAT, however, indicates that PARP-1 is not a general regulator of transcription factors, consistent with the recent evidence that PARP-1-dependent transcriptional regulation is gene and cell type-specific (Ha et al., 2002). In activated lymphocytes exposed to PARP-1 inhibitors, therefore, the unaffected activity of NFAT and other trans-activating factors can explain residual transcription of IL-2 (see Figure 6Bb) and clonal proliferation (Weltin et al., 1995). In myelin-immunized rats these events can be translated into an attenuated, yet not prevented, development of EAE.
Theoretically, PHE and BZD may have also affected EAE pathogenesis impairing lymphocyte proliferation by accumulation of DNA-strand breaks (Greer & Kaplan, 1986) or inhibiting NAD(P)+-dependent enzymes such as the newly identified PARPs (Smith, 2001; Chiarugi, 2002), mono(ADP-ribose) synthases (Banasik et al., 1992) and the ectoenzyme CD38, an ADP-ribosyl cyclase involved in lymphocyte signalling (Lund et al., 1998). However, the amelioration of EAE obtained with a selective inhibitor of PARP-1 structurally unrelated to PHE and BZD such as 5-iodo-6-amino-1,2-benzopyrone (Scott et al., 2001), along with the present evidence that BA did not reproduce any of the pharmacological effects of its parent compound strengthen the hypothesis that the synthesis of poly(ADP-ribose) assisted lymphocyte activation and development of EAE.
In conclusion, this study demonstrates that inhibitors of PARP-1 reduce transcription of pro-inflammatory cytokines in lymphocytes and attenuate EAE in rats. It is worth noting that from both a molecular and clinical point of view these effects resemble those of corticosteroids (de bosscher et al., 2000), among the most effective and widely used remedies for the treatment of multiple sclerosis. Considering the devastating effects of the autoimmune attack on the CNS, a possible therapeutic synergism between PARP-1 inhibitors and corticosteroids undoubtedly merits further investigation.
Acknowledgments
This work was supported in part by the ‘Project for Young Investigator' of the University of Florence. The support of Prof John Walsh is gratefully acknowledged.
Abbreviations
- AP-1
activator protein-1
- BA
benzoic acid
- BSA
bovine serum albumin
- BZD
benzamide
- COX-2
cyclooxygenase-2
- EAE
experimental allergic encephalomyelitis
- IL
interleukin
- IFNγ
interferon-γ
- MHC II
major histocompatibility complex II
- NF-κB
nuclear factor-κB
- PARP-1
poly(ADP-ribose) polymerase-1
- PBS
phosphate buffered saline
- PHE
6(5H)-phenanthridinone
- p.i.
post immunization
- RT
room temperature
- iNOS
inducible nitric oxide synthase
- TNFα
tumour necrosis factor-α
- HS
horse serum
References
- AKIYAMA T., TAKASAWA S., NATA K., KOBAYASHI S., ABE M., SHERVANI N.J., IKEDA T., NAKAGAWA K., UNNO M., MATSUNO S., OKAMOTO H. Activation of Reg gene, a gene for insulin-producing β-cell regeneration: Poly(ADP-ribose) polymerase binds Reg promoter and regulates the transcription by autopoly(ADP-ribosyl)ation. Proc. Natl. Acad. Sci. 2001;98:48–53. doi: 10.1073/pnas.240458597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BANASIK M., KOMURA H., SHIMOYAMA M., UEDA K. Specific inhibitors of poly(ADP-ribose) synthetase and mono(ADP-ribosyl)transferase. J. Biol. Chem. 1992;267:1569–1575. [PubMed] [Google Scholar]
- BAR-OR A., OLIVEIRA E.M.L., ANDERSON D.E., HAFLER D.A. Molecular pathogenesis of multiple sclerosis. J. Neuroimmunol. 1999;100:252–259. doi: 10.1016/s0165-5728(99)00193-9. [DOI] [PubMed] [Google Scholar]
- BERGER N.A. Poly (ADP-ribose) in the cellular response to DNA damage. Radiat. Res. 1985;101:4–15. [PubMed] [Google Scholar]
- BERTON G., SORIO C., LAUDANNA C., MENEGAZZI M., CARCERERI DE PRATI A., SUZUKI H. Activation of human-derived macrophages by interferon-γ is accompanied by increase of poly(ADP-ribose) polymerase activity. Biochim. Biophys. Acta. 1991;1091:101–109. doi: 10.1016/0167-4889(91)90228-p. [DOI] [PubMed] [Google Scholar]
- BROOMHEAD C., HUDSON L. Administration of adenosine diphosphate-ribosyl transferase antagonist allows in vivo control of anti-dinitrophenyl response. Int. Arch. Allergy Appl. Immunol. 1985;77:385–393. doi: 10.1159/000233845. [DOI] [PubMed] [Google Scholar]
- BUTLER J., ORDAHL C.P. Poly(ADP-ribose) polymerase binds with transcription enhancer factor 1 to MCAT1 elements to regulate muscle-specific transcription. Mol. Cell Biol. 1999;19:296–306. doi: 10.1128/mcb.19.1.296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANG W.J., ALVAREZ-GONZALEZ R. The sequence-specific DNA binding of NF-κB is reversibly regulated by automodification-reaction of poly(ADP-ribose) polymerase-1. J. Biol. Chem. 2001;276:47664–47670. doi: 10.1074/jbc.M104666200. [DOI] [PubMed] [Google Scholar]
- CHIARUGI A. PARP-1: killer or conspirator? The suicide hypothesis revisited. Trends Pharmacol. Sci. 2002;23:122–129. doi: 10.1016/S0165-6147(00)01902-7. [DOI] [PubMed] [Google Scholar]
- CHIARUGI A., COZZI A., BALLERINI C., MASSACESI L., MORONI F. Kynurenine 3-monooxygenase activity and neurotoxic kynurenine metabolites increase in the central nervous system of rats with experimental allergic encephalomyelitis. Neuroscience. 2001a;102:687–695. doi: 10.1016/s0306-4522(00)00504-2. [DOI] [PubMed] [Google Scholar]
- CHIARUGI A., MELI E., MORONI F. Similarities and differences in the neuronal death processes activated by 3OH-kynurenine and quinolinic acid. J. Neurochem. 2001b;77:1310–1316. doi: 10.1046/j.1471-4159.2001.00335.x. [DOI] [PubMed] [Google Scholar]
- D'AMOURS D., DESNOYERS S., POIRIER G.G. Poly(ADP-ribosyl)ation reactions in the regulation of nuclear functions. Biochem. J. 1999;342:249–268. [PMC free article] [PubMed] [Google Scholar]
- DE BOSSCHER K., VANDEN BERGHE W., HAEGEMAN G. Mechanisms of anti-inflammatory action and of immunosuppression by glucocorticoids: negative interference of activated glucocorticoid receptor with transcription factors. J. Neuroimm. 2000;109:16–22. doi: 10.1016/s0165-5728(00)00297-6. [DOI] [PubMed] [Google Scholar]
- EXLEY R., GORDON J., CLEMENS M.J. Induction of B-cell differentiation antigens in interferon- or phorbol ester-treated Daudi cells is impaired by inhibitors of ADP-ribosyltransferase. Proc. Natl. Acad. Sci. 1987;84:6467–6470. doi: 10.1073/pnas.84.18.6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FLUGEL A., BERKOWICZ T., RITTER T., LAUBER M., JENNE D.E., LI Z., WILLEM M., WEKERLE H. Migratory activity and functional changes of green fluorescent effector cells before and during experimental autoimmune encephalomyelitis. Immunity. 2001;14:547–560. doi: 10.1016/s1074-7613(01)00143-1. [DOI] [PubMed] [Google Scholar]
- GAAL J.C., SMITH K.R., PEARSON C.K. Cellular euthanasia mediated by a nuclear enzyme: a central role for nuclear ADP-ribosylation in cellular metabolism. Trends Biochem. Sci. 1987;12:129–130. [Google Scholar]
- GOTO S., XUE R., SUGO N., SAWADA M., BLIZZARD K.K., POITRAS M.F., JOHNS D.C., DAWSON T.M., DAWSON V.L., CRAIN B.J., TRAISTMAN R.G., MORI S., HURN P.D. Poly(ADP-ribose) polymerase impairs early and long-term experimental stroke recovery. Stroke. 2002;33:1101–1106. doi: 10.1161/01.str.0000014203.65693.1e. [DOI] [PubMed] [Google Scholar]
- GREER W.L., KAPLAN J.G. Early nuclear events in lymphocyte proliferation. The role of DNA strand break repair and ADP ribosylation. Exp. Cell Res. 1986;166:399–415. doi: 10.1016/0014-4827(86)90486-6. [DOI] [PubMed] [Google Scholar]
- HA H.C., HESTER L.D., SNYDER S.H. Poly(ADP-ribose) polymerase-1 dependence of stress-induced transcription factors and associated gene expression in glia. Proc. Natl. Acad. Sci. 2002;99:3270–3275. doi: 10.1073/pnas.052712399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HA H.C., SNYDER S.H. Poly(ADP-ribose) polymerase is a mediator of necrotic cell death by ATP depletion. Proc. Natl. Acad. Sci. 1999;69:13978–13982. doi: 10.1073/pnas.96.24.13978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HASSA P.O., COVIC M., HASAN S., IMHOF R., HOTTINGER M.O. The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappaB coactivator function. J. Biol. Chem. 2001;276:45588–45597. doi: 10.1074/jbc.M106528200. [DOI] [PubMed] [Google Scholar]
- HASSA P.O., HOTTINGER M.O. A role of poly(ADP-ribose) polymerase in NF-κB transcriptional activation. Biol. Chem. 1999;380:953–959. doi: 10.1515/BC.1999.118. [DOI] [PubMed] [Google Scholar]
- HERCEG Z., WANG Z.Q. Functions of poly(ADP-ribose) polymerase (PARP) in DNA repair, genomic integrity and cell death. Biochem. J. 2001;477:97–110. doi: 10.1016/s0027-5107(01)00111-7. [DOI] [PubMed] [Google Scholar]
- JIJON H.B., CHURCHILL T., MALFAIR D., WESSLER A., JEWELL L.D., PARSON H.G., MADSEN K. Inhibition of poly(ADP-ribose) polymerase attenuates inflammation in a model of chronic colitis. Am. J. Physiol. 2000;279:G641–G651. doi: 10.1152/ajpgi.2000.279.3.G641. [DOI] [PubMed] [Google Scholar]
- KANNAN P., YU Y.H., WANKHADE S., TAINSKY M.A. Poly(ADP-ribose)polymerase is a coactivator for AP2-mediated transcriptional activation. Nucl. Ac. Res. 1999;27:866–874. doi: 10.1093/nar/27.3.866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KUHNLE S., NICOTERA P., WENDEL A., LEIST M. Prevention of endotoxin-induced lethality, but not liver apoptosis in poly(ADP-ribose) polymerase-deficient mice. Biochem. Biophys. Res. Comm. 1999;263:433–438. doi: 10.1006/bbrc.1999.1393. [DOI] [PubMed] [Google Scholar]
- LE PAGE C., SANCEAU J., DRAPIER J.C., WIETZERBIN J. Inhibitors of ADP-ribosylation impair inducible nitric oxide synthase gene transcription through inhibition of NF-κB activation. Biochem. Biophys. Res. Comm. 1998;243:451–457. doi: 10.1006/bbrc.1998.8113. [DOI] [PubMed] [Google Scholar]
- LEIST M., SINGLE B., KUNSTLE G., VOLBRACHT C., ENTZE H., NICOTERA P. Apoptosis in the absence of poly(ADP-ribose)polymerase. Biochem. Biophys. Res. Comm. 1997;233:518–522. doi: 10.1006/bbrc.1997.6491. [DOI] [PubMed] [Google Scholar]
- LUND F.E., COCKAYNE D.A., RANDAL T.D., SOLVASON N., SCHUBER F., HOWARD M.C. CD38: a new paradigm in lymphocyte activation and signal transduction. Immunol. Rev. 1998;161:79–93. doi: 10.1111/j.1600-065x.1998.tb01573.x. [DOI] [PubMed] [Google Scholar]
- MCNERNEY R., DARLING D., JOHNSTONE A.P. Differential control of proto-oncogene c-myc and c-fos expression in lymphocytes and fibroblasts. Biochem. J. 1987;245:605–608. doi: 10.1042/bj2450605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCNERNEY R., TAVASOLLI M., SHALL S., BRAZINSKI A., JOHNSTONE A.P. Changes of mRNA levels of poly(ADP-ribose) polymerase during activation of human lymphocytes. Biochim. Biophys. Acta. 2001;1009:185–187. doi: 10.1016/0167-4781(89)90099-7. [DOI] [PubMed] [Google Scholar]
- MORONI F., MELI E., PERUGINELLI F., CHIARUGI A., COZZI A., PICCA R., ROMAGNOLI P., PELLICCIARI R., PELLEGRINI-GIAMPIETRO D.E. Poly(ADP-ribose) polymerase inhibitors attenuate necrotic but not apoptotic neuronal death in experimental models of cerebral ischemia. Cell Death Diff. 2001;8:921–932. doi: 10.1038/sj.cdd.4400884. [DOI] [PubMed] [Google Scholar]
- NIE J., SAKAMOTO S., SONG D., QU Z., OTA K., TANIGUICHI T. Interaction of Oct-1 and automodification domain of poly(ADP-ribose) synthetase. FEBS Lett. 1998;424:27–32. doi: 10.1016/s0014-5793(98)00131-8. [DOI] [PubMed] [Google Scholar]
- NIRODI C., NAGDAS S., GYGI S.P., OLSON G., RUEDI A., RICHMOND A. A role for poly(ADP-ribose) polymerase (PARP) in the transcriptional regulation of the melanoma growth stimulatory activity (CXCL1) gene expression. J. Biol. Chem. 2001;276:9366–9374. doi: 10.1074/jbc.M009897200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLIVER F.J., MENISSIER-DE MURCIA J., NACCI C., DECKER P., ANDRIANTSITOHANIA R., MULLER S., DE LA RUBIA G., STOCLET J.C., DE MURCIA G. Resistance to endotoxic shock as a consequence of defective NF-κB activation in poly(ADP-ribose) polymerase-1 deficient mice. EMBO J. 1999;18:4446–4454. doi: 10.1093/emboj/18.16.4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUAN N., WHITESIDE M., HERKENHAM M. Cyclooxygenase 2 mRNA expression in rat brain after peripheral injection of lipopolysaccharides. Brain Res. 1998;802:189–197. doi: 10.1016/s0006-8993(98)00402-8. [DOI] [PubMed] [Google Scholar]
- SCOTT G.S., HAKE P., KEAN R.B., VIRAG L., SZABO C., HOOPER D.C. Role of poly(ADP-ribose) synthetase activation in the development of experimental allergic encephalomyelitis. J. Neuroimm. 2001;117:78–86. doi: 10.1016/s0165-5728(01)00329-0. [DOI] [PubMed] [Google Scholar]
- SCOVASSI I.A., POIRIER G.G. Poly(ADP-ribosylation) and apoptosis. Mol. Cell. Biochem. 1999;199:125–137. doi: 10.1023/a:1006962716377. [DOI] [PubMed] [Google Scholar]
- SHAH G.M., POIRIER D., DUCHAINE G., DESNOYERS S., BROCHU G., LAGEAUX J., VERREAULT A., HOFLACK J.-C., KIRKLAND J.B., POIRIER G.G. Methods for biochemical study of poly(ADP-ribose) metabolism in vitro and in vivo. Anal. Biochem. 1995;227:1–13. doi: 10.1006/abio.1995.1245. [DOI] [PubMed] [Google Scholar]
- SHALL S., DE MURCIA G. Poly (ADP-ribose) polymerase-1: what we have learned from the deficient mouse model. Mutation Res. 2000;460:1–15. doi: 10.1016/s0921-8777(00)00016-1. [DOI] [PubMed] [Google Scholar]
- SIMBULAN-ROSENTHAL C.M., LY D.H., ROSENTHAL D.S., KONOPKA G., LUO R., WANG Z.Q., SCHULTZ P.G., SMULSON M.E. Misregulation of gene expression in primary fibroblasts lacking poly(ADP-ribose) polymerase. Proc. Natl. Acad. Sci. 2000;97:11274–11279. doi: 10.1073/pnas.200285797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SLATTERY E., DIGNAM J.D., MATSUI T., ROEDER R.G. Purification and analysis of a factor which suppresses nick-induced transcription by RNA polymerase II and its identity with poly(ADP-ribose) polymerase. J. Biol. Chem. 1983;285:5955–5959. [PubMed] [Google Scholar]
- SMITH K.J., KAPOOR R., FELTS P.A. Demyelination: the role of reactive oxygen and nitrogen species. Brain Pathol. 1999;9:69–92. doi: 10.1111/j.1750-3639.1999.tb00212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH S. The world according to PARP. Trends Biochem. Sci. 2001;26:174–179. doi: 10.1016/s0968-0004(00)01780-1. [DOI] [PubMed] [Google Scholar]
- SMULSON M.E., SIMBULAN-ROSENTHAL C.M., BOULARES A.H., YAKOVLEV A.G., STOICA B.A., IYER R., LUO R., HADDAD B., WANG Z.Q., PANG T., JUNG M., DRITSCHILO A., ROSENTHAL D.S. Roles of Poly(ADP-ribosyl)ation and PARP in apoptosis, DNA repair, genomic stability and functions of p53 and E2F-1. Advan. Enzyme Regul. 2000;40:183–215. doi: 10.1016/s0065-2571(99)00024-2. [DOI] [PubMed] [Google Scholar]
- SZABO C. Role of poly(ADP-ribose) synthetase in inflammation. Eur. J. Pharmacol. 1998;350:1–19. doi: 10.1016/s0014-2999(98)00249-0. [DOI] [PubMed] [Google Scholar]
- SZABO C., DAWSON V.L. Role of poly(ADP-ribose) synthetase in inflammation and ischemia-reperfusion. Trends Pharmacol. Sci. 1998;19:287–298. doi: 10.1016/s0165-6147(98)01193-6. [DOI] [PubMed] [Google Scholar]
- ULLRICH O., DIESTEL A., EYUPOGLU I.Y., NITSCH R. Regulation of microglial expression of integrins by poly(ADP-ribose) polymerase-1. Nat. Cell Biol. 2001;3:1035–1042. doi: 10.1038/ncb1201-1035. [DOI] [PubMed] [Google Scholar]
- VISPÈ S., YUNG T.M.C., RITCHOT J., SERIZAWA H., SATOH M.S. A cellular defense pathway to regulate transcription through poly(ADP-ribosyl)ation in response to DNA damage. Proc. Natl. Acad. Sci. 2000;97:9886–9891. doi: 10.1073/pnas.170280397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VYAS D.R., MCCARTHY J.J., TSIKA G.L., TSIKA R.W. Multiprotein complex formation at the β myosin heavy chain distal muscle CAT element correlates with slow muscle expression but not mechanical overload responsiveness. J. Biol. Chem. 2001;276:1173–1184. doi: 10.1074/jbc.M007750200. [DOI] [PubMed] [Google Scholar]
- WANG X., OHNISHI K., TAKAHASHI A., OHNISHI T. Poly(ADP-ribosyl)ation is required for p53-dependent signal transduction induced by radiation. Oncogene. 1998;17:2819–2825. doi: 10.1038/sj.onc.1202216. [DOI] [PubMed] [Google Scholar]
- WELTIN D., PICARD K., AUPEIX K., VARIN M., OTH D., MARCHAL J., DUFOUR P., BICHOFF P. Immunosuppressive activities of 6(5)-phenanthridinone, a new poly(ADP-ribose) polymerase inhibitor. Int. J. Immunopharmacol. 1995;17:265–271. doi: 10.1016/0192-0561(95)00007-o. [DOI] [PubMed] [Google Scholar]
- XIAO B.-G., LINK H. Antigen specific autoreactive T cells with a focus on multiple sclerosis and experimental allergic encephalomyelitis. Cell. Mol. Life Sci. 1999;56:5–21. doi: 10.1007/s000180050002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG Z., HILDEBRANDT E.F., SIMBULAN-ROSENTHAL C.M., ANDERSON M.G. Sequence-Specific Binding of Poly(ADP-Ribose) Polymerase-1 to the Human T Cell Leukemia Virus Type-1 Tax Responsive Element. Virology. 2002;296:107–116. doi: 10.1006/viro.2002.1385. [DOI] [PubMed] [Google Scholar]
- ZIEGLER M., OEI S.L. A cellular survival switch: poly(ADP-ribosyl)ation stimulates DNA repair and silences transcription. BioEssay. 2001;23:543–548. doi: 10.1002/bies.1074. [DOI] [PubMed] [Google Scholar]
- ZINGARELLI B., SALZMAN A.L., SZABO C. Genetic disruption of poly (ADP-ribose) synthetase inhibits the expression of P-selectin and intercellular adhesion molecule-1 in myocardial ischemia/reperfusion injury. Circ. Res. 1998;83:85–94. doi: 10.1161/01.res.83.1.85. [DOI] [PubMed] [Google Scholar]