

Abstract
Neurofibrillary tangles (NFTs) are the most common intraneuronal inclusion in the brains of patients with neurodegenerative diseases and have been implicated in mediating neuronal death and cognitive deficits. Here, we found that mice expressing a repressible human tau variant developed progressive age-related NFTs, neuronal loss, and behavioral impairments. After the suppression of transgenic tau, memory function recovered, and neuron numbers stabilized, but to our surprise, NFTs continued to accumulate. Thus, NFTs are not sufficient to cause cognitive decline or neuronal death in this model of tauopathy.
Neurofibrillary tangles are composed of filaments of hyperphosphorylated tau (1–4), a microtubule-associated protein (5). They correlate well with cognitive deficits (6–8) and neuron loss (9), and they have been implicated in mediating neurodegeneration and dementia in Alzheimer’s disease (AD) (10–13) and other tauopathies (14). Transgenic mice expressing human tau have consistently demonstrated neurological deficits and neuron loss appearing with NFTs (15–19). The association between NFTs, neuron loss, and brain dysfunction in humans and mice has led to the belief that NFTs invariably cause brain dysfunction and neurodegeneration. However, this idea has never been rigorously tested. We hypothesized that examining the effects of suppressing the tau transgene would elucidate the dependence of functional changes on the structural and biochemical abnormalities related to abnormal tau expression for two reasons: It would provide an opportunity to evaluate the dependence of specific structural and functional abnormalities on the continuous expression of the tau transgene, and it would distinguish reversible from irreversible lesions.
We created transgenic mice expressing mutant tau that could be suppressed with doxycycline (fig. S1A). A responder transgene was generated consisting of a tetracycline-operon–responsive element (TRE) placed upstream of a cDNA encoding human four-repeat tau with the P301L mutation (4R0N tauP301L) that is linked to hereditary tauopathy (20). An activator transgene in a second mouse line consisted of the tet-off open reading frame (21) placed downstream of Ca2+–calmodulin kinase II promoter elements, which resulted in expression from the TRE that was restricted to forebrain structures (22). Mice harboring responder or activator transgenes were bred to generate bigenic progeny containing both transgenes. As expected, transgenic tau mRNA expression in bigenic mice was largely restricted to structures in the forebrain (Fig. 1A).
Fig. 1.
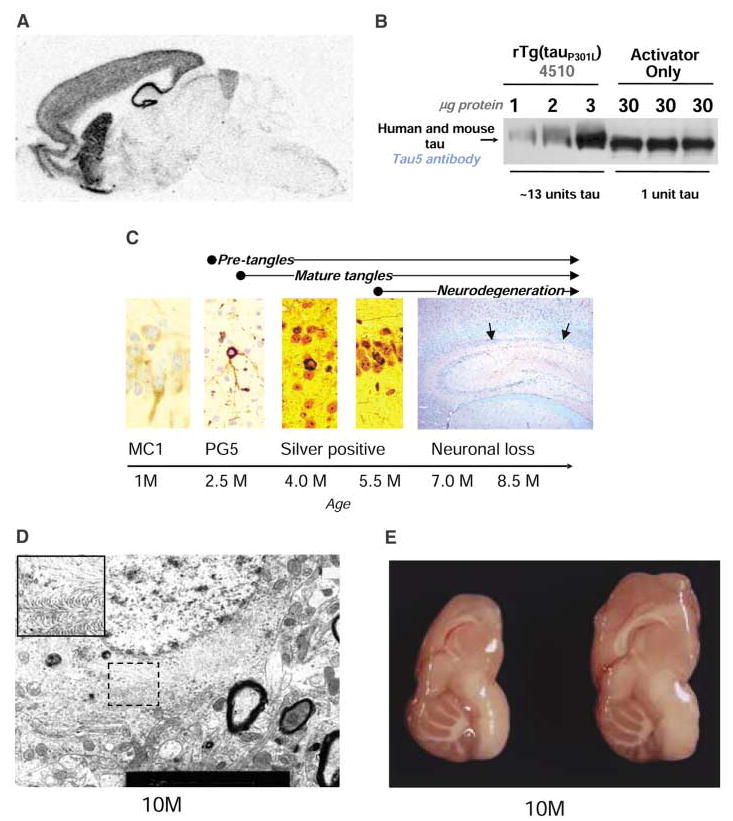
Molecular and neuropathological characterization of rTg(tauP301L)4510 mice. (A) The location of tauP301L mRNA was analyzed by in situ hybridization and found to be largely restricted to the hippocampus, cortex, olfactory bulb, and striatum. (B) The amount of tauP301L protein in total forebrain homogenates of a 2.5-month-old rTg(tauP301L)4510 mouse was ~13 times the endogenous tau in a littermate expressing the activator only, assessed by immunoblotting. (C) Tau pathology was evaluated by using a panel of antibodies, including tau conformation–specific MC1 and tau phosphoserine 409 PG5, and histological methods, including modified Bielschowsky and Nissl with periodic acid Schiff stains. These analyses revealed conformational changes, followed by nonargyrophilic, hyperphosphorylated pretangles and culminating in abundant argyrophilic tangles and neuronal loss (CA1 hippocampal subfield is between the arrows). (D) Tangle pathology in a 10-month-old mouse, examined by electron microscopy, consists of straight filaments, which sometimes formed a herringbone pattern (inset) identical to that previously reported in the JNPL3 mouse (10). Scale bar, 2 μm. (E) Gross forebrain atrophy, with preservation of hindbrain structures, in a 10-month-old tauP301L-positive transgenic mouse (left) compared with a nontransgenic littermate (right).
To ascertain the relation between tauP301L dosage and the development of neurofibrillary pathology, we examined two lines of mice expressing ~7 and ~13 units of tauP301L (one unit is equivalent to the level of endogenous murine tau) (Fig. 1B). The rate at which mice developed neurofibrillary pathology was directly related to the amount of tauP301L expression. Mice with ~7 units of tauP301L showed accumulation of hyperphosphorylated tau in cortical neurons (pretangles) by 14.5 months of age, but did not develop argyrophilic tangle-like inclusions until 20 months of age. Mice with ~13 units of tauP301L, rTg(tauP301L)4510 (r for regulatable), developed pretangles, which we detected using immunohistochemistry with multiple antibodies to human phosphotau epitopes, as early as 2.5 months of age. Pathology progressed rapidly in these mice (Fig. 1C). Argyrophilic tanglelike inclusions appeared in the cortex by 4 months and in the hippocampal formation by 5.5 months. The neuronal inclusions in this line were composed of a mass of straight tau filaments and are henceforward referred to as NFTs (Fig. 1D). A significant loss in brain weight was evident by 5.5 months (Fig. 2A), as well as significant decreases (~60%) in total numbers of CA1 hippocampal neurons (Fig. 2B). The loss in brain weight and neurons progressed in 7- and 8.5-month-old mice, with ~23% of CA1 pyramidal cells remaining at 8.5 months. Gross atrophy of the forebrain was evident in a 10-month-old mouse (Fig. 1E). Thus, the expression of tauP301L was sufficient to produce an age-related loss of neurons and generalized forebrain atrophy, with concomitant abnormal accumulation of hyperphosphorylated tau lesions.
Fig. 2.
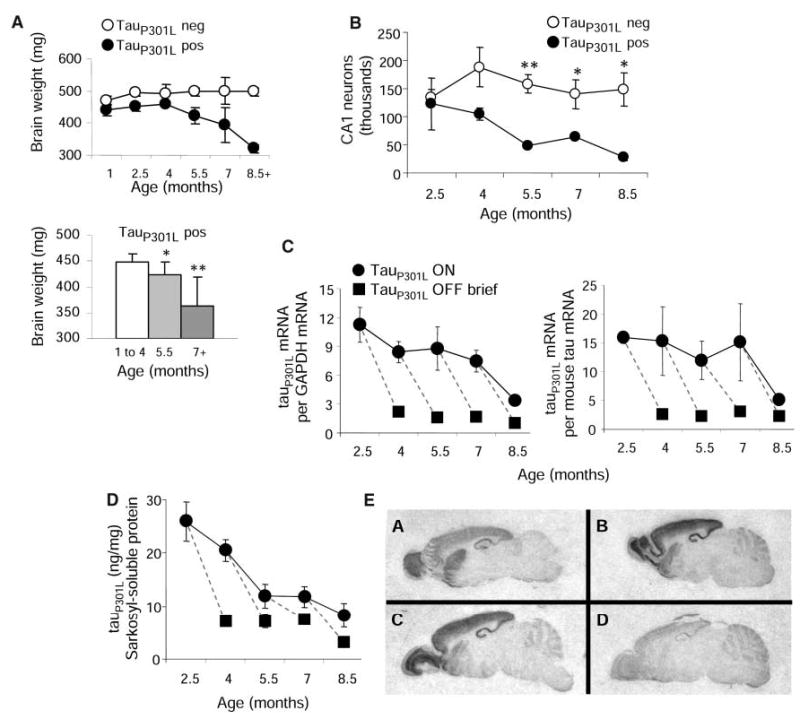
Neurodegeneration and suppression of transgenic tau. (A) Brain weights of tauP301L-positive mice <5 months were significantly lower than those of tauP301L-negative littermates (P < 0.001) (upper). Brain weights were significantly lower in 5.5-month-old and ≥7-month-old tauP301L-positive mice, relative to younger mice (lower). *P < 0.05; **P < 0.001. (B) CA1 neuron estimates in one hippocampal subfield (n = 3 per group) were significantly lower in tauP301L-positive mice 5.5 months of age and older. [ANOVA for genotype, F(1,16) = 23.97, P < 0.001]. *P < 0.05, **P < 0.01. (C) Transgenic tauP301L mRNA levels, whether normalized to GAPDH or to murine tau mRNA levels, declined with age and were dramatically suppressed after brief exposure (6 to 8 weeks) to doxycycline. (D) Sarkosyl-soluble tauP301L levels, measured in extracts from forebrain homogenates by using immunoblots, declined with age and were sbstantially reduced in animals receiving doxycycline. (E) In situ hybridization: A (top left) 7-unit tauP301L-expressing mouse; B (top right) 2-month-old 13-unit tauP301L-expressing mouse; C (lower left) 4-month-old 13-unit tauP301L-expressing mouse; D (lower right) 4-month-old 13-unit tauP301L-expressing mouse treated with doxycycline for 6 weeks. In this and subsequent figures, data are presented as means and SEM, and dashed lines compare groups of animals at the beginning and the end of doxycycline administration.
To test the effects of tauP301L expression in rTg(tauP301L)4510 mice on spatial reference memory, we examined performance using a version of the Morris water maze tailored to mice (23). There were no significant abnormalities during the probe trials in the water maze in 1.3-month-old rTg(tauP301L)4510 mice, the youngest mice we were able to test, indicating no major deficits in the retention of spatial memory at this age (fig. S2C). However, the retention of spatial memory became dramatically impaired as the mice aged (Fig. 3D and fig. S6B). Indeed, from 2.5 to 4 months of age and older, target quadrant occupancies fell in the probe trials from ~45% to random swimming (25%). Motor function assessed during the cued phase of the water maze (fig. S3D) and by a panel of motor function tests (fig. S4) revealed no significant deficits in mice up to 6 months of age. Thus, the retention of spatial reference memory declined dramatically in rTg(tauP301L)4510 mice in an age-dependent manner.
Fig. 3.
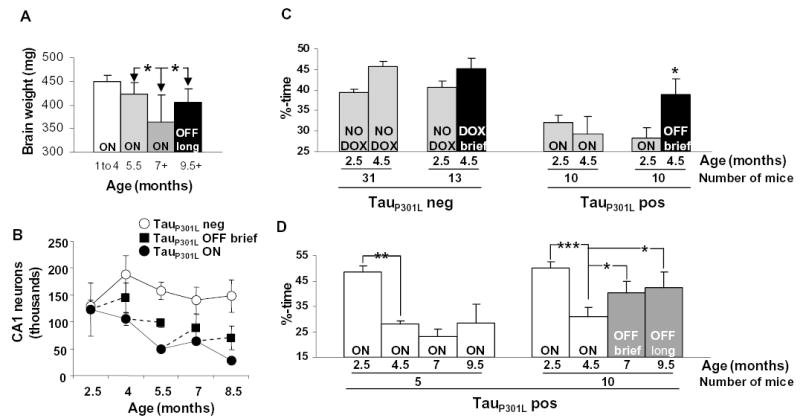
Neuron death ceases and memory recovers when transgenic tau expression is suppressed. (A) Brain weights in 9.5- to 10-month-old tauP301L-positive mice exposed to doxycycline for 4.5 months were significantly higher than brain weights in 7-month-old tauP301L-positive mice but were not higher than those of tauP301L-positive mice at 5.5 months, the age at which doxycycline was initiated. *P < 0.01. (B) CA1 neuron numbers in one hemisphere were estimated and found to stabilize after doxycycline treatment. (C) Twenty mice with target quadrant occupancies <39.5%, making up the half of a 2.5-month-old cohort having lower performance ratings, were tested in the water maze a second time at 4.5 months in the presence (n = 10) or absence of doxycycline (n = 10), which was initiated at 2.5 months. In tauP301L-positive mice, memory recovered significantly when transgenic tau was suppressed. In tauP301L-negative mice with target quadrant occupancies in the half of a cohort at 2.5 months of age having lower performance ratings in the presence (n = 13) or absence (n = 31) of doxycycline, a trend toward improved performance at 4.5 months was due to regression toward the mean (~48% in tauP301L-negative mice), as the trend was not apparent when the complete cohort was examined. *P < 0.05. (D) Memory was tested longitudinally at four ages in 15 mice in the top performing half of their cohort at 2.5 months of age (target quadrant occupancies >39.5%). Doxycycline was administered at 5.5 months to 10 mice, and control feed was administered to 5 mice. Significant improvements were found in tauP301L-positive mice treated with doxycycline, but not in tauP301L-positive mice maintained on the control diet. *P < 0.05, **P < 0.01, ***P < 0.0001.
However, we observed significantly longer path lengths in the hidden platform phase of the Morris water maze in 1.3-month-old rTg(tauP301L)4510 mice (fig. S2B), revealing a deficit in spatial navigation. This behavioral deficit was accompanied by a subtle, but significant, reduction in brain weights (~9%) in mice <5 months of age, compared with age-matched littermates lacking tauP301L (Fig. 2A). Thus, overexpression of tauP301L during brain development may cause subtle anatomical and cognitive, but not motor, abnormalities in rTg(tauP301L)4510 mice. It has been important in the study of behavioral deficits in transgenic mice overexpressing amyloid precursor proteins (APPs) to distinguish between age-independent and age-dependent abnormalities (23, 24): the former appear to result from the overexpression of APPs during brain development; the latter are related to biochemical or structural changes that occur as the mice age. Similarly, age-independent and age-dependent behavioral and pathological changes are found in rTg(tauP301L)4510 mice. We observed developmental, age-independent behavioral (spatial navigation deficit during the acquisition phase) and pathological (smaller brain size) abnormalities in rTg(tauP301L)4510 mice that are subtle when compared with the age-dependent functional (impaired retention of spatial reference memory) and structural (neurofibrillary pathology, neuron death, and gross atrophy) changes.
To investigate the potential differences in the plasticity and recovery of brain structure and function during the various stages of neurofibrillary pathology, we suppressed transgenic tau by feeding doxycycline to rTg(tauP301L)4510 mice. At ages ranging from 2.5 to 10 months (fig. S1B), doxycycline was administered for either a brief (6- to 8-week) or a long (4- to 4.5-month) period. We then performed extensive behavioral, biochemical, and histological analyses on the treated mice in comparison with animals that had not received doxycycline. Brief doxycycline treatment of rTg(tauP301L)4510 mice (for 6 to 8 weeks) at different ages reduced the total levels of mRNA to ~15% of the maximum tauP301L level, which decreased the amount of tauP301L mRNA relative to endogenous murine tau mRNA from ~15 units to ~2.5 units (Fig. 2C) and the amount of soluble tauP301L to ~27% of the maximum level (Fig. 2D). The levels of tauP301L mRNA and sarkosyl-soluble protein decreased with age, even without doxycycline (Fig. 2, C and D), which probably reflects a progressive, preferential loss of tauP301L-expressing neurons. The levels of tauP301L mRNA in the brain were uniformly suppressed by doxycycline (Fig. 2E).
After brief or long doxycycline treatment of the mice was complete, we investigated whether the progression of tau pathology was dependent on transgenic tau expression. The progression of tau pathology was dependent on transgenic tau expression at 2.5 months of age; when tauP301L was suppressed at 2.5 months of age, the pathological abnormalities ceased to progress in comparison with 4-month-old mice untreated during the same period (Fig. 4A). Although tauP301L suppression caused cessation of the progression of tau pathology, pretangle lesions (accumulations of nonargyrophilic hyperphosphorylated tau in the neuronal cell body) in 4-month-old mice treated with doxycycline begun at 2.5 months (4M2.5OFF) appeared to be stable, as their numbers were not reduced by transgene suppression even at this stage. The progression of the tau pathology became independent of transgenic tau expression at 4 months of age; when tauP301L was suppressed at ≥4 months NFT levels continued to increase until neuronal loss became extreme in old (8.5-month-old) mice. This was the case whether the amount of neurofibrillary pathology was assessed semiquantitatively by histopathology (Fig. 4A and fig. S5A), quantitatively using statistically unbiased stereological estimates of PHF-1–positive cells (Fig. 4B), biochemically by measuring a 64-kD sarkosyl-insoluble hyperphosphorylated tau species by immunoblot (Fig. 4C and fig. S5B), or biophysically by examining the number and length of sarkosyl-insoluble fibrils with electron microscopy (Fig. 4D). Indeed, once lesions were present in ≥4-month-old mice, the amounts of NFTs were similar whether transgenic tau was expressed continuously or suppressed with doxycycline. The transition from transgenic tau–dependent progression of tau pathology at 2.5 months to transgenic tau–independent progression of neurofibrillary pathology at 4 months was linked to the appearance of an insoluble 64-kD tau species (Fig. 4C and fig. S5B). The continued accumulation of the insoluble 64-kD tau species, which was clearly associated with increasing NFT numbers, even when transgenic tau was substantially reduced, suggests that it represents a stable, favorable state that probably acts as a sink for remaining tauP301L species. The 64-kD tau species is a hyperphosphorylated form of 4R0N tau, because the only tau species present after dephosphorylation comigrated with 4R0N recombinant tau (25). Importantly, the insoluble 64-kD tau species observed in this mouse model comigrates with a major hyperphosphorylated tau species observed in human NFTs in AD and other tauopathies.
Fig. 4.
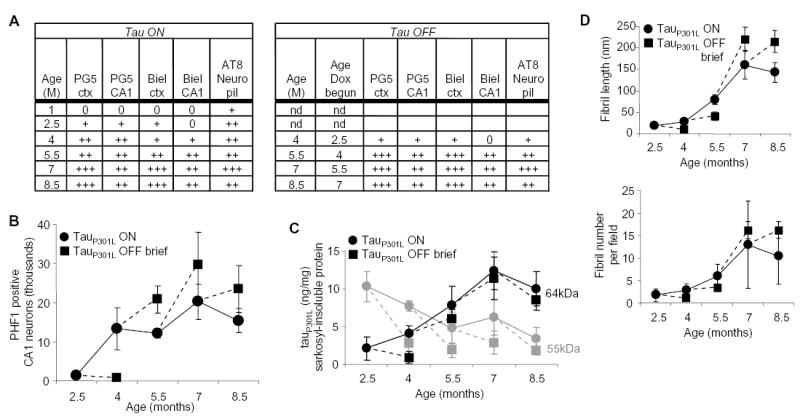
Accumulation of NFTs after transgenic tau suppression. (A) Semiquantitative grading of neurofibrillary pathology in tauP301L-positive mice. Tangles in three to five animals, at ages in months (M), evaluated with PG5, AT8, or Bielschowsky stain, in neocortical (ctx) and CA1 hippocampal (CA1) regions were graded using an adaptation of the CERAD (Consortium to Establish a Registry for AD) neuropsychological rating scales, by using area of most severe involvement, as follows: 0 (none), + (sparse, <5 NFTs/hpf), ++ (moderate, 5 to 25 NFTs/hpf), or +++ (severe, >25 NFTs/hpf). The neuropil scored 0 when AT8 staining was absent, + when only a mild granular background staining was noted, ++ when neuropil staining was moderate, and +++ when neuropil staining was severe and diffuse. (B) CA1 neurons staining with PHF1 antibody recognizing tau phosphoserines 396 and 404 did not diminish after transgene suppression for 6 weeks. (C) Concentrations of sarkosyl-insoluble tauP301L proteins were measured in extracts of forebrain homogenates by using immunoblots. With age, the predominant tau species shifted from a doublet of ~55-kD proteins to a 64-kD protein. Suppression of tauP301L reduced levels of the 55-kD species (gray symbols), but failed to halt the accumulation of the 64-kD species in mice ≥4 months of age (black symbols). (D) The number and length of fibrils in sarkosyl-insoluble fractions from extracts of forebrain homogenates increased with age. Suppressing transgenic tau in ≥4-month-old mice failed to prevent the increase in the amount or size of the fibrils.
We next determined the effects of transgenic tau suppression on neuronal loss and brain atrophy. To do this, we compared brain weights and numbers of CA1 neurons by unbiased stereological estimates in mice fed doxycycline or control feed. Doxycycline-treated mice were divided into groups according to brief treatment (6 to 8 weeks) and lengthy treatment (4 to 4.5 months) groups. Comparisons of brain weight between these two groups and the control group, adjusted for the possible confounding variables of gender and age-at-death, were obtained [ANCOVA, F(2,64) = 4.81, P = 0.011]. Although brief suppression of transgenic tau had no significant effect on loss of brain weight, longer suppression (from 5.5 to ~10 months) significantly protected against the loss of brain weight [t(45) = 3.47, P = 0.003] (Fig. 3A). Indeed, these 10M5.5OFF mice had brain weights similar to those of 5.5-month-old rTg(tauP301L)4510 mice. Moreover, CA1 neuron estimates were stabilized after even brief (6- to 8-week) tauP301L suppression (Fig. 3B). In all age groups, there were significantly more neurons in mice receiving doxycycline than in untreated age-matched control mice [ANCOVA, F(1,21) = 5.38, P = 0.03]. Thus, there was a cessation in the loss of neuron number and brain weight when tauP301L was suppressed, despite steady increases in NFTs.
To investigate the relation between NFT formation and memory function, we tested mice with transgenic tau–dependent (2.5-month) and transgenic tau–independent (≥4 months) progression of neurofibrillary pathology. First, we observed the cognitive effects of transgene suppression at 2.5 months, a model that halted tau pathology progression. In order to determine effects of transgene suppression specifically on impaired mice, doxycycline was fed to 2.5-month-old mice with established cognitive dysfunction. Thus, we administered doxycycline or control feed to 2.5-month-old mice that displayed deficits in the retention of spatial memory, with target quadrant occupancy <39.5%, made up of the lower half of a 2.5-month-old cohort. The performance of 4.5M2.5OFF mice improved [paired t(9) = 2.77, P = 0.02], in contrast to mice receiving control diet (Fig. 3C and fig. S6A). Analysis of target quadrant occupancies of tauP301L-expressing mice revealed a significant age-attesting by treatment interaction [RMANOVA, F(1,18) = 4.88, P = 0.04], which was not observed among control littermates lacking tauP301L that were in the bottom performing half of their 2.5-month-old cohort [RMANOVA, F(1,42) = 0.25, P = 0.62] (Fig. 3C). Similar results were obtained for path lengths in the hidden portion of the water maze (fig. S3A), whereas path lengths in the cued portion of the water maze were unaffected (fig. S3B), as predicted. Thus the ability to acquire and retain new spatial memories was restored when 2.5-month-old mice were treated with doxycycline, and the improvement in memory function was related to the suppression of tauP301L and not to a nonspecific effect of doxycycline on improving neurological function.
We also examined the effects on memory function of transgenic tau suppression at an age when the progression of NFT pathology was independent of tauP301L expression. We selected 15 high-performing mice with target quadrant occupancy >39.5% at 2.5 months, and examined them longitudinally at 4.5, 7, and 9.5 months of age. We observed a significant deterioration in the retention of spatial memory at 4.5 months [paired t(14) = 9.83, P < 0.0001] (Fig. 3D). We tested cognitive effects of transgene suppression at 5.5 months, a paradigm that had no effect on the progression of NFT pathology. Thus, doxycycline was administered to 10 mice starting at 5.5 months; the remaining five mice were maintained on control feed. Memory was tested in the water maze at 7 and 9.5 months. The ability to acquire and retain new spatial memories improved significantly in 7M5.5OFF mice [paired t(9) = 2.47, P = 0.04] and in 9.5M5.5OFF mice [paired t(9) = 2.91, P = 0.02] (Fig. 3D and fig. S6B). This was surprising because at 5.5 months, when transgenic tau was suppressed in these mice, there are already abundant NFTs and significant losses in both brain weight and hippocampal neurons in rTg(tauP301L)4510 mice (Fig. 1C; Fig. 2, A and B; Fig. 4, A and B). To confirm that the progression of tau pathology had become transgenic tau–independent in these mice, we showed that the amounts of the insoluble 64-kD tau species at the end of doxycycline treatment in 10-month-old mice were significantly higher (fig. S7A), that there was more extensive NFT pathology (fig. S7B), and that the numbers of PHF-1–positive CA1 neurons were greater (fig. S7C) than in 5.5-month-old mice. Any degree of neurodegeneration present at the start of doxycycline treatment in these mice may account for the incomplete recovery of the retention of spatial memory, but also makes the improvement in memory all the more remarkable. There was no significant change in path lengths in the hidden platform phase of testing in 7M5.5OFF or 9.5M5.5OFF mice, nor in 7MON or 9.5MON mice, compared with their own previous performance at 4.5 months (fig. S3C). No further deterioration in spatial navigation was observed in either group, despite the progressive accumulation of tangle pathology and the insoluble 64-kD tau species (Fig. 4 and figs. S5 and S7). Path lengths in the cued platform phase of testing remained intact (fig. S3D), and doxycycline had no effect on the acquisition or retention of new spatial memories in tauP301L-negative mice (fig. S3E), which indicated that the improved ability of tauP301L-positive mice to acquire and retain new memories was unrelated to nonspecific effects of doxycycline on cognitive or motor function.
That memory function improved in ≥4-month-old mice fed doxycycline despite ongoing accumulation of NFTs clearly implies dissociation between the processes that lead to memory loss and those that cause NFTs and that the NFTs remaining after tau suppression are not sufficient to disrupt cognitive function. The recovery of memory in this mouse model implies either that reversible neuronal dysfunction rather than irreversible structural degeneration is responsible for initial memory deficits or that neuronal remodeling of some form occurs after doxycycline treatment and allows recovery. Similarly, the cessation of neuron loss in ≥4-month-old mice fed doxycycline in the face of the persistent increase in NFTs suggests that NFTs do not invariably cause neuron death. Quantitative assessments show that neuronal loss far exceeds NFT number in human AD (9), which suggests that an NFT-independent method of neuronal death occurs both in the rTg(tauP301L)4510 line and in AD. However, whether NFTs are an incidental marker for the actual neurotoxic cascade in these mice and in human disease or represent a protective neuronal response, allowing sequestration of neurotoxic species into a less harmful stable form, as has been shown for the huntingtin protein (26), remains a crucial question. Finally, our results suggest that recovery of cognitive function may be possible in the early stages of the human tauopathies, including AD, although the clinical implications of our findings need to be interpreted with caution because of the limitations of modeling human disease in mice.
Supplementary Material
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/309/5733/476/DC1
Materials and Methods
Figs. S1 to S8
References and Notes
References
- 1.Grundke-Iqbal I, et al. Proc Natl Acad Sci USA. 1986;83:4913. doi: 10.1073/pnas.83.13.4913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kosik KS, Joachim CL, Selkoe DJ. Proc Natl Acad Sci USA. 1986;83:4044. doi: 10.1073/pnas.83.11.4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goedert M, Wischik CM, Crowther RA, Walker JE, Klug A. Proc Natl Acad Sci USA. 1988;85:4051. doi: 10.1073/pnas.85.11.4051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lee VM, Balin BL, Otvos L, Jr, Trojanowski JQ. Science. 1991;251:675. doi: 10.1126/science.1899488. [DOI] [PubMed] [Google Scholar]
- 5.Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW. Proc Natl Acad Sci USA. 1975;72:1858. doi: 10.1073/pnas.72.5.1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arriagada PV, Growdon JH, Hedley-Whyte ET, Hyman BT. Neurology. 1992;42:631. doi: 10.1212/wnl.42.3.631. [DOI] [PubMed] [Google Scholar]
- 7.Bancher C, Braak H, Fischer P, Jellinger KA. Neurosci Lett. 1993;162:179. doi: 10.1016/0304-3940(93)90590-h. [DOI] [PubMed] [Google Scholar]
- 8.Guillozet AL, Weintraub S, Mash DC, Mesulam MM. Arch Neurol. 2003;60:729. doi: 10.1001/archneur.60.5.729. [DOI] [PubMed] [Google Scholar]
- 9.Gomez-Isla T, et al. Ann Neurol. 1997;41:17. doi: 10.1002/ana.410410106. [DOI] [PubMed] [Google Scholar]
- 10.Lewis J, et al. Science. 2001;293:1487. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 11.Gotz J, Chen F, van Dorpe J, Nitsch RM. Science. 2001;293:1491. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 12.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Neuron. 2004;43:321. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 13.Bennett DA, Schneider JA, Wilson RS, Bienias JL, Arnold SE. Arch Neurol. 2004;61:378. doi: 10.1001/archneur.61.3.378. [DOI] [PubMed] [Google Scholar]
- 14.Spillantini MG, Goedert M. Trends Neurosci. 1998;21:428. doi: 10.1016/s0166-2236(98)01337-x. [DOI] [PubMed] [Google Scholar]
- 15.Ishihara T, et al. Neuron. 1999;24:751. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- 16.Lewis J, et al. Nat Genet. 2000;25:402. doi: 10.1038/78078. [DOI] [PubMed] [Google Scholar]
- 17.Gotz J, Chen F, Barmettler R, Nitsch RM. J Biol Chem. 2001;276:529. doi: 10.1074/jbc.M006531200. [DOI] [PubMed] [Google Scholar]
- 18.Tatebayashi Y, et al. Proc Natl Acad Sci USA. 2002;99:13896. doi: 10.1073/pnas.202205599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Andorfer C, et al. J Neurochem. 2003;86:582. doi: 10.1046/j.1471-4159.2003.01879.x. [DOI] [PubMed] [Google Scholar]
- 20.Hutton M, et al. Nature. 1998;393:702. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 21.Gossen M, Bujard H. Proc Natl Acad Sci USA. 1992;89:5547. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mayford M, et al. Science. 1996;274:1678. doi: 10.1126/science.274.5293.1678. [DOI] [PubMed] [Google Scholar]
- 23.Westerman MA, et al. J Neurosci. 2002;22:1858. doi: 10.1523/JNEUROSCI.22-05-01858.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen G, et al. Nature. 2000;408:975. doi: 10.1038/35050103. [DOI] [PubMed] [Google Scholar]
- 25.J. Lewis, M. Hutton, unpublished observations.
- 26.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Nature. 2004;431:805. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 27.We thank A. Michael for support; S. Iyadurai and G. Carlson for critical discussions; N. Nash, M. Sherman, A. Hanna, J. Knight, C. Zehr, S. Nelson, and D. Norton for technical help; and P. Sharpe for secretarial assistance. Supported by grants from the NIH (P01-AG15453, R01-026252 to K.H.A.; R01-AG26249, R01-AG08487 to B.H; T31-AG00277 to T.S.; R01-NS46355 to J.L.).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.