

Abstract
Protein–protein interactions play key roles in a range of biological processes, and are therefore important targets for the design of novel therapeutics. Unlike in the design of enzyme active site inhibitors, the disruption of protein–protein interactions is far more challenging, due to such factors as the large interfacial areas involved and the relatively flat and featureless topologies of these surfaces. Nevertheless, in spite of such challenges, there has been considerable progress in recent years. In this review, we discuss this progress in the context of mimicry of protein surfaces: targeting protein–protein interactions by rational design.
Keywords: protein–protein interaction, inhibitor, proteomimetics, surface, recognition
1. Introduction
Protein–protein interactions play crucial roles in a number of biological processes, such as viral self-assembly, cell proliferation, growth, differentiation, signal transduction and programmed cell death (Toogood 2002). As a result, their disruption can lead to novel therapeutic agents as well as tools to improve our understanding of biochemical pathways. Unlike traditional drug discovery in which small molecule substrates that bind enzyme active sites may be used as templates for the design of antagonists, the development of inhibitors of protein–protein interactions is a far more complicated process due to a number of particular challenges. First, the interfacial surface area necessary for specific recognition is typically large (approx. 750–1500 Å2), suggesting that large ligands may be required to compete effectively with the natural protein partner, as opposed to ‘drug-like’ small molecules that have enjoyed success in enzyme inhibition. Second, interaction surfaces are often shallow and relatively featureless, rather than well-defined binding pockets seen in enzyme active sites, rendering the design of selective inhibitors difficult. Third, the binding regions of protein–protein interactions are often non-contiguous, so that mimicry of these domains is not possible by simple synthetic peptides or peptidomimetics. In addition, the adaptivity of the protein surfaces involved in protein–protein interactions suggests that there may be binding conformations suitable for small molecules that are invisible in a single crystal structure. Finally, unlike with enzyme active sites where the activity of targeted ligands may simply be monitored by commercially available assays, novel and efficient screening assays must be developed.
Nevertheless, despite such challenges, there has been a number of advances in this area of research (Arkin & Wells 2004; Pagliaro et al. 2004; Zhao & Chmielewski 2005; Yin & Hamilton 2005; Fletcher & Hamilton 2005). The purpose of this manuscript is to review the recent progress in the recognition of protein surfaces and the disruption of protein–protein interactions with both low- and high-molecular-weight ligands developed through rational design. In particular, we will focus on a number of different strategies ranging from (metal-based) small molecules (whose success is likely due in part to the existence of protein ‘hot spots’; Delano 2002) to miniature folded proteins to the larger structures of porphyrins, calixarenes and cyclodextrins, and finally to the elegant field of proteomimetics: molecules that mimic the structures and functions of extended regions of proteins.
2. Small molecules and peptides
2.1 Hydrocarbon-stapled helices: disruption of the BID–Bcl-2 complex
A recent study by Verdine and co-workers describes very successful efforts towards targeting the interaction between BID and Bcl-2 by generating ‘hydrocarbon-stapled’ helices, based on the amphipathic α-helical BH3 domain of BID (Walensky et al. 2004). BID and Bcl-2 are pro- and anti-apoptotic proteins, respectively, and their interaction with each other regulates the process of apoptosis (cell death). Hence, modulation of the BID–Bcl-2 complex is an attractive target for developing anti-cancer therapeutics. Despite being attractive candidates for stabilizing or disrupting protein–protein interactions, peptides exhibit three inherent characteristics that limit their efficacies as such drugs in vivo. First, their secondary structure is lost; second, they are susceptible to proteolysis; and, third, cell membranes are poorly permeable, if at all, to peptides. Therefore, in an effort to prepare stabilized α-helices that are also more resilient to proteolytic enzymes and able to penetrate cells, Verdine et al. have generated a covalent cross-linking strategy. By incorporating α,α-disubstituted, non-natural amino acids with terminal olefinic side chains at positions i and (i+4) or i and (i+7) of the BH3 peptide, two reactive alkenes were positioned on the same face of the α-helix of a series of peptides. Subsequently, ruthenium-catalysed ring-closing metathesis furnished macrocyclic derivatives (scheme 1), termed ‘stabilized α-helix of Bcl-2 domains’ (SAHBs), with substantially increased α-helical content from 16% to as much as 87%. Two-dimensional 15N–1H heteronuclear single-quantum correlation (HSQC) NMR spectra of 15N-labelled Bcl-xL (Bcl-xL is a closely related Bcl-2 anti-apoptotic protein) recorded before and after the addition of peptide SAHBA were compared with the spectra generated before and after the addition of the unmodified BID BH3 peptide to 15N-labelled Bcl-xL, which demonstrated that the synthetic SAHBA peptide bound the targeted groove on the surface of Bcl-xL. These hydrocarbon-stapled SAHB peptides were then evaluated for their abilities to mimic the BH3 domain of BID through a fluorescence polarization binding assay. The authors reported a sixfold increase in binding affinity of peptide SAHBA for Bcl-2 (Kd=38.8 nM) compared to that of the unmodified BID BH3 peptide (Kd=269 nM), with concomitant protease resistance and cell permeability. Upon administration of constrained helix SAHBA to Jurkat T cell leukaemia cells in vitro, apoptosis was selectively activated, and the growth of human leukaemia xenografts in vivo was arrested.
Scheme 1.

Ring-closing metathesis of olefin-modified i and (i+4) residues to generate hydrocarbon-stapled helices. Reproduced with permission from Walensky et al. (2004).
2.2 Cyclic peptides: antagonism of the oestrogen receptor
In an alternative approach to Verdine's hydrocarbon-stapled α-helices, Burris & Wittliff et al. have engineered peptides bearing side chains that can be cross-linked in an effort to stabilize α-helical character. Using this technique, the authors have reported the selective inhibition of the oestrogen receptor α (ERα)-coactivator protein interaction (Leduc et al. 2003). With their aim being to mimic the α-helical Leu-X-X-Leu-Leu nuclear receptor box of the coactivator protein, the authors designed a peptide containing a cyclic amide (lactam) by condensing Lys and Glu side chains at positions i and (i+4), and cyclic peptides containing disulphide bonds, through the oxidation of Cys residues at positions i and (i+3). Interestingly, circular dichroism (CD) experiments demonstrated that only their disulphide peptides exhibited α-helical character, as monitored at 208 and 222 nm, and only in the presence of 7% of trifluoroethanol, a structure-inducing solvent. Furthermore, cyclic disulphide peptide PERM-1 displayed the highest affinity for the ERs, binding ERα with a Ki of 25 nM and ERβ, 390 nM, demonstrating a 15-fold selectivity for ERα over ERβ. X-ray studies confirmed that PERM-1 binds the ER ligand-binding domain in the predicted fashion with the binding process inducing α-helical structure in the peptide. Conversely, their lactam peptide, which exhibited no α-helical character, even in the presence of trifluoroethanol, bound the ERs an order of magnitude lower (ERα: Ki=0.22 μM; ERβ: Ki=4.8 μM). The observation that α-helical character of the otherwise unstructured PERM-1 peptide was induced upon binding ER has pertinent implications for ER-induced conformational changes in the coactivator itself. Universally, this offers more evidence for the adaptivity of protein surfaces involved in protein–protein interactions, and the inherent difficulty in designing molecules to target these dynamic interfaces.
2.3 Anti-HIV-1 agents
2.3.1 Antagonist of the gp120–CD4 interaction
Human immunodeficiency virus type 1 (HIV-1)/acquired immunodeficiency syndrome (AIDS) is a major cause of mortality in the world, and due to high mutation rates that leads to resistance to current therapeutics (Little et al. 2002), the development of new anti-HIV-1 agents is an area of active research. While current therapies tend to target the active sites of HIV reverse transcriptase and HIV protease, alternate targets such as the viral-cell surface fusion event remain largely unexplored. The entry of the HIV virus to cells proceeds via a three-step mechanism. First, the viral surface protein gp120 interacts with the cellular receptor CD4 (Dalgleish et al. 1984). This causes a conformational change in gp120 (Wyatt et al. 1998), which then interacts with a second receptor, known as a chemokine receptor (LaBranche et al. 2001), allowing the HIV protein gp41 to penetrate the cell membrane, causing membrane fusion and HIV cellular access (Yachou & Sekaly 1999). In an effort to inhibit the fusion process, there has been considerable research directed towards developing antagonists of the chemokine receptors CCR5 (Tremblay et al. 2002) and CXCR4 (Tamamura et al. 2000; Ruff et al. 2001). In particular, Enfuvirtide was approved by the US FDA in 2003 (Fletcher 2003; LaBonte et al. 2003). However, Gilbert and co-workers chose an alternative approach to inhibiting the fusion process by designing small molecules that were intended to bind to gp120 and prevent interaction with CD4 (Boussard et al. 2004). From the solved crystal structure of gp120 bound to CD4 (Kwong et al. 1998), Phe43 and Arg59 of CD4 appear to be make crucial interactions with gp120, therefore Gilbert et al. designed small molecules to mimic the phenyl ring and the guanidinium group, respectively. Their best hit, 1, had a fusion inhibition IC50 of 131 μM, although an enzyme-linked immunosorbent assay (ELISA) using a recombinant protein showed that the HIV-specific CD4–gp120 interaction was only weakly inhibited at a concentration of 5 mM, suggesting their anti-virals operate by a mode other than inhibition of the CD4–gp120 interaction.
2.3.2 HIV-1 protease dimerization inhibitors
The high mutation rate of HIV leads to rapid active site-directed drug resistance. Hence, in an effort to design inhibitors that are impervious to viral mutations, Chmielewski et al. have targeted the four-stranded, β-sheet dimerization interface of HIV-1 protease because this region has been shown to mutate less readily (Miller 2001). The feasibility of inhibiting HIV-1 protease dimerization with peptides derived from the dimerization surface was first demonstrated by Poorman et al. in the early 1990s (Zhang et al. 1991). Elaborating on this innovative research, Chmielewski and colleagues designed inhibitors intended to mimic the four interdigitating strands that form the dimerization interface by cross-linking N-terminus and C-terminus-derived peptides (Zutshi et al. 1997). Considerable research into the reduction of the high molecular complexity of their hits, most recently through a focused library approach, has culminated in the generation of several, potent, small-molecule dimerization inhibitors of HIV-1 protease (e.g. compound 2: Ki=71 nM; Shultz et al. 2004). More importantly, however, their protein surface inhibitors are equipotent with wild-type HIV-1 protease and a mutant form of the enzyme that is drug-resistant to active-site directed inhibitors, suggesting a promising complement, or perhaps alternative, to active-site directed inhibition in anti-HIV therapy (figure 1).
Figure 1.

Anti-HIV-1 agents: (1) gp120–CD4 inhibitor (IC50=131 μM); (2) HIV-1 protease dimerization inhibitor (Ki=71 nM).
2.4 PDZ domains
2.4.1 MAGI3–PTEN inhibitors
PDZ domains are key regions of proteins that are involved in a variety of protein–protein interactions, most notably those located within signalling complexes at the mammalian plasma membrane (Sheng & Sala 2001). Implicated in a number of associations with medicinally important proteins, the design of inhibitors of PDZ domains may lead to the treatment of diseases, such as metastatic breast cancer (Koo et al. 2002), prostate cancer (Chaib et al. 2001) and Parkinson's disease (Dev et al. 2003). MAGI3, which contains six PDZ domains, binds PTEN, a lipid/protein phosphatase that suppresses tumours (Wu et al. 2000a,b) using its second PDZ domain (MAGI3–PDZ2). Since there exists high structural homology between PDZ domains, Guy and co-workers (Fujii et al. 2003) designed a potent, irreversible inhibitor (3) of the MAGI3–PTEN interaction based on the crystal structure of postsynaptic density-95 kDa protein (PSD-95)-PDZ3 bound to the ligand CRIPT (Doyle et al. 1996). In the first report of a specifically targeted irreversible inhibitor of a protein–protein interaction, the design of 3 includes a weakly ionizable hydroxyl group, intended to capture the imidazole of the conserved histidine in the PDZ domain (His372). Fluorescence polarization showed that upon pre-treatment of MAGI3–PDZ2 with 3, ligand-binding by the fluorescently labelled PTEN carboxy terminal peptide was blocked in a dose-dependent manner. Furthermore, while the efficacy of the peptide ligand binding decreased with increasing concentrations of 3, the ligand affinity remained the same; this result is consistent with 3 acting as an irreversible inhibitor. Coupled with evidence of covalent modification, their results confirm 3 is indeed an irreversible inhibitor of MAGI3–PDZ2, most likely attaching itself to the domain through functionalization of His372.
2.4.2 PSD-95–NMDA receptor inhibitors
In order to disrupt the interaction of the third PDZ domain of the PSD-95 with the NMDA receptor, Spaller et al. have synthesized a series of lactam-constrained peptides, based on the C-terminal residues of CRIPT (Doyle et al. 1996; Niethammer et al. 1998), through bridging side chains, such as Lys and Glu (Li et al. 2004). Rather than directly linking the Lys ϵ-amino group with the γ-carboxylic acid of Glu, which the authors reasoned could lead to an unfavourable distortion of the peptide backbone away from an optimal binding geometry, Spaller and colleagues have covalently tethered the solvent-accessible P-1 and P-3 side chains with spacers of different lengths. Simultaneously, they maintained the identities of the crucial protein-binding P0 and P-2 side chains, thereby affording macrocyclic ligands 4 (figure 2). The lactam ring sizes of these peptides may be readily expanded or contracted through variation of the bridging component, thereby enabling some conformational variety. Isothermal calorimetry was used to determine the thermodynamic parameters of the inhibitor/PDZ interactions; many Kd's were less than 50 μM.
Figure 2.

Antagonists of PDZ domains: (3) MAGI3–PTEN inhibitor; (4) PSD-95–NMDA inhibitor.
2.5 Inhibition of E2-mediated cellular anchoring of hepatitis C virus
More than 170 million people are currently infected with hepatitis C virus (HCV), with the majority unable to clear the virus. These people tend to develop cirrhosis or liver cancer (Memon et al. 2002). Currently, there is no vaccine for HCV and the number of HCV-infected people is likely to triple by 2010. Todd et al. have recently created non-peptidic small molecules, exhibiting a novel bis-imidazole scaffold that are capable of reversibly disrupting the binding of the HCV envelope glycoprotein E2 to its receptor partner CD81, by mimicking the spatial and hydrophobic features of the solvent-exposed face of helix D of CD81 (VanCompernolle et al. 2003). Their most potent inhibitor of the HCV-E2–CD81 interaction was 5 with an EC50 of 38 μM (figure 3).
Figure 3.
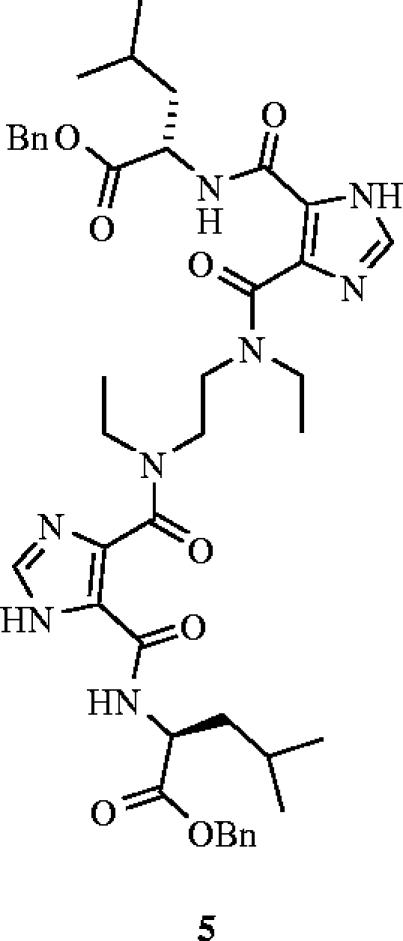
A novel bis-imidazole inhibitor (5) that prevents hepatitis C virus (HCV) cell entry through the disruption of the HCV-E2–CD81 interaction (EC50=38 μM).
2.6 Metal-based systems: the recognition of histidine
2.6.1 Carbonic anhydrase recognition by Cu2+ complexes
Each protein exhibits a unique distribution of charged, polar, aliphatic and aromatic amino acids residues on it surface; this pattern governs the binding and selectivity for surface receptor molecules. For example, various transition metal ions (e.g. Cu2+ and Ni2+) coordinate strongly to the imidazole side chain of histidine. In an effort to bind carbonic anhydrase (CA) selectively, Mallik and co-workers have described the design, synthesis and binding properties of transition metal complexes to target the histidine residues on the surface of CA (Fazal et al. 2001). Importantly, metal–ligand interactions can be stronger in water than hydrogen-bonds, electrostatic or other non-covalent forces. Indeed, simultaneous and complementary metal–ligand interactions can lead to tighter and more selective binding, highlighting the advantages that this particular strategy offers for protein surface recognition. Furthermore, the binding process may be monitored simply by the changes in the spectroscopic properties of the metal. Mallik et al. prepared receptors comprising three Cu2+-iminodiacetate (Cu2+-IDA) arms separated by different spacers (figure 4). Compound 6 was designed to match the surface distribution of the six histidine residues of CA, and was found to bind with a Ka of 2.99×105 M−1. The selectivity of 6 was demonstrated by weaker binding both to chicken egg albumin (CEA; Ka=1.0×103 M−1), which also presents six histidine residues on its surface but in a different pattern to CA, and to myoglobin (Mb; Ka=2.0×104 M−1), which has seven surface histidines. Receptor 7, with a shorter spacing in between the Cu2+-IDA groups, exhibited less potent binding to CA, with a Ka of 7.5×104 M−1, which was attributed to a less favourable enthalpic term. As anticipated, the more flexible phenolic ether 8 bound CA even more weakly (Ka=3.3×104 M−1), due in part to a considerable loss of entropy upon binding to CA. In addition, the authors reported that there was much less selectivity of 7 and 8 with CEA and Mb. Importantly, with the copper cations removed, none of the receptors showed binding to CA. These data demonstrate that the geometric fit and pre-organization of the Cu2+-IDA arms are important requisites for strong binding and good selectivity of transition metal complexes.
Figure 4.
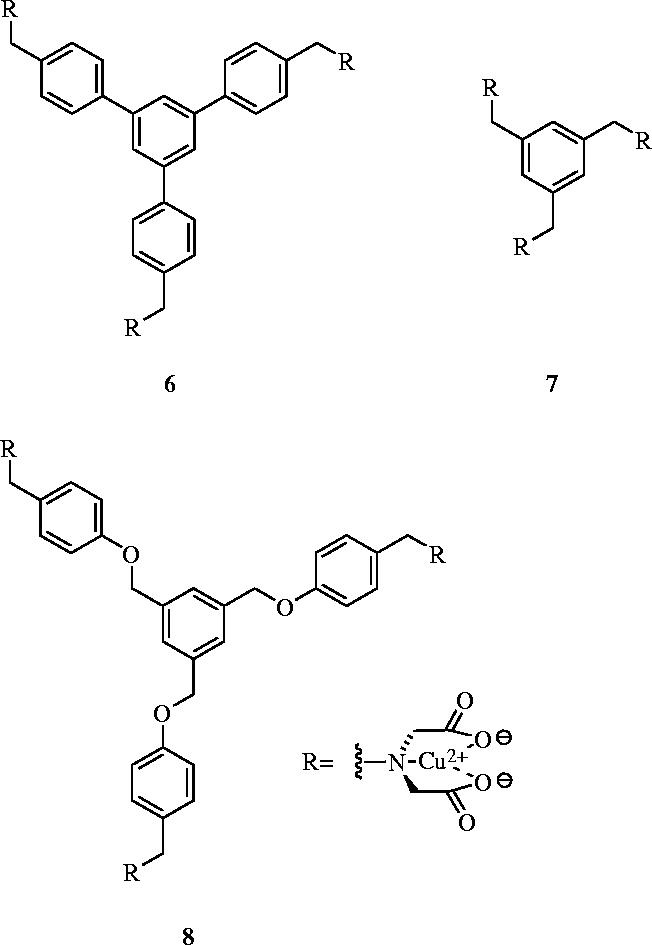
Cu2+-iminodiacetate derivatives that target the surface of carbonic anhydrase: 6, Ka=2.99×105 M−1; 7, Ka=7.5×104 M−1; 8, Ka=3.3×104 M−1.
2.6.2 α-Helix stabilization
More recently, Hamachi and co-workers have designed small molecules incorporating palladium or zinc that, in a sequence-selective fashion, recognize histidine or phosphorylated amino acid residues on peptide surfaces (Ojida et al. 2004a,b). The authors used CD spectroscopy to ascertain the effects on α-helicity of a series of 17-mer peptides upon incubation with their synthetic receptors. A mononuclear Pd(II)-ethylenediamine (Pd(en)) dinitrate complex cooperatively stabilized the α-helical conformation of peptides bearing two His residues i and i+(3 or 4) residues apart. On the other hand, a decrease in α-helicity was observed with peptides presenting a single His residue or two His residues further apart at positions i and i+(7 or 11). These results suggest that the cross-linking of two His residues stabilized the α-helical conformation, whereas binding of the receptor to a single His led to a destabilization of the α-helix. Complementary to their Pd(en) complex, the authors also prepared binuclear, dipicolylamine-based Zn(II) (Zn(dpa)) complexes that exhibited the greatest α-helical stabilization effect on peptides bearing His residues at positions i and (i+7) or i and (i+11). These data illustrate that, not only are Pd(en) and Zn(dpa) strong enough to bind and control peptide conformations in an aqueous environment, but that mono- and dinuclear complexes can recognize different surface distributions of histidines. It is anticipated that the structural modification of ethylenediamine, dpa and the linker length of the dinuclear complexes, should improve the binding selectivity of such metal complexes.
3. Nanoparticles: inhibition of α-chymotrypsin
Rotello et al. have developed mixed-monolayer-protected gold clusters (MMPCs) functionalized with mercaptoundecanoic acid (MUA) ligands, generating anionic nanoparticles to target the cationic patch that encircles the active site of α-chymotrypsin (ChT; Fischer et al. 2002). They found that these amphiphilic MMPCs inhibited the enzyme by a two-stage mechanism: first, a fast, reversible step wherein the active site was sterically blocked upon inhibitor binding, followed by a second, kinetically irreversible step in which the enzyme was slowly denatured (scheme 2a), as monitored by CD spectroscopy at 202 nm. The interaction of the MMPCs with ChT was reported to be very efficient , involving a stoichiometry of five protein molecules to one MMPC. Selectivity of ChT inhibition through this design of electrostatic complementarity was observed, since neither β-galactosidase, nor cellular retinoic acid binding protein nor the ChT-related serine protease elastase were inhibited by the MMPCs. Moreover, cationic MMPCs exhibited no activity towards ChT.
Scheme 2.

Ligands used for Au and CdSe nanoparticles, and schematic depiction of ChT-nanoparticle leading to: (a) two-step denaturation of ChT and (b) reversible binding with retention of ChT structure. Reproduced and edited with permission from Hong et al. (2004).
In further work, the authors demonstrated that the in situ modification of the nanoparticle surface by the addition of cationic surfactants led to the reversal of ChT inhibition (Fischer et al. 2003). Dynamic light scattering studies confirmed that ChT was released from the MMPC surface, while fluorescence and fluorescence anisotropy measurements suggested that a high degree of native structure was recovered. While still achieving enzyme inhibition, Rotello and co-workers sought to avert ChT denaturation from the outset. This they accomplished by incorporating oligo(ethylene glycol) (OEG) spacers into the MUA ligands in order to reduce their hydrophobicity, a property believed to be responsible for the observed denaturation of ChT (Hong et al. 2004). Tryptophan fluorescence and CD experiments of the resulting CdSe-based MMPC complexes with ChT suggested that the secondary structure of ChT remained intact (scheme 2b). ChT binding to such modified MMPCs was destroyed simply by raising the ionic strength with a concomitant restoration of enzymatic activity. More recently, Rotello and colleagues have developed UV light-inducible inhibitors of ChT with gold nanoparticles adorned with photocleavable phenacyl ester side chains (Fischer et al. 2004). One of these particular MMPCs, which had no effect on ChT activity in the absence of light, caused 90% inhibition of ChT after exposure to UV light for 360 min.
4. Miniature proteins
Schepartz et al. have recently described an elegant strategy, termed protein grafting, for the identification of miniature proteins (MPs) that bind protein (and nucleic acid) targets with high affinity and selectivity, thereby inhibiting protein–protein interactions (Chin & Schepartz 2001a,b; Rutledge et al. 2003; Montclare & Schepartz 2003; Golemi-Kotra et al. 2004; Gemperli et al. 2005). In this technique, which is often used in combination with molecular evolution, the residues that comprise a functional α-helical binding epitope are substituted (‘grafted’) onto the solvent-exposed, α-helical face of the small, yet well-folded avian pancreatic polypeptide (aPP). In an effort to develop novel anti-cancer agents, Schepartz & Chin have successfully used this procedure to disrupt the Bak–Bcl-2 and Bak–Bcl-xL interactions, where Bcl-2 and Bcl-xL are central antagonists of programmed cell death (apoptosis) and Bak is a death agonist (Chin & Schepartz 2001a). The authors prepared a series of MPs, of which their best hit, PPBH3-1, bound Bcl-2 up to 100-fold stronger than the key binding region of Bak (Bak72–87), with a corresponding Kd of 52±5 nM. The enhanced affinity was attributed to pre-organization of the otherwise unstructured Bak72–87 functional epitope. Furthermore, studies revealed that the PPBH3-1–Bcl-xL complex (Kd=7±2 nM) was sevenfold more stable than the PPBH3-1–Bcl-2 complex, suggesting that MP PPBH3-1 discriminates between the molecular surfaces of the related Bcl-2 and Bcl-xL proteins as well as Bak72–87. This successful application of protein grafting led Schepartz and co-workers to explore more challenging, shallower protein–protein interactions—Bcl-2 and Bcl-xL both contain a deep, hydrophobic cleft (approx. 7 Å at their deepest points)—and thereby ascertain the generality of their strategy for the disruption of protein–protein interactions. Towards this end, the authors investigated the interaction between the KIX domain of the transcriptional coactivator protein CBP (CREB-binding protein) and the kinase-inducible activation domain (KID) of the transcription factor CREB, and identified MPs that bind the KIX domain of CBP with high nanomolar to low micromolar affinity (Rutledge et al. 2003; Volkman et al. 2005). Moreover, their phosphopeptide PPKID4P, which bound CBP KIX with high affinity (Kd=562±41 nM), exhibited high specificity for the CBP KIX domain over the two unrelated biomolecules carbonic anhydrase (CA; Kd=106±12 μM) and calmodulin (CaM; Kd=52±12 μM), proteins that also bind hydrophobic or α-helical molecules.
Most recently, Schepartz et al. have developed their protein grafting technology further (Schneider et al. 2005). The authors grafted the cAMP-dependent protein kinase (PKA) recognition epitope of protein kinase inhibitor onto the α-helix of aPP, leading to a MP that recognizes PKA with nanomolar affinity. The C-terminal cysteine residue of the MP was then conjugated to the high affinity but non-selective kinase active site inhibitor K252a (scheme 3). The complementary effects of protein surface recognition and active site binding led to the identification of a potent protein-inhibitor conjugate (MP-K252a) that exhibits increased kinase specificity, relative to free K252a.
Scheme 3.

Increasing the kinase specificity of the kinase inhibitor K252a through conjugation of K252a to a miniature protein that features the cAMP-dependent protein kinase (PKA) recognition epitope. Reproduced and edited with permission from Schneider et al. (2005).
5. Porphyrins
5.1 Recognition of the surface of cytochrome c
Horse heart cytochrome c (cyt c) is a well-characterized, highly basic (pI=10) protein that plays key roles in electron transport and apoptosis (Scott & Mauk 1996). The principal region of cyt c that interacts with its protein partners, such as cytochrome oxidase and cytochrome c reductase, is a hydrophobic patch located at the exposed haem edge surface, that is surrounded by a series of cationic Arg and Lys residues. In addition to acting as structural elements involved in the recognition of protein partners, these residues are also important for the thermal stability of the protein (Hagihara et al. 1994).
Jain & Hamilton designed various tetraphenylporphyrin (TPP) derivatives with peripheral carboxylate groups to complement the distribution of functionality on the surface of cyt c (figure 5; Jain & Hamilton 2000). Dissociation constants (Kd's) of the porphyrins with cyt c were determined by a simple fluorescence-quenching assay, which relied upon the porphyrins binding in close proximity to the haem group. Receptors 9 and 10, differing only in the number of carboxylic acids, were used to probe the charge requirements for binding to the surface of cyt c. Compound 9 with eight free acids bound cyt c more than five times as tightly (Kd=860±90 nM) as 10 with only four free acids (Kd=160±20 nM), revealing the importance for electrostatic interactions between the receptor and the protein. A further eightfold enhancement in cyt c recognition was furnished by decorating the porphyrin periphery with an additional four phenyl groups, through the conjugation of a Tyr-Asp dipeptide residue to each core aromatic acid (11; Kd=20±5 nM). Remarkably, receptor 11 bound cyt c around 100 times as strongly as its natural protein partner cytochrome c peroxidase (Kd=2.4 μM). More recently, Aya & Hamilton reported a sub-nanomolar receptor of cyt c by enhancing the hydrophobicity of the porphyrin core and by increasing the number of peripheral carboxylic acids from 8 to 16 to give tetrabiphenylporphyrin-based 12 as one of the most potent, synthetic protein receptor ever designed (Kd=0.67±0.34 nM; Aya & Hamilton 2003). Furthermore, 12 exhibited good selectivity for cyt c, since Kd values for binding to the closely related protein cyt c551 and ferredoxin were 180 nM and 17 μM, respectively, indicating that complementary charge and size are essential for strong binding.
Figure 5.

Synthetic porphyrins that recognize the surface of cytochrome c: Kd's=860 nM (9), 160 nM (10), 20 nM (11) and 0.67 nM (12).
Reports that the thermal stability of cyt c was reduced upon its binding to cytochrome c peroxidase (Kresheck & Erman 1988) and cytochrome oxidase (Yu et al. 1985) encouraged Hamilton and co-workers to investigate if their porphyrins would behave similarly. Indeed, in the presence of 1.2 eq of 11, the melting point (Tm) of cyt c was reduced from 85 to 64 °C, as shown by a shift in the CD spectrum of cyt c at θ=222 nm (Jain & Hamilton 2002). Similar denaturing effects were seen with achiral porphyrin 13, which was developed in the same laboratories. However, the copper porphyrin dimer 14, led to a more dramatic denaturing effect, with 2 eq of 14 unravelling more than 60% of the α-helical secondary structure of cyt c at room temperature. Further studies indicated that 14 selectively compromised the thermal stability of cyt c through binding-induced disruption of tertiary and secondary structure (Jain & Hamilton 2002; Wilson et al. 2003), thereby leading to accelerated and catalytic proteolytic degradation (Groves et al. 2004). These results are especially interesting since they suggest an alternative to classical medicinal chemistry, with the concept of ‘conformational drugs’. Such drugs might operate by first binding the targeted protein, causing a conformational change and thereby rendering that protein unable to participate in its usual protein–protein interactions (figure 6).
Figure 6.
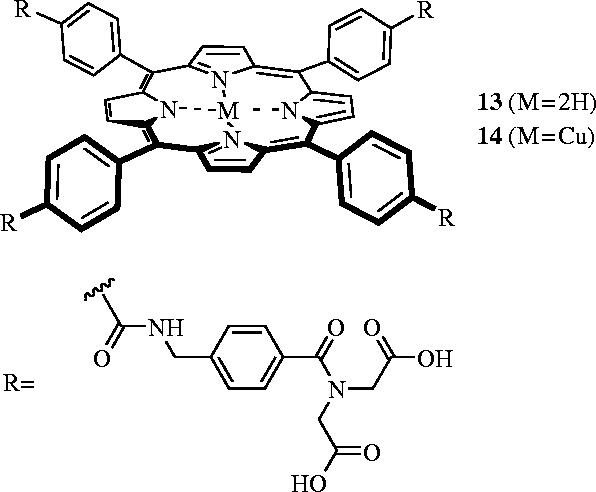
Synthetic porphyrins that denature cytochrome c.
5.2 Inhibitors of the human Kv1.3 potassium channel
Another application of protein surface recognition by rational design comes from the labs of Trauner et al. (Gradl et al. 2003). Potassium channels are crucial to the existence of organisms as diverse as archaebacteria and humans (Ruta et al. 2003), controlling the transmembrane-potential which, itself, governs such cellular functions as excitability, proliferation and secretion. In a similar manner to the design of receptors for the surface of cyt c (Jain & Hamilton 2000), Trauner and co-workers designed derivatives of TPP to match the fourfold symmetry of the human Kv1.3 potassium channel (Gradl et al. 2003). While a large variety of inhibitors of this homotetrameric protein have been identified both of synthetic origin, such as tetraethylammonium ion, and of natural origin, for instance toxins from spiders and scorpions, all of these inhibitors bind the central pore region of the channel; none of these inhibitors take full advantage of the inherent symmetry of the protein.
Trauner et al. proposed that the fourfold symmetry presented by porphyrin scaffolds could provide the necessary molecular architecture to bind all four channel subunits simultaneously, resulting in a strong polyvalency effect. Therefore, to complement the negative charges of the conserved aspartate residues (Asp381) located in ‘hot spots’ (Delano 2002) of each subunit, the authors prepared symmetrical porphyrin derivatives with positively charged peripheries. Competitive binding studies with 125I-hongotoxin1-Asp19Tyr/Tyr37Phe, which binds Kv1.3 channels with femtomolar affinity (Kd=0.046 pM), revealed a number of cationic porphyrins, with appropriate geometry, bound the protein with nanomolar affinities. Negatively charged porphyrins did not interfere with 125I-hongotoxin1-Asp19Tyr/Tyr37Phe. Furthermore, electrophysiological assays with cationic porphyrin 15 demonstrated that the ionic current through Shaker channels, the archetypical Kv1x channel, was reduced. Interestingly, however, the current was not totally blocked, suggesting that the porphyrin may not bind the protein precisely as desired (figure 7b). Further work is underway to determine the exact nature of the binding. Nonetheless, the modular composition of these receptors allows for the facile preparation of a number of derivatives that may allow discrimination among closely related potassium channels, which could ultimately lead to novel therapeutics to treat cardiac diseases, diabetes and epilepsy.
Figure 7.

(a) A cationic porphyrin (15) that binds the human Kv1.3 channel with a Ki of 20 nM; (b) overlay of the likely interaction of porphyrin 15 with the human Kv1.3 channel. Part (b) is reproduced with permission from Gradl et al. (2003).
5.3 Anti-FGF and anti-VEGF agents
Angiogenesis, the growth of new blood vessels, is necessary for the development of solid tumours (Folkman & Klagsbrun 1987). One of the chief classes of angiogenesis-promoting proteins is the heparin-binding human fibroblast growth factors (FGFs), including aFGF (acidic FGF) and bFGF (basic FGF), which exhibit a high affinity for heparin sulphate proteoglycans (Burgess & Maciag 1989). Using a porphyrin scaffold, Yayon et al. have successfully disrupted the bFGF–FGFR (fibroblast growth factor receptor) interaction, as well as the interaction between the vascular endothelial growth factor (VEGF) and its receptor (VEGFR), leading to inhibition of endothelial cell proliferation, tumour progression and metastasis (Aviezer et al. 2000). Similarly to bFGF, VEGF is a potent angiogenic factor, and these growth factors act synergistically on endothelial cell activation and differentiation (Ferrara 1999). A high-throughput screening system initially identified the cationic tetra(methylpyridinium)porphyrin (TMPP) 16 as a potent inhibitor of bFGF binding to its receptor. Further studies showed that TMPP was able to block the binding of 125I-labelled bFGF to FGFR with an IC50 value of 1 μM. Additionally, TMPP demonstrated affinity for VEGF and blocked its interaction with VEGFR with an IC50 value of approximately 10 μM. Selectivity of TMPP was shown by the lack of inhibition of the binding of the non-heparin-binding epidermal growth factor (EGF) to its tyrosine kinase receptor EGFR, suggesting that TMPP may operate by interfering with the heparin-binding domain.
In an effort to ascertain the structural requirements necessary for inhibitory activity of bFGF and VEGF, Yayon and co-workers developed a series of porphyrin analogues of TMPP (16). First, only positively charged porphyrins exhibited activity; neutral or negatively charged porphyrins had no effect. In addition, upon varying the position of the N-methyl group of the peripheral pyridinium rings from the para to the ortho and meta positions and by including 2,3,5,6-tetrafluorophenyl spacers in between the core porphyrin and the peripheral pyridinium rings, the authors arrived at tetracationic porphyrins 17 and 18. While 17 was 10-fold less active at disrupting the bFGF–FGFR interaction than TMPP (IC50=1 μM versus 10 μM), compound 18 was found to be significantly more active, indicating the position of charge to be important. More interestingly, the non-symmetrical and tricationic porphyrin derivative 19, in which one N-methylpyridinium group was replaced with a pentafluorophenyl group, was found to inhibit the bFGF–FGFR interaction in vitro with an IC50 of 20 nM. This is approximately a 50-fold improvement over TMPP. However, in the Lewis lung carcinoma tumour model, 19 exhibited limited suppression of tumour metastasis in mice, whereas 18 was more active than TMPP. Therefore, in a structure–activity approach, Yayon et al. designed a novel, water-soluble corrole analogue (20) of TMPP, displaying three positive charges as in 19 but the same side chains as in 18, and they tested the activities of 20 both in vitro and in vivo. Porphyrin derivative 20 appeared to exhibit the best pharmacological effects of both 18 and 19, since 20 was around 10-fold more active than TMPP in vitro, and fivefold more potent in vivo, inhibiting lung metastasis formation at a concentration of only 5 mg kg−1 mouse body weight. It is unclear at this stage as to the exact mechanism by which TMPP and other related porphyrins inhibit the growth factor-receptor binding and activation, other than the fact that TMPP disrupts the formation of the ternary bFGF–heparin–FGFR complex, likely through interacting with a heparin-binding site (figure 8).
Figure 8.

Cationic porphyrins as anti-FGF and anti-VEGF agents.
6. Calixarenes
Platelet-derived growth factor (PDGF) is a potent inducer of growth and motility in several cell types, inducing such processes as cell proliferation, angiogenesis and wound healing, while inhibiting apoptosis (Heldin et al. 1998). In addition, PDGF has been directly implicated in cancer, where overexpression of PDGF and/or its cell-surface receptor PDGFR is observed in many carcinomas (Kumar et al. 1998). Upon binding to its receptor, PDGF undergoes dimerization, followed by receptor autophosphorylation then recruitment of tumour cells by means of the resulting phosphotyrosine of SH2-containing signalling proteins, leading to the biological responses associated with PDGF. The over-activity of PDGF in cancerous diseases has led researchers to investigate approaches to block the binding of PDGF to its receptor, PDGFR. In particular, Hamilton and Sebti et al. have designed synthetic molecules that bind the surface of PDGF, disrupting its interaction with PDGFR, and thereby eliciting anti-tumour and anti-angiogenesis effects (Blaskovich et al. 2000). Specifically, the authors have reported a calix[4]arene derivative, GFB-111 (21), that targets loops I and III of PDGF, the regions of the protein that bind its receptor.
The upper rim of calix[4]arenes displays four positions that can be coupled to different substituents. Moreover, the nature and symmetry of such recognition elements may be varied to furnish a diverse family of molecules that can selectively target the highly irregular surfaces, comprised of charged, polar and hydrophobic residues, that are presented by proteins. Hamilton and co-workers prepared a series of synthetic receptors in which four peptide loop domains are attached to a central calix[4]arene scaffold. Each peptide loop is based on a cyclic hexapeptide in which two residues have been replaced by a 3-aminomethylbenzoate dipeptide mimetic, which also contains a 5-amino substituent for anchoring the peptide to the scaffold. Through the attachment of various peptide loops, the authors have prepared a series of calix[4]arenes expressing negatively and positively charged regions as well as hydrophobic regions on the same face of the calix[4]arene and over a surface area of approximately 500 Å2, designed to bind to complementary regions on PDGF. A cell-based screening assay with NIH 3T3 cells was used to identify molecules that were capable of blocking PDGF-BB-induced tyrosine autophosphorylation of PDGFR (PDGF-BB is a homodimer of two proteins that comprise two β strands with three intramolecular disulphide bridges). The region of PDGF that binds PDGFR is composed primarily of cationic and hydrophobic residues, therefore GBF-111 (21) was designed to be complementary to this area of PDGF, with other calix[4]arenes designed to probe the requirement for charge and hydrophobicity. Indeed, 21, with fourfold symmetry and the peptide loop Gly-Asp-Gly-Tyr, offering negative charge (Asp) and hydrophobicity (Tyr), was the most potent antagonist of PDGFR, with an IC50 value of 250 nM. The importance of negative charge and hydrophobicity in the peptide loop were confirmed by a higher IC50 value of 40 μM for Gly-Lys-Gly-Lys (22) and a moderate IC50 value of 2.5 μM for Gly-Asp-Gly-Asp (23). Gel electrophoresis confirmed that 21 binds PDGF, and the authors demonstrated that 21 displaces 125I-PDGF from PDGFR in a dose-dependent manner. Taken together, these data suggest that 21 indeed binds the targeted region of PDGF. Furthermore, cell-based studies showed that 21 selectively inhibits PDGF-stimulated DNA synthesis, arrests the growth of human tumours in nude mice and inhibits angiogenesis in vivo. Further biological evaluation is currently underway.
In additional work, Hamilton et al. observed that the highly negatively charged calix[4]arene 23 is capable of disrupting another protein–protein interaction (Wei et al. 2001). In a fluorescence titration assay, 23 competed effectively with cyt c peroxidase for binding cyt c, forming a 1 : 1 complex with a Kd of 30±10 nM. Further studies demonstrated that 23 could disrupt the complex of cyt c–Apaf-1, a crucial intermediate in the apoptosis pathway (Purring et al. 1999), upon the addition of 23 at an antagonist : complex ratio of around 200 : 1 (figure 9).
Figure 9.

Calix[4]arene derivatives that inhibit the (i) PDGF–PDGFR (21: IC50=250 nM) and (ii) cyt c–cyt c peroxidase (23: Kd=30 nM) interactions.
Tryptase has been recognized as playing a key role in the pathogenesis of asthma and psoriasis, hence is an important target for developing therapeutics of these diseases (Clark et al. 1995). Cunsolo et al. have described the design and synthesis of calix[8]arene derivatives to target tryptase selectively. They demonstrated that their positively charged peptidocalixarenes behave as competitive inhibitors of recombinant human tryptase, likely binding the targeted region of the negatively charged Asp residues near the active sites of the tetrameric protein (Mecca et al. 2004).
The enzyme transglutaminase, which catalyses the transfer of an acyl group from the γ-carboxamide of the protein-bound glutamine residue to the free, ϵ-amino group of the protein-bound lysine residue, has recently been implicated in Huntington's disease (Kim et al. 2002). Neri and co-workers have prepared a series of peptidocalix[4]arene diversomers (isomers comprising the same components arranged in different orders) to bind the surface of transglutaminase and thereby disrupt is interaction with its usual substrates (Francese et al. 2005). Again, competition assays suggested that their inhibitors bind to a region of the protein (‘hot spot’) other than the active site, exerting their inhibitory effects either by causing a conformational change in the protein or by hindering the approach of the enzyme's substrate.
7. Cyclodextrins
Breslow and co-workers have reported that β-cyclodextrin (β-CD) dimers can selectively inhibit the assembly of protein subunits into their active, multimeric aggregates by binding hydrophobic, surface residues (Leung et al. 2000). β-CD dimers had previously been shown to bind the hydrophobic side chains in polypeptides selectively (Breslow et al. 1998), and Breslow et al. hoped that these results may be extended to bind hydrophobic residues on the interfacial regions of multimeric proteins, and thereby inhibit protein aggregation, hence protein function. A series of 11 cyclodextrin dimers with a variety of linkers and cyclodextrin cavity orientation were prepared, and the abilities of these analogues to disrupt protein aggregation were assessed by monitoring enzyme activity. Seven enzymes were studied, one monomer, four dimers and three tetramers. From these only the dimeric citrate synthase (CS) and the tetrameric lactate dehydrogenase (LDH) were inhibited, even at cyclodextrin dimer concentrations as high as 480 mM. Unaffected enzymes included adenosine deaminase, phosphohexose isomerase and sorbitol dehydrogenase (which, like LDH, also uses NADH as a substrate). Compounds containing β- or α-CD groups oriented with their cavities away from the linker were inactive in the enzyme assays, as were those β-CD compounds with their cavities facing each other and separated by ether spacers. However, β-CD compounds linked by pyridyl-2,6-dicarboxylates as diester 24 and diamide 25, selectively disrupted the aggregation of CS (IC50=140 μM) and LDH (IC50=30 μM). The authors showed that neither pyridine, β-CD, pyridine-2,6-dicarboxylic acid nor a β-CD dimer linked with isophthalic acid exhibited any inhibitory activity towards CS or LDH, demonstrating the importance of the correctly spaced and oriented cavities of the β-CD cavities as well as the nature of the linking unit. Although the location of binding to the protein surfaces, and the associated selectivity, remains unclear, the authors have identified a number of potential hydrophobic, aromatic side chains that could be captured by the cavities of β-CD dimers 24 and 25, and thereby prevent protein aggregation (figure 10).
Figure 10.
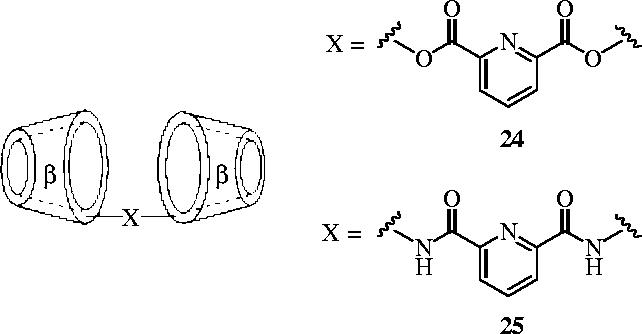
β-Cyclodextrin dimers that inhibit the self-assembly of dimeric citrate synthase (CS) and tetrameric lactate dehydrogenase (LDH).
8. Proteomimetics: molecules that mimic the structures and functions of extended regions of proteins
8.1 α-Helical mimetics
Hamilton and co-workers have introduced a terphenyl scaffold as structural and functional mimetics of α-helices, in which the 3,2′,2″-substituents on the phenyl rings project functionality in a spatial orientation that mimics the i, (i+3 or 4) and (i+7) residues—all of which are located on the same face—of an α-helix (figure 11). Their first application of this strategy targeted the interaction between calmodulin (CaM) and an α-helical domain of smooth muscle myosin light-chain kinase (Orner et al. 2001). Following a modular synthesis developed on the basis of sequential Negishi coupling reactions, Orner et al. prepared a series of 3,2′,2″-tris-substituted-terphenyl derivatives, of which the most potent antagonist of CaM was 26 (IC50=9 nM). Their next target was the HIV-1 transmembrane envelope glycoprotein, gp41. By designing terphenyl-based proteomimetics of a hydrophobic, α-helical 4–3 heptad repeat, Ernst and co-workers disrupted the assembly of the fusion-competent hexameric core of gp41, resulting in reduced levels of HIV-1 entry into host cells (Ernst et al. 2002). Specifically, titration of terphenyl 27 into a solution containing a model system of the gp41 six-helix bundle, composed of two peptides (N36 and C34) from the N- and C-heptad repeats, led to a reduction in the CD signal at θ=222 and 208 nm, corresponding to a loss in α-helicity of the hexameric bundle. Furthermore, ELISA assays with antagonist 27 and the N36/C34 helix bundle indicated that 27 effectively disrupted N36/C34 complexation with an IC50 value of 13.18±2.54 μg ml−1. Finally, through a dye-transfer cell fusion assay, the authors confirmed that the HIV-1 fusion mechanism was inhibited by 27, as suggested by the CD and ELISA experiments, with an IC50 of 15.70±2.54 μg ml−1.
Figure 11.
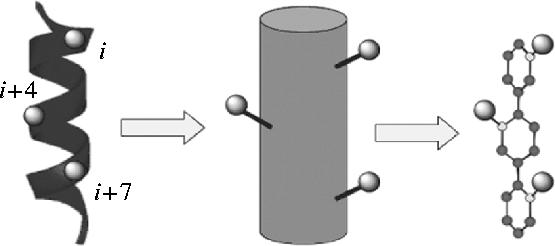
Analogy between the α-helix and the terphenyl scaffold.
More recently, Yin and colleagues have designed terphenyl-based helical mimetics to target the p53–HDM2 interaction (Yin et al. 2005a). The human double minute 2 (HDM2) protein regulates the cellular levels of p53, a protein that is known to play a key role in the apoptosis pathway (Lane 1992). In over 50% of cancerous tumours, the p53 protein is found in a mutated or inactive state (Hollstein et al. 1994), while over-expression of HDM2 has been implicated in human osteogenic and soft-tissue sarcomas (Oliner et al. 1992). Therefore, disruption of the p53–HDM2 interaction represents an important target for the development of anti-cancer drugs. X-ray crystallography of the p53–HDM2 complex showed that three hydrophobic residues (F19, W23 and L26) located on one face of the p53 helix are necessary for binding (Kussie et al. 1996). Thus, a series of 3,2′,2″-substituted terphenyls were successfully designed to mimic the helical region of p53 and bind HDM2 (Yin et al. 2005a). Fluorescence polarization assays revealed that their most potent antagonist of HDM2 was 28 (Ki=0.182±0.02 μM), and 15N–1H HSQC NMR experiments confirmed their α-helical mimetics bound the targeted region of HDM2. Moreover, 28 exhibited 14- and 82-fold selectivities over the Bak–Bcl-xL and Bak–Bcl-2 interactions, respectively (figure 12).
Figure 12.
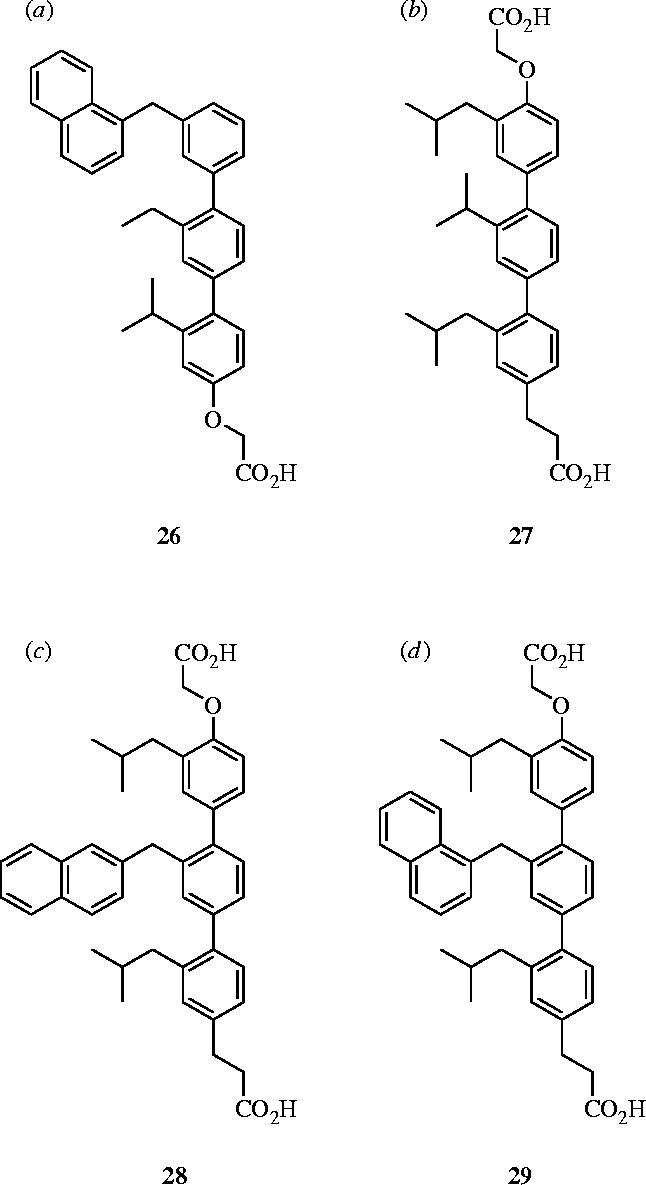
Terphenyl-based α-helical mimetics: (a) antagonist of calmodulin, 26: IC50=9 nM; (b) inhibitor of hexameric gp41 self-assembly, 27: IC50=13.2 μg ml−1; (c) antagonist of HDM2, 28: Ki=182 nM; (d) antagonist of Bcl-xL, 29: Ki=114 nM.
Another protein–protein interaction targeted within the Hamilton laboratories was the Bak–Bcl-xL interaction. The Bak–Bcl-xL complex structure, determined by NMR spectroscopy, shows that an α-helical region of Bak, specifically the BH3 domain (residues 72–87), binds to a hydrophobic cleft on the surface of Bcl-xL with a Kd of 340 nM (Sattler et al. 1997). Alanine scanning of the Bak peptide indicated that the key residues for binding are Val74, Leu78, Ile81 and Ile85, projecting in an i, (i+4), (i+7) and (i+11) arrangement from one face of the α-helix. In addition, it was observed Asp83 forms an ion-pair with a lysine residue of Bcl-xL. With the structural requirements identified, Kutzki et al. designed a series of terphenyls, containing alkyl or aryl substituents, as well as terminal carboxylic acids to mimic the ion-pair interaction, and assessed their binding affinities for Bcl-xL with a fluorescence polarization assay (Kutzki et al. 2002). Their most potent inhibitor was 29 with a Kd value of 114 nM; the importance of the positions of the three substituents was confirmed by scrambling experiments, which led to a 30-fold decrease in affinity (Kd=2.70 μM). Similarly, the significance of the hydrophobic substituents was confirmed by studies with an analogue of 29 that lacks the naphthyl and isobutyl groups (Kd=27.4 μM), and the importance of the carboxylic acids of 29 was determined by partial removal or replacement with positively charged groups, in both cases leading to substantial loss of affinity. Furthermore, HSQC NMR experiments with 29 confirmed that this series of terphenyls targets the hydrophobic cleft on Bcl-xL that is known to bind the Bak helix.
Hamilton and co-workers developed their repertoire of α-helical mimetics further with a trispyridylamide scaffold, again to target the Bak–Bcl-xL interaction (Ernst et al. 2003). This structure assumes a preferred conformation with all O-substituents projected on the same side of the molecule, as confirmed by X-ray crystallography. The pyridyl nitrogen lone pair interacts with the amide hydrogen which itself interacts with an oxygen ether lone pair, forming a hydrogen-bonding network that results in efficient mimicry of the α-helical BH3 domain of Bak. The authors have thus reported low micromolar range trispyridylamide-based antagonists of Bcl-xL, for example compound 30 bound with a Ki of 2.3 μM (Ernst et al. 2003). With an alternative, less synthetically challenging terephthalamide scaffold, Yin & Hamilton have developed simpler and more water soluble, sub-micromolar inhibitors (Ki=0.78 μM) of the Bak–Bcl-xL interaction that are also α-helical mimetics (Yin & Hamilton 2004). Whole cell assays with HEK293 cells revealed that terephthalamide 31 led to the disruption of the Bax (a Bak-analogue)–Bcl-xL interaction with an IC50 value of 35.0 μM (Yin et al. 2005b; figure 13).
Figure 13.
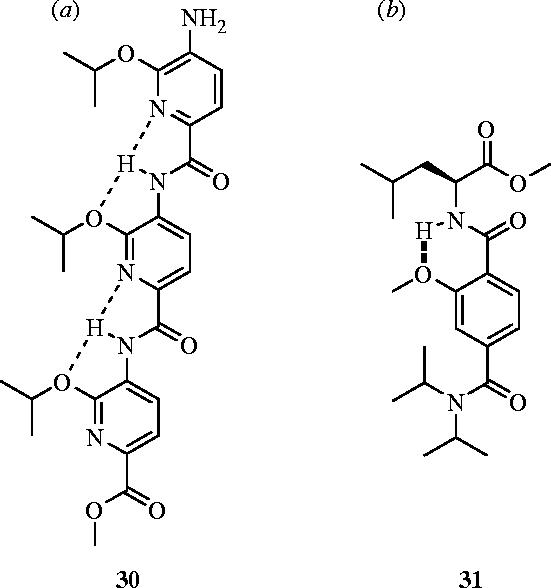
Alternative α-helical mimetics that antagonize Bcl-xL: (a) trispyridylamide 30 (Ki=2.3 μM) and (b) terephthalamide 31 (IC50=35.0 μM).
β-Peptides, which differ from their α-peptide counterparts by one additional backbone carbon atom that confers resistance to metabolism and proteolysis (Schreiber et al. 2002), may fold into helices, sheets and turns, hence have the potential as mimics of regions of proteins. Recently, Schepartz and colleagues prepared a set of β3-peptides that exhibited significant helix-14 character in water (Kritzer et al. 2004). Moreover, one of these oligomers was shown to recognize the p53-binding cleft of HDM2 with nanomolar affinity. Robinson et al. have also targeted the p53–HDM2 interaction by designing β-hairpins that mimic an α-helix (Fasan et al. 2004). Through the d-Pro-l-Pro-mediated pre-organization of eight-residue loops into regular β-hairpins, the authors designed a series of synthetic, cyclic peptides that successfully mimicked the α-helical region of p53, inhibiting the p53–HDM2 interaction with nanomolar affinity (32: IC50=140 nM) (figure 14).
Figure 14.
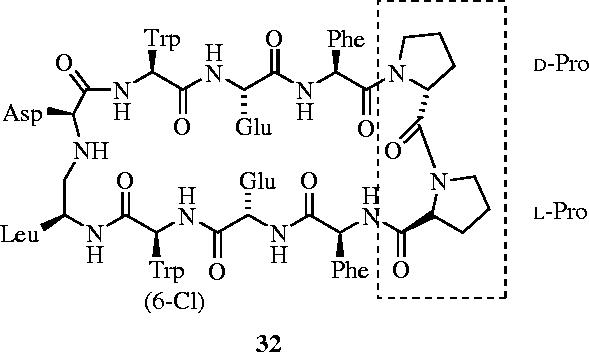
Inhibition of the p53–HDM2 interaction through mimicry of the α-helical region of p53 with β-hairpin 32 (IC50=140 nM).
Gellman and co-workers have investigated the design of α-helical mimetics of the Bak 16-mer for binding to Bcl-xL (Sadowsky et al. 2005). While the preparation of 12-helix and 14-helix structures generated through β-peptide scaffolds and the 11-helix and 14/15-helix structures formed by the 1 : 1 alternation of α- and β-amino acid residues (‘α/β peptides’) along the backbone generated only very weak Bcl-xL ligands (IC50>500 mM), a chimeric approach proved very successful. The authors generated a 15-mer (α/β+α)-chimeric peptide, comprising nine residues of an α/β peptide, followed by six α-residues that are related to the C-terminal segment of the Bak 16-mer peptide. This peptide bound with a Ki of 1.9 nM, which was further optimized to Ki=0.7 nM. These results suggest that combining different foldamer scaffolds to generate chimeric compounds is an effective approach to improving the potency of inhibitors.
8.2 β-Strand/β-sheet mimetic
The protein–protein interaction between leukocyte functional antigen-1 (LFA-1) and intercellular adhesion molecule-1 (ICAM-1) is paramount to lymphocyte and immune system function (Oppenheimer-Marks & Lipsky 1996), through mediating the adhesion, extravasation, migration and proliferation of lymphocytes. Fisher et al. have demonstrated that a discontinuous epitope comprising the residues Glu34, Lys39, Met64, Tyr66, Asn68 and Gln73, spanning three different β-sheets across the face of the protein surface (Casasnovas et al. 1998) within the first domain of ICAM-1, is essential for its interaction with LFA-1 (Fisher et al. 1997). In a non-traditional drug discovery approach, Gadek and colleagues decided to use ICAM-1, the native ligand of LFA-1, as their ‘lead’ towards identifying an antagonist of LFA-1; this process began with the emulation of Glu34 and Lys39 (Gadek et al. 2002). The authors sought a flexible Arg-X-Asp amino acid sequence and discovered that kistrin, a disintegrin protein containing the Arg-Gly-Asp sequence (Adler et al. 1993), inhibited the binding of LFA-1 and ICAM-1 in vitro with an IC50 of 700 nM in an ELISA assay, likely through intended mimicry of the critical Glu34 and Lys39 residues of ICAM-1. Alanine mutagenesis of kistrin identified a linear Arg-Gly-Asp-Met-Pro epitope (Dennis et al. 1993), which was then investigated further as a structure–activity relationship (SAR) study by means of a series of cyclic peptides. This led to cyclic disulphide, H2N-Cys-Gly-Tyr(m)-Asp-Met-Pro-Cys-CO2H (Tyr(m)=meta-tyrosine), as their lead (IC50=1.6 μM).
Meanwhile, in a related program, compound 33 was identified as an inhibitor of the LFA-1–ICAM-1 interaction, with an IC50 value of 1.4 μM (figure 15). Subsequent mimicry of the Tyr(m) residue in their peptide lead with a meta-phenol group developed 33 into 34 with a concomitant 30-fold increase in potency for antagonism of the LFA-1–ICAM-1 interaction. Further SAR studies led to increasingly more potent inhibitors 35 (IC50=3.7 nM) and 36 (IC50=1.4 nM), as determined by an ELISA assay. Due to similar potencies for 35 and 36 in the ELISA assay, a mixed lymphocyte reaction assay was next conducted, which demonstrated that 36 was more than 400 times more potent than 35. Furthermore, the authors showed that 36 inhibited LFA-1-mediated lymphocyte proliferation and adhesion in vitro. These results suggest that Gadek et al. have successfully mimicked ICAM-1 through the transfer of the ICAM-1 epitope to a small molecule; crystal structures of LFA-1 with ICAM-1 and with compounds 34, 35 or 36 will provide definitive proof of the design.
Figure 15.

LFA-1–ICAM-1 inhibitors: IC50s=1.4 μM (33), 47 nM (34), 3.7 nM (35) and 1.4 nM (36).
Additionally, based on the 21-mer IB peptide of ICAM-1, Siahann et al. have prepared cyclic peptides that inhibit the LFA-1–ICAM-1-mediated T-cell adhesion to epithelial cells (Anderson et al. 2004). Their results suggest that the Pro-Arg-Gly sequence may be important for binding to LFA-1.
8.3 β-Turn mimetic
Toll-like receptors (TLRs) and the interleukin-1 receptor superfamily (IL-1Rs), which share a conserved, cytoplasmic Toll/IL-1R/resistance (TIR) domain (Gay & Keith 1991), are crucial to both innate and adaptive immunity for host defence (Anderson 2000). Upon ligand binding, IL-1RI and IL-1R accessory protein (IL-1RAcP) monomers function as pro-inflammatory mediators through dimerization and recruitment of the adaptor protein MyD88 via homotypic binding of the TIR domains of IL-1RI and MyD88 (Medzhitov et al. 1998). The structural basis for TIR-mediated homotypic interactions has been reported by Xu et al., who have solved X-ray crystal structures of the TIR domains of TLR1 and TLR2 (Xu et al. 2000). Both structures comprise a large, conserved surface patch of a central, five-stranded parallel β-sheet that is surrounded by five α-helices. Structural and functional studies conducted by Xu and co-workers, through creating point mutations, indicated that residues located in the BB-loop of this surface patch are critical for receptor signalling (Xu et al. 2000).
Recently, Bartfai and Rebek et al. described a small molecule β-turn mimetic that disrupts the interaction between IL-1RI and MyD88 (Bartfai et al. 2003). The authors prepared analogues of the central and protruding three amino acid residues of the BB-loop, which mimics the (Phe/Tyr)-(Val/Leu/Ile)-(Pro/Gly) sequence, and studied their effects on IL-1RI signalling in vitro and in vivo. Their best hit, hydrocinnamoyl-l-valyl pyrrolidine (37), inhibited IL-1β-induced phosphorylation of the mitogen-activated protein kinase p38 in EL4 thymoma cells, with statistical significance at concentrations of 37 above 10 μM. Sandwich ELISA assays demonstrated that compound 37 inhibits the IL-1β mediated association of IL-1RI and MyD88 in both EL4 cells and in freshly isolated lymphocytes from mouse spleen. Moreover, Bartfai and colleagues have shown that the disruption of the IL-1RI–MyD88 interaction is selective over other members of the Toll receptor superfamily. Finally, the inhibitory effects of 37 on IL-1β-signaling in vivo were confirmed by the observation of a significant attenuation of the IL-1β-induced fever response (200 mg kg−1, i.p.) in mice (figure 16).
Figure 16.
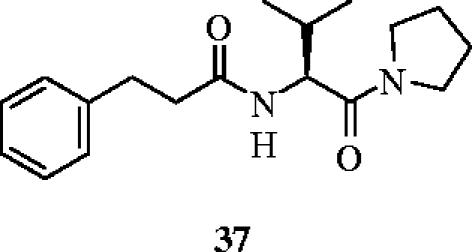
A small molecule, β-turn mimetic that disrupts the interaction between IL-1RI and MyD88.
9. Conclusions
Despite the challenges of disrupting protein–protein interactions with synthetic agents, there has been considerable progress in recent years. It is perhaps surprising to many that small molecules have proven successful at modulating protein–protein interactions. This success is likely due to the existence of protein ‘hot spots’ and has served to demonstrate that large molecules are not necessarily required to target protein surfaces. In particular, small molecule inhibitors of the dimerization of HIV-1 protease have led to the identification of molecules that are equipotent across both the wild-type of HIV-1 and a mutant strain that is resistant to active-site directed inhibitors. This result may have important implications in the directions of future anti-HIV therapies. Protein grafting has led to inhibitors of a number of interactions, such as the Bak–Bcl-xL complex, which is important in apoptosis, hence cancer. Other successful approaches by rational design have targeted cyt c, the human Kv1.3 potassium channel, the growth factors FGF and VEGF, and the self-assembly of protein aggregates. Anionic porphyrins designed to bind the principal protein–protein interaction surface of cyt c have led to the identification of synthetic protein denaturants. Furthermore, the field of proteomimetics has also produced some successful inhibitors of protein–protein interactions involved in such events as HIV cell entry, apoptosis and immune system function, through the efficient mimicry of secondary structural elements: α-helices, β-turns and β-sheets. Taken together, the results reported herein confirm that the development of novel therapeutics to exploit the large, diverse and highly functionalized surfaces involved in protein–protein interactions is making encouraging progress.
With the sudden increase in proteomics and genomics, there is an ever-increasing number of novel therapeutic targets based on protein–protein interactions. While structures such as porphyrins, calixarenes and cyclodextrins would be expected to exhibit poor drug-like properties, some of the proteomimetic inhibitors reviewed herein may have applications in the clinic. In addition, success with small molecule-mediated disruption of protein–protein interactions bodes well for the identification of ‘drug-like’ inhibitors of such targets similar to those observed for decades in enzyme inhibition. Through the combination of NMR and/or X-ray structural analysis with virtual screening methods and the continued development of more efficient screening assays, it is hoped that more potent and more selective inhibitors of protein–protein interactions will be identified, leading to novel, bioavailable therapeutics.
Acknowledgments
The authors gratefully acknowledge the NIH for support of their work in this area. We thank Debarati Mazumder and Christopher Cummings for critical reading of this manuscript.
References
- Adler M, Carter P, Lazarus R.A, Wagner G. Cysteine pairing in the glycoprotein IIbIIIa antagonist kistrin using NMR, chemical analysis, and structure calculations. Biochemistry. 1993;32:282–289. doi: 10.1021/bi00052a036. doi:10.1021/bi00052a036 [DOI] [PubMed] [Google Scholar]
- Anderson K.V. Toll signaling pathways in the innate immune response. Curr. Opin. Immunol. 2000;12:13–19. doi: 10.1016/s0952-7915(99)00045-x. doi:10.1016/S0952-7915(99)00045-X [DOI] [PubMed] [Google Scholar]
- Anderson M.E, Yakovleva T, Hu Y, Siahaan T.J. Inhibition of ICAM-1/LFA-1-mediated heterotypic T-cell adhesion to epithelial cells: design of ICAM-1 cyclic peptides. Bioorg. Med. Chem. Lett. 2004;14:1399–1402. doi: 10.1016/j.bmcl.2003.09.100. doi:10.1016/j.bmcl.2003.09.100 [DOI] [PubMed] [Google Scholar]
- Arkin M.R, Wells J.A. Small-molecule inhibitors of protein–protein interactions: progressing towards the dream. Nat. Rev. Drug Discov. 2004;3:301–317. doi: 10.1038/nrd1343. doi:10.1038/nrd1343 [DOI] [PubMed] [Google Scholar]
- Aviezer D, Cotton S, David M, Segev A, Khaselev N, Galili N, Gross Z, Yayon A. Porphyrin analogues as novel antagonists of fibroblast growth factor and vascular endothelial growth factor receptor binding that inhibit endothelial cell proliferation, tumor progression, and metastasis. Cancer Res. 2000;60:2973–2980. [PubMed] [Google Scholar]
- Aya T, Hamilton A.D. Tetrabiphenylporphyrin-based receptors for protein surfaces show sub-nanomolar affinity and enhance unfolding. Bioorg. Med. Chem. Lett. 2003;13:2651–2654. doi: 10.1016/s0960-894x(03)00551-1. doi:10.1016/S0960-894X(03)00551-1 [DOI] [PubMed] [Google Scholar]
- Bartfai T, Behrens M.M, Gaidarova S, Pemberton J, Shivanyuk A, Rebek J., Jr A low molecular weight mimic of the Toll/IL-1 receptor/resistance domain inhibits IL-1 receptor-mediated responses. Proc. Natl Acad. Sci. USA. 2003;100:7971–7976. doi: 10.1073/pnas.0932746100. doi:10.1073/pnas.0932746100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaskovich M.A, Lin Q, Delarue F.L, Sun J, Park H.S, Coppola D, Hamilton A.D, Sebti S.M. Design of GFB-111, a platelet-derived growth factor binding molecule with antiangiogenic and anticancer activity against human tumors in mice. Nat. Biotechnol. 2000;18:1065–1070. doi: 10.1038/80257. doi:10.1038/80257 [DOI] [PubMed] [Google Scholar]
- Boussard C, Klimkait T, Mahmood N, Pritchard M, Gilbert I.H. Design, synthesis and evaluation of potential inhibitors of HIV gp120–CD4 interactions. Bioorg. Med. Chem. Lett. 2004;14:2673–2676. doi: 10.1016/j.bmcl.2004.02.091. doi:10.1016/j.bmcl.2004.02.091 [DOI] [PubMed] [Google Scholar]
- Breslow R, Yang Z.W, Ching R, Trojandt G, Odobel F. Sequence selective binding of peptides by artificial receptors in aqueous solution. J. Am. Chem. Soc. 1998;120:3536–3537. doi:10.1021/ja973991y [Google Scholar]
- Burgess W.H, Maciag T. The heparin-binding (fibroblast) growth factor family of proteins. Annu. Rev. Biochem. 1989;58:575–606. doi: 10.1146/annurev.bi.58.070189.003043. doi:10.1146/annurev.bi.58.070189.003043 [DOI] [PubMed] [Google Scholar]
- Casasnovas J.M, Stehle T, Liu J.H, Wang J.H, Springer T.A. A dimeric crystal structure for the N-terminal two domains of intercellular adhesion molecule-1. Proc. Natl Acad. Sci. USA. 1998;95:4134–4139. doi: 10.1073/pnas.95.8.4134. doi:10.1073/pnas.95.8.4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaib H, Rubin M.A, Mucci N.R, Li L, Taylor J.M.G, Day M.L, Rhim J.S, Macoska J.A. Activated in prostate cancer: a PDZ domain-containing protein highly expressed in human primary prostate tumors. Cancer Res. 2001;61:2390–2394. [PubMed] [Google Scholar]
- Chin J.W, Schepartz A. Design and evolution of a miniature Bcl-2 binding protein. Angew. Chem. Int. Ed. Engl. 2001a;40:3806–3809. doi: 10.1002/1521-3773(20011015)40:20<3806::AID-ANIE3806>3.0.CO;2-B. doi:10.1002/1521-3773(20011015)40:20<3806::AID-ANIE3806>3.0.CO;2-B [DOI] [PubMed] [Google Scholar]
- Chin J.W, Schepartz A. Concerted evolution of structure and function in a miniature protein. J. Am. Chem. Soc. 2001b;123:2929–2930. doi: 10.1021/ja0056668. doi:10.1021/ja0056668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J.M, Abraham W.M, Fishman C.E, Forteza R, Ahmed A, Cortes A, Warne R.L, Moore W.R, Tanaka R.D. Tryptase inhibitors block allergen-induced airway and inflammatory responses in allergic sheep. Am. J. Respir. Crit. Care Med. 1995;152:2076–2083. doi: 10.1164/ajrccm.152.6.8520778. [DOI] [PubMed] [Google Scholar]
- Dalgleish A.G, Beverley P.C, Clapham P.R, Crawford D.H, Greaves M.F, Weiss R.A. The CD4 (T4) antigen is an essential component of the receptor for the AIDS retrovirus. Nature. 1984;312:763–767. doi: 10.1038/312763a0. doi:10.1038/312763a0 [DOI] [PubMed] [Google Scholar]
- Delano W.L. Unravelling hot spots in binding interfaces: progress and challenges. Curr. Opin. Struct. Biol. 2002;12:14–20. doi: 10.1016/s0959-440x(02)00283-x. doi:10.1016/S0959-440X(02)00283-X [DOI] [PubMed] [Google Scholar]
- Dennis M.S, Carter P, Lazarus R.A. Binding interactions of kistrin with platelet glycoprotein IIb–IIIa: analysis by site-directed mutagenesis. Proteins. 1993;15:312–321. doi: 10.1002/prot.340150308. doi:10.1002/prot.340150308 [DOI] [PubMed] [Google Scholar]
- Dev K.K, van der Putten H, Sommer B, Rovelli G. Part I: parkin-associated proteins and Parkinson's disease. Neuropharmacology. 2003;45:1–13. doi: 10.1016/s0028-3908(02)00337-4. doi:10.1016/S0028-3908(02)00337-4 [DOI] [PubMed] [Google Scholar]
- Doyle D.A, Lee A, Lewis J, Kim E, Sheng M, MacKinnon R. Crystal structures of a complexed and peptide-free membrane protein-binding domain: molecular basis of peptide recognition by PDZ. Cell. 1996;85:1067–1076. doi: 10.1016/s0092-8674(00)81307-0. doi:10.1016/S0092-8674(00)81307-0 [DOI] [PubMed] [Google Scholar]
- Ernst J.T, Kutzki O, Debnath A.K, Jiang S, Lu H, Hamilton A.D. Design of a protein surface antagonist based on alpha-helix mimicry: inhibition of gp41 assembly and viral fusion. Angew. Chem. Int. Ed. Engl. 2002;41:278–281. doi: 10.1002/1521-3773(20020118)41:2<278::aid-anie278>3.0.co;2-a. doi:10.1002/1521-3773(20020118)41:2<278::AID-ANIE278>3.0.CO;2-A [DOI] [PubMed] [Google Scholar]
- Ernst J.T, Becerril J, Park H.S, Yin H, Hamilton A.D. Design and application of an alpha-helix-mimetic scaffold based on an oligoamide-foldamer strategy: antagonism of the Bak BH3/Bcl-xL complex. Angew. Chem. Int. Ed. Engl. 2003;42:535–539. doi: 10.1002/anie.200390154. doi:10.1002/anie.200390154 [DOI] [PubMed] [Google Scholar]
- Fasan R, Dias R.L, Moehle K, Zerbe O, Vrijbloed J.W, Obrecht D, Robinson J.A. Using a beta-hairpin to mimic an alpha-helix: cyclic peptidomimetic inhibitors of the p53–HDM2 protein–protein interaction. Angew. Chem. Int. Ed. Engl. 2004;43:2109–2112. doi: 10.1002/anie.200353242. doi:10.1002/anie.200353242 [DOI] [PubMed] [Google Scholar]
- Fazal M.A, Roy B.C, Sun S, Mallik S, Rodgers K.R. Surface recognition of a protein using designed transition metal complexes. J. Am. Chem. Soc. 2001;123:6283–6290. doi: 10.1021/ja003193z. doi:10.1021/ja003193z [DOI] [PubMed] [Google Scholar]
- Ferrara N. Molecular and biological properties of vascular endothelial growth factor. J. Mol. Med. 1999;77:527–543. doi: 10.1007/s001099900019. doi:10.1007/s001099900019 [DOI] [PubMed] [Google Scholar]
- Fischer N.O, McIntosh C.M, Simard J.M, Rotello V.M. Inhibition of chymotrypsin through surface binding using nanoparticle-based receptors. Proc. Natl Acad. Sci. USA. 2002;99:5018–5023. doi: 10.1073/pnas.082644099. doi:10.1073/pnas.082644099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer N.O, Verma A, Goodman C.M, Simard J.M, Rotello V.M. Reversible “irreversible” inhibition of chymotrypsin using nanoparticle receptors. J. Am. Chem. Soc. 2003;125:13 387–13 391. doi: 10.1021/ja0352505. doi:10.1021/ja0352505 [DOI] [PubMed] [Google Scholar]
- Fischer N.O, Paulini R, Drechsler U, Rotello V.M. Light-induced inhibition of chymotrypsin using photocleavable monolayers on gold nanoparticles. Chem. Commun. (Camb.) 2004:2866–2867. doi: 10.1039/b408972c. [DOI] [PubMed] [Google Scholar]
- Fisher K.L, Lu J, Riddle L, Kim K.J, Presta L.G, Bodary S.C. Identification of the binding site in intercellular adhesion molecule 1 for its receptor, leukocyte function-associated antigen 1. Mol. Biol. Cell. 1997;8:501–515. doi: 10.1091/mbc.8.3.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher C.V. Enfuvirtide, a new drug for HIV infection. Lancet. 2003;361:1577–1578. doi: 10.1016/S0140-6736(03)13323-5. doi:10.1016/S0140-6736(03)13323-5 [DOI] [PubMed] [Google Scholar]
- Fletcher S, Hamilton A.D. Protein surface recognition and proteomimetics: mimics of protein surface structure and function. Curr. Opin. Chem. Biol. 2005;9:632–638. doi: 10.1016/j.cbpa.2005.10.006. doi:10.1016/j.cbpa.2005.10.006 [DOI] [PubMed] [Google Scholar]
- Folkman J, Klagsbrun M. Angiogenic factors. Science. 1987;235:442–447. doi: 10.1126/science.2432664. [DOI] [PubMed] [Google Scholar]
- Francese S, Cozzolino A, Caputo I, Esposito C, Martino M, Gaeta C, Troisi F, Neri P. Transglutaminase surface recognition by peptidocalix[4]arene diversomers. Tetrahedron Lett. 2005;46:1611–1615. doi:10.1016/j.tetlet.2005.01.078 [Google Scholar]
- Fujii N, Haresco J.J, Novak K.A, Stokoe D, Kuntz I.D, Guy R.K. A selective irreversible inhibitor targeting a PDZ protein interaction domain. J. Am. Chem. Soc. 2003;125:12 074–12 075. doi: 10.1021/ja035540l. doi:10.1021/ja035540l [DOI] [PubMed] [Google Scholar]
- Gadek T.R, et al. Generation of an LFA-1 antagonist by the transfer of the ICAM-1 immunoregulatory epitope to a small molecule. Science. 2002;295:1086–1089. doi: 10.1126/science.295.5557.1086. doi:10.1126/science.295.5557.1086 [DOI] [PubMed] [Google Scholar]
- Gay N.J, Keith F.J. Drosophila Toll and IL-1 receptor. Nature. 1991;351:355–356. doi: 10.1038/351355b0. doi:10.1038/351355b0 [DOI] [PubMed] [Google Scholar]
- Gemperli A.C, Rutledge S.E, Maranda A, Schepartz A. Paralog-selective ligands for bcl-2 proteins. J. Am. Chem. Soc. 2005;127:1596–1597. doi: 10.1021/ja0441211. doi:10.1021/ja0441211 [DOI] [PubMed] [Google Scholar]
- Golemi-Kotra D, Mahaffy R, Footer M.J, Holtzman J.H, Pollard T.D, Theriot J.A, Schepartz A. High affinity, paralog-specific recognition of the Mena EVH1 domain by a miniature protein. J. Am. Chem. Soc. 2004;126:4–5. doi: 10.1021/ja037954k. [DOI] [PubMed] [Google Scholar]
- Gradl S.N, Felix J.P, Isacoff E.Y, Garcia M.L, Trauner D. Protein surface recognition by rational design: nanomolar ligands for potassium channels. J. Am. Chem. Soc. 2003;125:12 668–12 669. doi: 10.1021/ja036155z. doi:10.1021/ja036155z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves K, Wilson A.J, Hamilton A.D. Catalytic unfolding and proteolysis of cytochrome C induced by synthetic binding agents. J. Am. Chem. Soc. 2004;126:12 833–12 842. doi: 10.1021/ja0317731. [DOI] [PubMed] [Google Scholar]
- Hagihara Y, Tan Y, Goto Y. Comparison of the conformational stability of the molten globule and native states of horse cytochrome c. Effects of acetylation, heat, urea and guanidine-hydrochloride. J. Mol. Biol. 1994;237:336–348. doi: 10.1006/jmbi.1994.1234. doi:10.1006/jmbi.1994.1234 [DOI] [PubMed] [Google Scholar]
- Heldin C.H, Ostman A, Ronnstrand L. Signal transduction via platelet-derived growth factor receptors. Biochim. Biophys. Acta. 1998;1378:F79–113. doi: 10.1016/s0304-419x(98)00015-8. [DOI] [PubMed] [Google Scholar]
- Hollstein M, et al. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res. 1994;22:3551–3555. [PMC free article] [PubMed] [Google Scholar]
- Hong R, Fischer N.O, Verma A, Goodman C.M, Emrick T, Rotello V.M. Control of protein structure and function through surface recognition by tailored nanoparticle scaffolds. J. Am. Chem. Soc. 2004;126:739–743. doi: 10.1021/ja037470o. doi:10.1021/ja037470o [DOI] [PubMed] [Google Scholar]
- Jain R.K, Hamilton A.D. Protein surface recognition by synthetic receptors based on a tetraphenylporphyrin scaffold. Org. Lett. 2000;2:1721–1723. doi: 10.1021/ol005871s. doi:10.1021/ol005871s [DOI] [PubMed] [Google Scholar]
- Jain R.K, Hamilton A.D. Designing protein denaturants: synthetic agents induce cytochrome c unfolding at low concentrations and stoichiometries. Angew. Chem. Int. Ed. Engl. 2002;41:641–643. doi:10.1002/1521-3773(20020215)41:4<641::AID-ANIE641>3.0.CO;2-1 [Google Scholar]
- Kim S.Y, Jeitner T.M, Steinert P.M. Transglutaminases in disease. Neurochem. Int. 2002;40:85–103. doi: 10.1016/s0197-0186(01)00064-x. doi:10.1016/S0197-0186(01)00064-X [DOI] [PubMed] [Google Scholar]
- Koo T.H, Lee J.J, Kim E.M, Kim K.W, Kim H.D, Lee J.H. Syntenin is overexpressed and promotes cell migration in metastatic human breast and gastric cancer cell lines. Oncogene. 2002;21:4080–4088. doi: 10.1038/sj.onc.1205514. doi:10.1038/sj.onc.1205514 [DOI] [PubMed] [Google Scholar]
- Kresheck G.C, Erman J.E. Calorimetric studies of the thermal denaturation of cytochrome c peroxidase. Biochemistry. 1988;27:2490–2496. doi: 10.1021/bi00407a035. doi:10.1021/bi00407a035 [DOI] [PubMed] [Google Scholar]
- Kritzer J.A, Lear J.D, Hodsdon M.E, Schepartz A. Helical b-peptide inhibitors of the p53–HDM2 interaction. J. Am. Chem. Soc. 2004;126:9468–9469. doi: 10.1021/ja031625a. doi:10.1021/ja031625a [DOI] [PubMed] [Google Scholar]
- Kumar R, Yoneda J, Bucana C.D, Fidler I.J. Regulation of distinct steps of angiogenesis by different angiogenic molecules. Int. J. Oncol. 1998;12:749–757. doi: 10.3892/ijo.12.4.749. [DOI] [PubMed] [Google Scholar]
- Kussie P.H, Gorina S, Marechal V, Elenbaas B, Moreau J, Levine A.J, Pavletich N.P. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. doi:10.1126/science.274.5289.948 [DOI] [PubMed] [Google Scholar]
- Kutzki O, Park H.S, Ernst J.T, Orner B.P, Yin H, Hamilton A.D. Development of a potent Bcl-x(L) antagonist based on alpha-helix mimicry. J. Am. Chem. Soc. 2002;124:11 838–11 839. doi: 10.1021/ja026861k. doi:10.1021/ja026861k [DOI] [PubMed] [Google Scholar]
- Kwong P.D, Wyatt R, Robinson J, Sweet R.W, Sodroski J, Hendrickson W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature. 1998;393:648–659. doi: 10.1038/31405. doi:10.1038/31405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBonte J, Lebbos J, Kirkpatrick P. Enfuvirtide. Nat. Rev. Drug Discov. 2003;2:345–346. doi: 10.1038/nrd1091. doi:10.1038/nrd1091 [DOI] [PubMed] [Google Scholar]
- LaBranche C.C, Galasso G, Moore J.P, Bolognesi D.P, Hirsch M.S, Hammer S.M. HIV fusion and its inhibition. Antiviral Res. 2001;50:95–115. doi: 10.1016/s0166-3542(01)00130-9. doi:10.1016/S0166-3542(01)00130-9 [DOI] [PubMed] [Google Scholar]
- Lane D.P. Cancer—p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. doi:10.1038/358015a0 [DOI] [PubMed] [Google Scholar]
- Leduc A.M, Trent J.O, Wittliff J.L, Bramlett K.S, Briggs S.L, Chirgadze N.Y, Wang Y, Burris T.P, Spatola A.F. Helix-stabilized cyclic peptides as selective inhibitors of steroid receptor–coactivator interactions. Proc. Natl Acad. Sci. USA. 2003;100:11 273–11 278. doi: 10.1073/pnas.1934759100. doi:10.1073/pnas.1934759100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung D.K, Yang Z, Breslow R. Selective disruption of protein aggregation by cyclodextrin dimers. Proc. Natl Acad. Sci. USA. 2000;97:5050–5053. doi: 10.1073/pnas.97.10.5050. doi:10.1073/pnas.97.10.5050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li T, Saro D, Spaller M.R. Thermodynamic profiling of conformationally constrained cyclic ligands for the PDZ domain. Bioorg. Med. Chem. Lett. 2004;14:1385–1388. doi: 10.1016/j.bmcl.2003.09.103. doi:10.1016/j.bmcl.2003.09.103 [DOI] [PubMed] [Google Scholar]
- Little S.J, et al. Antiretroviral-drug resistance among patients recently infected with HIV. N. Engl. J. Med. 2002;347:385–394. doi: 10.1056/NEJMoa013552. doi:10.1056/NEJMoa013552 [DOI] [PubMed] [Google Scholar]
- Mecca T, Consoli G.M, Geraci C, Cunsolo F. Designed calix[8]arene-based ligands for selective tryptase surface recognition. Bioorg. Med. Chem. 2004;12:5057–5062. doi: 10.1016/j.bmc.2004.07.037. doi:10.1016/j.bmc.2004.07.037 [DOI] [PubMed] [Google Scholar]
- Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway C.A., Jr MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol. Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. doi:10.1016/S1097-2765(00)80136-7 [DOI] [PubMed] [Google Scholar]
- Memon M.I, Memon M.A. Hepatitis C: an epidemiological review. J. Viral Hepat. 2002;9:84–100. doi: 10.1046/j.1365-2893.2002.00329.x. doi:10.1046/j.1365-2893.2002.00329.x [DOI] [PubMed] [Google Scholar]
- Miller V. International perspectives on antiretroviral resistance. Resistance to protease inhibitors. J. Acquir. Immune Defic. Syndr. 2001;26(Suppl. 1):S34–S50. doi: 10.1097/00042560-200103011-00005. doi:10.1097/00042560-200103011-00005 [DOI] [PubMed] [Google Scholar]
- Montclare J.K, Schepartz A. Miniature homeodomains: high specificity without an N-terminal arm. J. Am. Chem. Soc. 2003;125:3416–3417. doi: 10.1021/ja028628s. doi:10.1021/ja028628s [DOI] [PubMed] [Google Scholar]
- Niethammer M, Valtschanoff J.G, Kapoor T.M, Allison D.W, Weinberg T.M, Craig A.M, Sheng M. CRIPT, a novel postsynaptic protein that binds to the third PDZ domain of PSD-95/SAP90. Neuron. 1998;20:693–707. doi: 10.1016/s0896-6273(00)81009-0. doi:10.1016/S0896-6273(00)81009-0 [DOI] [PubMed] [Google Scholar]
- Ojida A, Miyahara Y, Kohira T, Hamachi I. Recognition and fluorescence sensing of specific amino acid residue on protein surface using designed molecules. Biopolymers. 2004a;76:177–184. doi: 10.1002/bip.10574. doi:10.1002/bip.10574 [DOI] [PubMed] [Google Scholar]
- Ojida A, Mito-oka Y, Sada K, Hamachi I. Molecular recognition and fluorescence sensing of monophosphorylated peptides in aqueous solution by bis(zinc(II)-dipicolylamine)-based artificial receptors. J. Am. Chem. Soc. 2004b;126:2454–2463. doi: 10.1021/ja038277x. doi:10.1021/ja038277x [DOI] [PubMed] [Google Scholar]
- Oliner J.D, Kinzler K.W, Meltzer P.S, George D.L, Vogelstein B. Amplification of a gene encoding a p53-associated protein in human sarcomas. Nature. 1992;358:80–83. doi: 10.1038/358080a0. doi:10.1038/358080a0 [DOI] [PubMed] [Google Scholar]
- Oppenheimer-Marks N, Lipsky P.E. Adhesion molecules as targets for the treatment of autoimmune diseases. Clin. Immunol. Immunopathol. 1996;79:203–210. doi: 10.1006/clin.1996.0069. doi:10.1006/clin.1996.0069 [DOI] [PubMed] [Google Scholar]
- Orner B.P, Ernst J.T, Hamilton A.D. Toward proteomimetics: terphenyl derivatives as structural and functional mimics of extended regions of an alpha-helix. J. Am. Chem. Soc. 2001;123:5382–5383. doi: 10.1021/ja0025548. doi:10.1021/ja0025548 [DOI] [PubMed] [Google Scholar]
- Pagliaro L, Felding J, Audouze K, Nielsen S.J, Terry R.B, Krog-Jensen C, Butcher S. Emerging classes of protein–protein interaction inhibitors and new tools for their development. Curr. Opin. Chem. Biol. 2004;8:442–449. doi: 10.1016/j.cbpa.2004.06.006. doi:10.1016/j.cbpa.2004.06.006 [DOI] [PubMed] [Google Scholar]
- Purring C.B, Zou H, Wang X, McLendon G. Stoichiometry, free energy, and kinetic aspects of cytochrome c: Apaf-1 binding in apoptosis. J. Am. Chem. Soc. 1999;121:7435–7436. doi:10.1021/ja991235h [Google Scholar]
- Ruff M.R, Melendez-Guerrero L.M, Yang Q.E, Ho W.Z, Mikovits J.W, Pert C.B, Ruscetti F.A. Peptide T inhibits HIV-1 infection mediated by the chemokine receptor-5 (CCR5) Antiviral Res. 2001;52:63–75. doi: 10.1016/s0166-3542(01)00163-2. doi:10.1016/S0166-3542(01)00163-2 [DOI] [PubMed] [Google Scholar]
- Ruta V, Jiang Y, Lee A, Chen J, MacKinnon R. Functional analysis of an archaebacterial voltage-dependent K+ channel. Nature. 2003;422:180–185. doi: 10.1038/nature01473. doi:10.1038/nature01473 [DOI] [PubMed] [Google Scholar]
- Rutledge S.E, Volkman H.M, Schepartz A. Molecular recognition of protein surfaces: high affinity ligands for the CBP KIX domain. J. Am. Chem. Soc. 2003;125:14 336–14 347. doi: 10.1021/ja034508o. doi:10.1021/ja034508o [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowsky J.D, Schmitt M.A, Lee H.S, Umezawa N, Wang S, Tomita Y, Gellman S.H. Chimeric (alpha/beta+alpha)-peptide ligands for the BH3-recognition cleft of Bcl-XL: critical role of the molecular scaffold in protein surface recognition. J. Am. Chem. Soc. 2005;127:11 966–11 968. doi: 10.1021/ja053678t. doi:10.1021/ja053678t [DOI] [PubMed] [Google Scholar]
- Sattler M, et al. Structure of Bcl-xL–Bak peptide complex: recognition between regulators of apoptosis. Science. 1997;275:983–986. doi: 10.1126/science.275.5302.983. doi:10.1126/science.275.5302.983 [DOI] [PubMed] [Google Scholar]
- Schneider T.L, Mathew R.S, Rice K.P, Tamaki K, Wood J.L, Schepartz A. Increasing the kinase specificity of K252a by protein surface recognition. Org. Lett. 2005;7:1695–1698. doi: 10.1021/ol050179o. doi:10.1021/ol050179o [DOI] [PubMed] [Google Scholar]
- Schreiber J.V, Frackenpohl J, Moser F, Fleischmann T, Kohler H.P, Seebach D. On the biodegradation of beta-peptides. Chembiochem. 2002;3:424–432. doi: 10.1002/1439-7633(20020503)3:5<424::AID-CBIC424>3.0.CO;2-0. doi:10.1002/1439-7633(20020503)3:5<424::AID-CBIC424>3.0.CO;2-0 [DOI] [PubMed] [Google Scholar]
- Scott R.A, Mauk A.G. University Science Books; Sausalito, CA: 1996. Cytochrome c: a multidisciplinary approach. [Google Scholar]
- Sheng M, Sala C. PDZ domains and the organization of supramolecular complexes. Annu. Rev. Neurosci. 2001;24:1–29. doi: 10.1146/annurev.neuro.24.1.1. doi:10.1146/annurev.neuro.24.1.1 [DOI] [PubMed] [Google Scholar]
- Shultz M.D, Ham Y.W, Lee S.G, Davis D.A, Brown C, Chmielewski J. Small-molecule dimerization inhibitors of wild-type and mutant HIV protease: a focused library approach. J. Am. Chem. Soc. 2004;126:9886–9887. doi: 10.1021/ja048139n. doi:10.1021/ja048139n [DOI] [PubMed] [Google Scholar]
- Tamamura H, Omagari A, Oishi S, Kanamoto T, Yamamoto N, Peiper S.C, Nakashima H, Otaka A, Fujii N. Pharmacophore identification of a specific CXCR4 inhibitor, T140, leads to development of effective anti-HIV agents with very high selectivity indexes. Bioorg. Med. Chem. Lett. 2000;10:2633–2637. doi: 10.1016/s0960-894x(00)00535-7. doi:10.1016/S0960-894X(00)00535-7 [DOI] [PubMed] [Google Scholar]
- Toogood P.L. Inhibition of protein–protein association by small molecules: approaches and progress. J. Med. Chem. 2002;45:1543–1558. doi: 10.1021/jm010468s. doi:10.1021/jm010468s [DOI] [PubMed] [Google Scholar]
- Tremblay C.L, Giguel F, Kollmann C, Guan Y, Chou T.C, Baroudy B.M, Hirsch M.S. Anti-human immunodeficiency virus interactions of SCH-C (SCH 351125), a CCR5 antagonist, with other antiretroviral agents in vitro. Antimicrob. Agents Chemother. 2002;46:1336–1339. doi: 10.1128/AAC.46.5.1336-1339.2002. doi:10.1128/AAC.46.5.1336-1339.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- VanCompernolle S.E, Wiznycia A.V, Rush J.R, Dhanasekaran M, Baures P.W, Todd S.C. Small molecule inhibition of hepatitis C virus E2 binding to CD81. Virology. 2003;314:371–380. doi: 10.1016/s0042-6822(03)00406-9. doi:10.1016/S0042-6822(03)00406-9 [DOI] [PubMed] [Google Scholar]
- Volkman H.M, Rutledge S.E, Schepartz A. Binding mode and transcriptional activation potential of high affinity ligands for the CBP KIX domain. J. Am. Chem. Soc. 2005;127:4649–4658. doi: 10.1021/ja042761y. doi:10.1021/ja042761y [DOI] [PubMed] [Google Scholar]
- Walensky L.D, Kung A.L, Escher I, Malia T.J, Barbuto S, Wright R.D, Wagner G, Verdine G.L, Korsmeyer S.J. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004;305:1466–1470. doi: 10.1126/science.1099191. doi:10.1126/science.1099191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei Y, McLendon G.L, Hamilton A.D, Case M.A, Purring C.B, Lin Q, Park H.S, Lee C.S, Yu T. Disruption of protein–protein interactions: design of a synthetic receptor that blocks the binding of cytochrome c to cytochrome c peroxidase. Chem. Commun. (Camb.) 2001;17:1580–1581. doi: 10.1039/b104142h. [DOI] [PubMed] [Google Scholar]
- Wilson A.J, Groves K, Jain R.K, Park H.S, Hamilton A.D. Directed denaturation: room temperature and stoichiometric unfolding of cytochrome C by a metalloporphyrin dimer. J. Am. Chem. Soc. 2003;125:4420–4421. doi: 10.1021/ja028574m. doi:10.1021/ja028574m [DOI] [PubMed] [Google Scholar]
- Wu X, et al. Evidence for regulation of the PTEN tumor suppressor by a membrane-localized multi-PDZ domain containing scaffold protein MAGI-2. Proc. Natl Acad. Sci. USA. 2000a;97:4233–4238. doi: 10.1073/pnas.97.8.4233. doi:10.1073/pnas.97.8.4233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Dowbenko D, Spencer S, Laura R, Lee J, Gu Q, Lasky L.A. Interaction of the tumor suppressor PTEN/MMAC with a PDZ domain of MAGI3, a novel membrane-associated guanylate kinase. J. Biol. Chem. 2000b;275:21 477–21 485. doi: 10.1074/jbc.M909741199. doi:10.1074/jbc.M909741199 [DOI] [PubMed] [Google Scholar]
- Wyatt R, Kwong P.D, Desjardins E, Sweet R.W, Robinson J, Hendrickson W.A, Sodroski J.G. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature. 1998;393:705–711. doi: 10.1038/31514. doi:10.1038/31514 [DOI] [PubMed] [Google Scholar]
- Xu Y, Tao X, Shen B, Horng T, Medzhitov R, Manley J.L, Tong L. Structural basis for signal transduction by the Toll/interleukin-1 receptor domains. Nature. 2000;408:111–115. doi: 10.1038/35040600. doi:10.1038/35047056 [DOI] [PubMed] [Google Scholar]
- Yachou A, Sekaly R.P. Binding of soluble recombinant HIV envelope glycoprotein, rgp120, induces conformational changes in the cellular membrane-anchored CD4 molecule. Biochem. Biophys. Res. Commun. 1999;265:428–433. doi: 10.1006/bbrc.1999.1686. doi:10.1006/bbrc.1999.1686 [DOI] [PubMed] [Google Scholar]
- Yin H, Hamilton A.D. Terephthalamide derivatives as mimetics of the helical region of Bak peptide target Bcl-xL protein. Bioorg. Med. Chem. Lett. 2004;14:1375–1379. doi: 10.1016/j.bmcl.2003.09.096. doi:10.1016/j.bmcl.2003.09.096 [DOI] [PubMed] [Google Scholar]
- Yin H, Hamilton A.D. Strategies for targeting protein–protein interactions with synthetic agents. Angew. Chem. Int. Ed. Engl. 2005;44:4130–4163. doi: 10.1002/anie.200461786. doi:10.1002/anie.200461786 [DOI] [PubMed] [Google Scholar]
- Yin H, Lee G.I, Park H.S, Payne G.A, Rodriguez J.M, Sebti S.M, Hamilton A.D. Terphenyl-based helical mimetics that disrupt the p53/HDM2 interaction. Angew. Chem. Int. Ed. Engl. 2005;44:2704–2707. doi: 10.1002/anie.200462316. doi:10.1002/anie.200462316 [DOI] [PubMed] [Google Scholar]
- Yin H, Lee G.I, Sedey K.A, Rodriguez J.M, Wang H.G, Sebti S.M, Hamilton A.D. Terephthalamide derivatives as mimetics of helical peptides: disruption of the Bcl-x(L)/Bak interaction. J. Am. Chem. Soc. 2005b;127:5463–5468. doi: 10.1021/ja0446404. doi:10.1021/ja0446404 [DOI] [PubMed] [Google Scholar]
- Yu C.A, Gwak S.H, Yu L. Studies on protein–lipid interactions in cytochrome c oxidase by differential scanning calorimetry. Biochim. Biophys. Acta. 1985;812:656–664. doi: 10.1016/0005-2736(85)90258-5. [DOI] [PubMed] [Google Scholar]
- Zhang Z.-Y, Poorman R.A, Maggiora L.L, Heinrikson R.L, Kezdy F.J. Dissociative inhibition of dimeric enzymes. Kinetic characterization of the inhibition of HIV-1 protease by its COOH-terminal. J. Biol. Chem. 1991;266:15 591–15 594. [PubMed] [Google Scholar]
- Zhao L, Chmielewski J. Inhibiting protein–protein interactions using designed molecules. Curr. Opin. Struct. Biol. 2005;15:31–34. doi: 10.1016/j.sbi.2005.01.005. doi:10.1016/j.sbi.2005.01.005 [DOI] [PubMed] [Google Scholar]
- Zutshi R, Franciskovich J, Shultz M, Schweitzer B, Bishop P, Wilson M, Chmielewski J. Targeting the dimerization interface of HIV-1 protease: inhibition with cross-linked interfacial peptides. J. Am. Chem. Soc. 1997;119:4841–4845. doi:10.1021/ja962496j [Google Scholar]