

Abstract
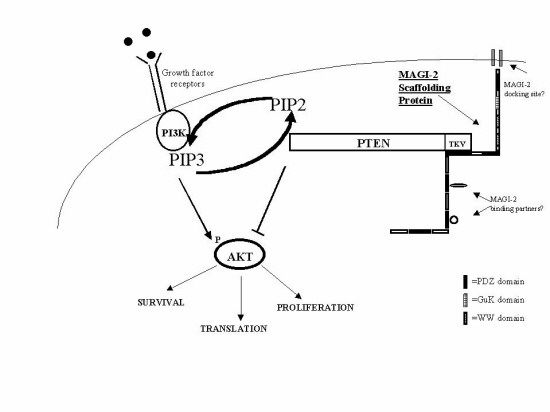
PTEN is a tumor suppressor gene mutated in human cancers. Although many mutations target the phosphatase domain, others create a truncated protein lacking the C-terminal PDZ-binding motif or a protein that extends beyond the PDZ-binding motif. Using the yeast two-hybrid system, we isolated a membrane-associated guanylate kinase family protein with multiple PDZ domains [AIP-1 (atrophin interacting protein 1), renamed MAGI-2 (membrane associated guanylate kinase inverted-2)]. MAGI-2 contains eight potential protein–protein interaction domains and is localized to tight junctions in the membrane of epithelial cells. PTEN binds to MAGI-2 through an interaction between the PDZ-binding motif of PTEN and the second PDZ domain of MAGI-2. MAGI-2 enhances the ability of PTEN to suppress Akt activation. Furthermore, certain PTEN mutants have reduced stability, which is restored by adding the minimal PDZ-binding motif back to the truncated protein. We propose that MAGI-2 improves the efficiency of PTEN signaling through assembly of a multiprotein complex at the cell membrane.
Somatic mutations of the PTEN tumor suppressor gene (also known as MMAC1 or TEP1) occur in many human malignancies (1–3). Germ-line mutations of PTEN are found in patients with two cancer predisposition syndromes (4, 5), and mice with heterozygous disruption of PTEN develop multiple tumors (6–8). Expression of PTEN suppresses the growth of glioblastoma cells (9). PTEN encodes a cytoplasmic phosphatase with both protein and lipid phosphatase activity, and many mutations cause loss of enzymatic function (10–12). Candidate substrates for PTEN include focal adhesion kinase and phosphatidylinositol (PI) lipids phosphorylated at the 3′ position by PI3-kinase (11, 13). Recent work has implicated the PI3-kinase/Akt pathway as a target of PTEN in cancer cells (14–19).
PTEN contains a 220-aa C-terminal region that is also a target of mutations in tumors. Many frameshift mutations lead to premature truncation of the protein in exons 8 or 9 (12). Because the last 4 aa of PTEN encode a PDZ domain-binding motif, all C-terminal mutations would be expected to disrupt this protein–protein interaction. PDZ domains are protein–protein interaction domains that bind to consensus motifs (S/TXV) in the C terminus of partner proteins or, alternatively, to other PDZ domains or β-hairpin finger motifs present internally in the partner protein (20, 21). PDZ domains are found in many types of proteins, including a family of membrane associated scaffold proteins known as MAGUKs (membrane-associated guanylate kinases). MAGUKs generally contain 3–5 PDZ domains, a catalytically inactive guanylate kinase domain, and several Src homology or WW domains, all of which function primarily as protein–protein interaction modules. Well-characterized MAGUK proteins such as PSD-95 (postsynaptic density) are believed to play a critical role in signal transduction through clustering of associated proteins at critical structures in the membrane such as synapses, ion channels, and tight junctions (22, 23). The multi-PDZ domain Drosophila scaffold protein InaD, which functions in photoreceptor signal transduction, enhances stability of its partner proteins through PDZ domain-mediated interactions (24). It is proposed that MAGUKs function as scaffold proteins to assemble multiprotein signaling complexes and enhance their stability, thereby increasing the efficiency of signal transduction (25).
To test the hypothesis that PTEN binds to a PDZ domain-containing protein, we performed a yeast two-hybrid screen to isolate such proteins. We identified the multi-PDZ domain-containing MAGUK protein AIP-1 [atrophin interacting protein; renamed MAGI-2 (membrane associated guanylate kinase inverted-2)]. MAGI-2 originally was isolated based on its interaction with atrophin-1, a protein containing polyglutamine repeats in patients with a neurological disorder known as dentatorubral and pallidoluysian atrophy (26). Here we show that PTEN binds to MAGI-2 through an interaction between the C terminus of PTEN and the second PDZ domain of MAGI-2. MAGI-2 enhances the efficiency of PTEN signaling and PTEN mutants that fail to bind MAGI-2 show defects in Akt regulation. We propose that: (i) MAGI-2 functions as a scaffold protein to regulate PTEN activity through assembly of a multiprotein signaling complex, and (ii) the truncated PTEN proteins found in some tumors are loss of function mutants because of the combined effects of protein instability and failure to bind MAGI-2.
Materials and Methods
Plasmids.
Wild-type PTEN and PTEN mutants were FLAG-tagged at the N terminus and cloned into pcDNA3 or the retroviral vector pSRαMSVtkNeo (16). Full-length hemagglutinin (HA)-MAGI-2 was constructed by adding an HA epitope at the N terminus of a full-length MAGI-2 cDNA (26). The individual MAGI-2 PDZ domain constructs were cloned into pcDNA3.
Protein Analysis.
The yeast two-hybrid screen was performed by using the C124S PTEN mutant cloned into pAS2.1 (CLONTECH). Independent clones (2 × 106) from a normal human prostate cDNA library fused to the Gal4 activation domain (CLONTECH) were screened.
In vitro transcription/translation reactions were performed by using rabbit reticulocyte lysate (Promega) in the presence of [35S]methionine (Amersham Pharmacia). The product was diluted 1:100 in buffer containing 20 mM Hepes (pH 7.4), 150 mM NaCl, 10% glycerol, protease inhibitors, and 0.1% Triton X-100 and incubated with glutathione S-transferase (GST) fusion proteins bound to glutathione beads for 45 min at room temperature.
293T cells were lysed in 1% Triton buffer 24 h after transfection. Lysates were diluted 1:10 in buffer containing no detergent, incubated with anti-FLAG M2 antibody overnight, then incubated with protein A Sepharose beads for 2 h, run on an SDS/PAGE gel and blotted with anti-HA antibody 12CA5 to detect MAGI-2 or anti-FLAG antibody to detect PTEN.
Fresh mouse brain was isolated and homogenized in 2 mM EDTA, 25 mM Hepes (pH 7.5), 150 mM NaCl, 0.5% NP-40, and 10% glycerol. MAGI-2 was immunoprecipitated by using rabbit polyclonal antisera raised against the WW domains of S-SCAM (synaptic scaffolding molecule), the rat homologue of MAGI-2 (27). PTEN was immunoprecipitated by using rabbit polyclonal antisera (28). Immunoblots were analyzed by using rabbit polyclonal antisera raised against a GST-MAGI-2 fusion protein that spans the WW domains and first two PDZ domains.
Pulse–chase experiments were performed in 293T cells by transfection with appropriate plasmids. Cells were cultured for 24 h with 500 μCi of [35S]methionine in methionine-free media, then chased with unlabeled complete media for 0–72 h. Cells were lysed, and PTEN was immunoprecipitated by using anti-FLAG antibody.
Results
Isolation of the MAGUK Protein MAGI-2 as a PTEN-Interacting Protein.
To identify candidate proteins that bind PTEN, we performed a yeast two-hybrid screen using a PTEN/Gal4 fusion protein as bait. We isolated two different cDNAs from overlapping regions of AIP-1. Both clones interacted with PTEN/Gal4 but not with a control lamin C/Gal4 protein in the yeast two-hybrid assay, as measured by growth in nutrient depleted (His−) media and by expression of β-galactosidase (not shown). AIP-1 is a member of the MAGUK family of proteins. It contains one guanylate kinase domain, two WW domains, and six PDZ domains (Fig. 1A) and is expressed at high levels in brain and at lower levels in all tissues tested, including prostate (26). The closest human homologue of AIP-1 is AIP-3 (also called BAP1), which also was isolated on the basis of its interaction with atrophin-1 (26, 29). The rodent homologues of AIP-1 and AIP-3, respectively are S-SCAM (synaptic scaffolding molecule) (27), which binds to a proline-rich adaptor protein, SAPAP, involved in membrane targeting of neuronal cell proteins, and MAGI-1 (30). To clarify the nomenclature for this growing family of inverted repeat MAGUKs, we propose that AIP-1 be renamed MAGI-2.
Figure 1.

PTEN binds MAGI-2 through a PDZ domain-mediated interaction. (A) Structure of MAGI-2. The two clones isolated in the two-hybrid screen (clones 4.1 and 20.1) are indicated by arrows. PDZ0 indicates a probable PDZ domain that does not have the consensus GLGF sequence. The numbering of PDZ domains 1–5 is based on the previously published nomenclature for MAGI-1 (30). GuK, guanylate kinase domain. (B) 35S-labeled in vitro transcribed/translated MAGI-2 protein from clone 20.1 and clones of individual PDZ domains 2 or 4 was pulled down with GST alone or full-length PTEN-GST beads. Input represents 20% of the protein used. The bottom panel shows Coomassie staining of protein bound to beads. (C) 293T cells were transfected with wild-type (wt) or mutant FLAG-PTEN constructs and HA-MAGI-2. Lysates were immunoprecipitated (IP) with anti-FLAG antibody and immunoblotted with anti-HA antibody 12CA5 to detect MAGI-2 or anti-FLAG antibody to detect PTEN. (D) Transfection and immunoprecipitation (IP) were performed in 293T cells as indicated in C. (E) MAGI-2 was immunoprecipitated (IP) from the homogenate of fresh mouse brain (lane 4) by using antisera raised against the WW domains of S-SCAM (synaptic scaffolding molecule), the rat homologue of MAGI-2 (27). PTEN was immunoprecipitated (lane 5) by using polyclonal antisera (28) or an independently derived PTEN antibody (19) (not shown). Anti-Akt polyclonal antisera (New England Biolabs) (lane 3), pre-IP lysate (lane 6), and no lysate (lane 7) were used as controls. Immunoblots were analyzed by using rabbit polyclonal antisera against GST-MAGI-2.
MAGI-2/PTEN Complexes.
Sequence analysis of the two MAGI-2 clones that bound PTEN showed that the smallest clone isolated (20.1) encodes PDZ domains 2–4, suggesting that one or more of these three PDZ domains is responsible for binding to PTEN. We tested this hypothesis by asking whether a GST-PTEN fusion protein could bind to individual PDZ domains isolated from MAGI-2. As expected, protein translated in vitro from clone 20.1 bound to GST-PTEN beads but not with GST alone (Fig. 1B), confirming the results from the two-hybrid experiment. Furthermore, GST-PTEN bound to PDZ domain 2 specifically.
To determine whether binding between MAGI-2 and PTEN also occurs in cells, we performed coimmunoprecipitation experiments. A full-length HA-MAGI-2 clone was transfected into 293T cells together with wild-type PTEN or PTEN C124S constructs, both of which contain a FLAG epitope. HA-MAGI-2 protein was detected in anti-FLAG immunoprecipitates of wild-type PTEN and PTEN C124S, but not in cells that were not transfected with PTEN (Fig. 1C). We asked whether PTEN C-terminal truncation mutants that lack the PDZ binding motif bind to MAGI-2. We created two truncation mutants, PTEN 1–337 and PTEN 1–377, which lack 66 or 26 aa from the C terminus of PTEN based on mutations identified in human tumors (1, 31). HA-MAGI-2 coprecipitated with wild-type PTEN but not with PTEN 1–337 or PTEN 1–377 (Fig. 1D). These results provide evidence that the MAGI-2/PTEN complex is formed by direct interaction between PDZ domain 2 of MAGI-2 and the PDZ binding motif of PTEN.
We performed coimmunoprecipitation experiments of the endogenous proteins from mouse brain tissue (Fig. 1E). 293T cells expressing HA-MAGI-2 served to identify the size of MAGI-2. First, we confirmed that endogenous MAGI-2 protein was successfully immunoprecipitated by using antisera directed against the WW domains and immunoblotted with a second antibody raised against a GST fusion protein spanning the WW domains and PDZ domains 1 and 2. Endogenous MAGI-2 also was detected by immunoblotting of whole-cell mouse brain lysates. If either MAGI-2 antibody was preincubated with the immunogen before immunoblotting, no signal corresponding to MAGI-2 was observed (not shown). We observed no MAGI-2 signal when mouse brain lysate was not included in the immunoprecipitation, eliminating the possibility that the 180-kDa band represents Ig. MAGI-2 was detected in immunoprecipitates by using two different antisera directed against PTEN but not by using antisera against the unrelated protein Akt, indicating that native MAGI-2/PTEN complexes exist in cells. The identity of the 100-kDa protein in the MAGI-2 and PTEN immunoprecipitates is unknown but may represent a MAGI-2 isoform or another MAGUK family member capable of binding to PTEN.
PTEN and MAGI-2 Are Localized at Tight Junctions.
Because PTEN regulates the PI3-kinase/Akt pathway through lipid phosphatase activity, it presumably requires membrane localization to gain access to its substrates. We addressed this issue in MDCK kidney epithelial cells because these cells grow as a polarized monolayer in vitro and form tight junctions, where many MAGUK proteins are localized. Using confocal microscopy, we observed intense staining of FLAG-PTEN in discrete membranous regions at the site of cellular projections (Fig. 2). Diffuse staining also was observed in the cytoplasm, as previously reported (3, 28). HA-MAGI-2 was localized primarily at the membrane, but diffuse nuclear staining also was seen in some cells. Studies using an antibody against the tight junction protein ZO-1 gave a membrane staining pattern similar to MAGI-2, supporting the notion that MAGI-2 is localized to tight junctions, like other MAGUKs. Dual color staining of transfected 293 cells confirmed that PTEN and MAGI-2 colocalize at the membrane (not shown). The fact that the MAGI-2/PTEN complex is present at tight junctions is noteworthy in light of evidence that 3′ phosphoinositides and activated Akt also are localized in this region of the membrane (32). The significance of the nuclear staining is unknown but suggests that a fraction of MAGI-2 protein may shuttle between the membrane and the nucleus. Recent precedent for regulation of MAGUKs in this fashion comes from the Drosophila discs large protein (DLG) whose membrane localization is regulated by phosphorylation (33).
Figure 2.

Subcellular localization of PTEN and MAGI-2. MDCK cells were plated onto fibronectin-treated cover slips, then transfected with FLAG-PTEN or HA-MAGI-2. After 24 h the cells were washed, fixed in paraformaldehyde, permeabilized in 0.2% Triton X-100, and blocked in 3% BSA in PBS containing 1 mM CaCl2 and 1 mM MgCl2. Cells were incubated in primary antibody in blocking buffer for 1 h at 37°C. Cells were washed in blocking solution plus 0.2% Triton. Cells were incubated with secondary antibody in blocking buffer for 45 min, washed, and then visualized by using a Zeiss LSM310 laser scanning confocal microscope.
MAGI-2 Enhances the Efficiency of PTEN in Repression of Akt.
The confocal microscopy studies demonstrate that MAGI-2 and PTEN both are localized to the plasma membrane at the appropriate site for access to lipid substrates and regulation of Akt. If MAGI-2 assembles a signaling complex, we reasoned that overexpression of MAGI-2 should enhance the efficiency of PTEN, particularly when present at limiting amounts. We have observed that PTEN represses Akt activation in 293T cells (16), and that plasmid doses as low as 100 ng are sufficient to achieve maximal suppression of the pathway (not shown). We examined the effect of MAGI-2 in this assay by cotransfection with limiting doses of PTEN. In the absence of additional MAGI-2, 100 ng of PTEN plasmid suppressed Akt kinase activity more than 20-fold, whereas 1 ng had minimal activity, indicating that PTEN was no longer saturating at this dose. When MAGI-2 plasmid was added, additional suppression of Akt was observed at the 1-ng PTEN dose but not at 100 ng (Fig. 3). MAGI-2 had no effect on Akt activity in the absence of transfected PTEN or when the PTEN 1–377 mutant, which does not bind MAGI-2, was used. These data demonstrate that MAGI-2 enhances the activity of low levels of PTEN. The fact that this effect is not seen at higher PTEN plasmid doses is most likely because of saturating amounts of PTEN protein at the membrane when expressed at supraphysiologic levels.
Figure 3.

Effect of MAGI-2 on PTEN activity. (A) 293T cells were transfected with MAGI-2, HA-Akt, and different concentrations of wild-type or mutant PTEN plasmid as indicated. The Akt kinase activity was measured by immunoprecipitation using an anti-HA antibody, followed by a kinase assay using histone H2B as substrate. MAGI-2 and HA-Akt expression was verified by immunoblotting. (B) Quantitative results from two independent experiments are shown.
The PTEN PDZ Binding Motif Is Required for Complete Akt Suppression.
If binding to MAGI-2 plays a role in PTEN function, then C-terminal PTEN truncation mutants that cannot bind MAGI-2 should be defective in Akt regulation. We previously have shown that stable expression of wild-type PTEN blocks serum-induced Akt activation (16). Consistent with our model, PTEN 1–377 (Fig. 4A) and PTEN 1–337 (not shown) were unable to suppress serum-induced Akt activation in this assay. However, the level of PTEN 1–377 and PTEN 1–337 proteins was consistently lower than wild-type PTEN in multiple cell lines and in different cell types. This finding could not be explained by abnormalities in transcription or translation because both mutants were expressed at levels comparable to wild-type PTEN in transient transfection experiments (Fig. 1D). Therefore, we hypothesized that reduced expression was caused by a change in protein stability. To eliminate any role of the growth suppressive effect of PTEN (and potential counterselection against expression), the PTEN 1–377 truncation was engineered into the catalytically inactive C124S mutant, which is not growth suppressive. In three independent experiments, the half-life of full-length PTEN C124S protein was 48–72 h, compared with 12–24 h for PTEN C124S 1–377 (Fig. 4B).
Figure 4.

The PTEN PDZ-binding motif is required for complete Akt suppression. (A) Rat-1 fibroblasts stably expressing wild-type (WT) FLAG-PTEN, FLAG-PTEN 1–377, or Neo control were serum-starved overnight, then challenged with serum for 30 min. Phospho-Akt, total Akt, and PTEN levels were measured by immunoblot (16). (B) Pulse–chase experiments were performed in 293T cells by transfection with indicated plasmids. PTEN was immunoprecipitated by using anti-FLAG antibody and visualized by autoradiography. Signals quantitated by a PhosphorImager are shown relative to time 0 of the chase. (C) NIH 3T3 cells stably expressing indicated PTEN proteins were harvested after serum stimulation at indicated times and processed as described in A, except anti-PTEN antibody (Santa Cruz Biotechnology) was used to measure PTEN expression.
Although the PTEN 1–377 mutant is clearly defective in the Akt suppression assay, the instability of the protein makes it difficult to implicate loss of MAGI-2 binding as the mechanism for loss of function. We noted that PTEN 1–394, which lacks only the last 9 aa, is expressed at levels comparable to wild-type PTEN (Fig. 4C). Using the serum-induced Akt activation assay, we observed that Akt suppression was not as complete in cells expressing PTEN 1–394 compared with wild-type PTEN at multiple time points (Fig. 4C). Similar results were obtained in three independent experiments, and control immunoblots confirmed that levels of both Akt and PTEN protein were comparable in all lanes at all time points. These results indicate that PTEN 1–394 is a partial loss of function mutant and establish a role for the PDZ-binding motif in maintaining the efficiency of PTEN-mediated Akt repression.
The PDZ Binding Motif of PTEN Affects Protein Stability.
Studies of the Drosophila scaffold protein InaD indicate that the stability of its partner proteins are enhanced through PDZ domain-mediated binding (24). To determine whether the stability of the PTEN 1–377 mutant is affected by PDZ domain binding, we added the 7 aa from the C terminus of PTEN (HTQITKV), which includes the PDZ-binding motif, back to PTEN C124S 1–377 (now called PTEN C124S Δ377–396). Addition of this motif restored the PTEN-MAGI-2 interaction in the coimmunoprecipitation assay (Fig. 5B). Remarkably, addition of this motif was sufficient to restore stable, high levels of PTEN C124S 1–377 protein expression in PTEN-positive and PTEN-negative cells (Fig. 5C) and to increase the half-life of PTEN C124S 1–377 in pulse–chase experiments (not shown).
Figure 5.

The PDZ binding motif of PTEN affects protein stability. (A) Schematic diagram of PTEN mutants. WT, wild type. (B) Coimmunoprecipitation experiments were performed in 293T cells transfected with the indicated plasmids as described in Fig. 1. IP, immunoprecipitation. (C) Rat-1 (Upper) and LNCaP (Lower) cells were infected with retrovirus expressing the indicated PTEN mutant, then selected in G418. Lysates were analyzed for expression of FLAG-PTEN at multiple time points. Results from the 25-day time point are shown.
Discussion
Our data support a model for regulation of PTEN function through binding to the MAGUK family protein MAGI-2 (see Fig. 6, which is published as supplementary material on the PNAS web site, www.pnas.org). Because MAGI-2 expression is restricted to certain tissues whereas PTEN is expressed ubiquitously, there may be other PDZ domain-containing proteins that bind PTEN (34). This model is conceptually similar to the regulation of photoreceptor signaling protein levels in the fly eye by the multi-PDZ domain scaffold protein InaD (24). InaD assembles a series of proteins involved in transmission of photoreceptor signals, each binding a distinct PDZ domain, into a multisubunit signaling complex. InaD gene deletion causes a slowed rate of photoreceptor signal transduction. Flies with InaD alleles containing mutations in individual PDZ domains express reduced levels of the respective binding protein, consistent with the notion that binding to the PDZ domain protects the signaling protein from degradation. Because MAGI-2 contains six potential PDZ domains and two WW domains, it is likely to bind other proteins in addition to PTEN. Because WW domains are known to bind proline-rich motifs as well as phospho-serine/threonine residues (35, 36), potential MAGI-2-binding proteins are not restricted to those containing PDZ domain-binding motifs. Thus, MAGI-2 may function as a scaffold protein that could assemble a multisubunit signaling complex.
Many mutations in tumors affect the C terminus of PTEN, including three independent missense mutations that target the stop codon and result in 8 aa added to the PDZ-binding motif (37–39). These mutations are predicted to disrupt binding to PDZ domains. We provide experimental evidence implicating MAGI-2 in PTEN function by showing that expression of MAGI-2 enhances the ability of PTEN to suppress Akt activation. We also demonstrate that PTEN mutants that fail to bind MAGI-2 are defective in their ability to completely suppress Akt activation. It is important to note that these mutants have partial rather than complete loss of function, a phenotype consistent with the concept that scaffold proteins optimize activity through a signaling pathway but are not essential. Studies using C-terminal PTEN truncation mutants report conflicting results regarding the role of the PDZ-binding motif (9, 40, 41). However, all of these experiments rely on overexpression of the mutant PTEN protein, which may mask any loss of function phenotype that results from failure to bind a scaffold protein. The most definitive strategy to address this issue is through targeted mutation in the mouse germ line. Our data showing partial loss of function would predict that a PDZ-binding mutant “knockin” may have a less severe phenotype than the embryonic lethality observed with complete PTEN knockouts. The recently determined crystal structure of PTEN shows the presence of a lipid binding C2 domain postulated to play a role in membrane localization (42). Data presented here are consistent with the hypothesis that both the C2 domain and the PDZ-binding motif contribute to the recruitment and maintenance of membrane association of PTEN.
Our studies also have uncovered a role for the C terminus in maintaining protein stability and are in agreement with a recent report (41). We postulate that these truncations cause loss of function because of a shortened protein half-life as well as an inability to dock to a PDZ-domain scaffold protein like MAGI-2. Studies of PTEN-deficient embryonic stem cells indicate that a 50% reduction in PTEN protein level may be sufficient to produce a tumor prone phenotype (18). Our data with the PTEN 1–394 mutant establish that the PDZ-binding motif is not required for stability, yet adding the PDZ-binding motif back to an unstable PTEN truncation mutant is sufficient to restore stability. Alternative considerations include the potential role of phosphorylation in regulating protein stability. More work is needed to define the variables that control PTEN stability.
Inactivation of PTEN through protein destabilization is reminiscent of other tumor suppressors such as p53 and p27Kip1, which have reduced stability in some cancer cells (43, 44). Although similar in concept, the PTEN example discussed here differs in that the genetic abnormality occurs in PTEN itself rather than in the protein degradation machinery. Certain human tumors have reduced PTEN protein levels without gene mutation or loss of mRNA expression (45). Growing evidence indicates that PTEN inactivation can occur through a range of mechanisms, including gene deletion or mutation, loss of mRNA expression, and reduced protein stability.
Loss of function mutations in the Drosophila MAGUK protein discs large (DLG) cause outgrowths of the imaginal disk, leading to the concept that DLG functions as a tumor suppressor (46). Our observation that MAGI-2 enhances PTEN function raises the possibility that MAGI-2 itself may act as a tumor suppressor. MAGI-2 has been localized to chromosome 7q21 (26), a region that is deleted in uterine leiomyomas, prostate cancer, and glioblastoma (47–50). Definitive tests of this hypothesis will require analysis of MAGI-2 in clinical material and/or the creation of MAGI-2 loss-of-function mutations.
Supplementary Material
Acknowledgments
We thank Yoshimi Takai and William Sellers for antibodies, and Matt Schibler, Chris Tran, Jing Ou, Hongbing Wang, and Jiayu Liao for technical assistance. We are grateful to Michael Carey, Genhong Cheng, Kristen Senechal, Ke Shuai, Ayyappan Rajasekaran, and Owen Witte for advice and Lisa Herrera-Dove for manuscript preparation. This work is supported by grants from the Department of Defense, National Cancer Institute, and the Association for the Cure of Cancer of the Prostate. C.L.S. is a Scholar of the Leukemia Society of America.
Abbreviations
- MAGI
membrane associated guanylate kinase inverted
- PI
phosphatidylinositol
- MAGUK
membrane-associated guanylate kinase
- AIP
atrophin interacting protein
- HA
hemagglutinin
- GST
glutathione S-transferase
Note Added in Proof
The Murine MAGI-2 recently was cloned as described in ref. 51.
References
- 1.Li J, Yen C, Liaw D, Podsypanina K, Bose S, Wang S I, Puc J, Miliaresis C, Rodgers L, McCombie R, et al. Science. 1997;275:1943–1947. doi: 10.1126/science.275.5308.1943. [DOI] [PubMed] [Google Scholar]
- 2.Steck P A, Pershouse M A, Jasser S A, Yung W K, Lin H, Ligon A H, Langford L A, Baumgard M L, Hattier T, Davis T, et al. Nat Genet. 1997;15:356–362. doi: 10.1038/ng0497-356. [DOI] [PubMed] [Google Scholar]
- 3.Li D M, Sun H. Cancer Res. 1997;57:2124–2129. [PubMed] [Google Scholar]
- 4.Liaw D, Marsh D J, Li J, Dahia P L, Wang S I, Zheng Z, Bose S, Call K M, Tsou H C, Peacocke M, et al. Nat Genet. 1997;16:64–67. doi: 10.1038/ng0597-64. [DOI] [PubMed] [Google Scholar]
- 5.Marsh D J, Dahia P L, Zheng Z, Liaw D, Parsons R, Gorlin R J, Eng C. Nat Genet. 1997;16:333–334. doi: 10.1038/ng0897-333. [DOI] [PubMed] [Google Scholar]
- 6.Di Cristofano A, Pesce B, Cordon-Cardo C, Pandolfi P P. Nat Genet. 1998;19:348–355. doi: 10.1038/1235. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki A, de la Pompa J L, Stambolic V, Elia A J, Sasaki T, del Barco Barrantes I, Ho A, Wakeham A, Itie A, Khoo W, et al. Curr Biol. 1998;8:1169–1178. doi: 10.1016/s0960-9822(07)00488-5. [DOI] [PubMed] [Google Scholar]
- 8.Podsypanina K, Ellenson L H, Nemes A, Gu J, Tamura M, Yamada K M, Cordon-Cardo C, Catoretti G, Fisher P E, Parsons R. Proc Natl Acad Sci USA. 1999;96:1563–1568. doi: 10.1073/pnas.96.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Furnari F B, Lin H, Huang H S, Cavenee W K. Proc Natl Acad Sci USA. 1997;94:12479–12484. doi: 10.1073/pnas.94.23.12479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Myers M P, Stolarov J P, Eng C, Li J, Wang S I, Wigler M H, Parsons R, Tonks N K. Proc Natl Acad Sci USA. 1997;94:9052–9057. doi: 10.1073/pnas.94.17.9052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maehama T, Dixon J E. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- 12.Ali I U, Schriml L M, Dean M. J Natl Cancer Inst. 1999;91:1922–1932. doi: 10.1093/jnci/91.22.1922. [DOI] [PubMed] [Google Scholar]
- 13.Tamura M, Gu J, Matsumoto K, Aota S, Parsons R, Yamada K M. Science. 1998;280:1614–1617. doi: 10.1126/science.280.5369.1614. [DOI] [PubMed] [Google Scholar]
- 14.Stambolic V, Suzuki A, de la Pompa J L, Brothers G M, Mirtsos C, Sasaki T, Ruland J, Penninger J M, Siderovski D P, Mak T W. Cell. 1998;95:29–39. doi: 10.1016/s0092-8674(00)81780-8. [DOI] [PubMed] [Google Scholar]
- 15.Myers M P, Pass I, Batty I H, Van der Kaay J, Stolarov J P, Hemmings B A, Wigler M H, Downes C P, Tonks N K. Proc Natl Acad Sci USA. 1998;95:13513–13518. doi: 10.1073/pnas.95.23.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu X, Senechal K, Neshat M S, Whang Y E, Sawyers C L. Proc Natl Acad Sci USA. 1998;95:15587–15591. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Haas-Kogan D, Shalev N, Wong M, Mills G, Yount G, Stokoe D. Curr Biol. 1998;8:1195–1198. doi: 10.1016/s0960-9822(07)00493-9. [DOI] [PubMed] [Google Scholar]
- 18.Sun H, Lesche R, Li D M, Liliental J, Zhang H, Gao J, Gavrilova N, Mueller B, Liu X, Wu H. Proc Natl Acad Sci USA. 1999;96:6199–6204. doi: 10.1073/pnas.96.11.6199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ramaswamy S, Nakamura N, Vazquez F, Batt D B, Perera S, Roberts T M, Sellers W R. Proc Natl Acad Sci USA. 1999;96:2110–2115. doi: 10.1073/pnas.96.5.2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Songyang Z, Fanning A S, Fu C, Xu J, Marfatia S M, Chishti A H, Crompton A, Chan A C, Anderson J M, Cantley L C. Science. 1997;275:73–77. doi: 10.1126/science.275.5296.73. [DOI] [PubMed] [Google Scholar]
- 21.Hillier B J, Christopherson K S, Prehoda K E, Bredt D S, Lim W A. Science. 1999;284:812–815. [PubMed] [Google Scholar]
- 22.Gomperts S N. Cell. 1996;84:659–662. doi: 10.1016/s0092-8674(00)81043-0. [DOI] [PubMed] [Google Scholar]
- 23.Craven S E, Bredt D S. Cell. 1998;93:495–498. doi: 10.1016/s0092-8674(00)81179-4. [DOI] [PubMed] [Google Scholar]
- 24.Tsunoda S, Sierralta J, Sun Y, Bodner R, Suzuki E, Becker A, Socolich M, Zuker C S. Nature (London) 1997;388:243–249. doi: 10.1038/40805. [DOI] [PubMed] [Google Scholar]
- 25.Pawson T, Scott J D. Science. 1997;278:2075–2080. doi: 10.1126/science.278.5346.2075. [DOI] [PubMed] [Google Scholar]
- 26.Wood J D, Yuan J, Margolis R L, Colomer V, Duan K, Kushi J, Kaminsky Z, Kleiderlein J J, Sharp A H, Ross C A. Mol Cell Neurosci. 1998;11:149–160. doi: 10.1006/mcne.1998.0677. [DOI] [PubMed] [Google Scholar]
- 27.Hirao K, Hata Y, Ide N, Takeuchi M, Irie M, Yao I, Deguchi M, Toyoda A, Sudhof T C, Takai Y. J Biol Chem. 1998;273:21105–21110. doi: 10.1074/jbc.273.33.21105. [DOI] [PubMed] [Google Scholar]
- 28.Whang Y E, Wu X, Suzuki H, Reiter R E, Tran C, Vessella R L, Said J W, Isaacs W B, Sawyers C L. Proc Natl Acad Sci USA. 1998;95:5246–5250. doi: 10.1073/pnas.95.9.5246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shiratsuchi T, Futamura M, Oda K, Nishimori H, Nakamura Y, Tokino T. Biochem Biophys Res Commun. 1998;247:597–604. doi: 10.1006/bbrc.1998.8603. [DOI] [PubMed] [Google Scholar]
- 30.Dobrosotskaya I, Guy R K, James G L. J Biol Chem. 1997;272:31589–31597. doi: 10.1074/jbc.272.50.31589. [DOI] [PubMed] [Google Scholar]
- 31.Rasheed B K, Stenzel T T, McLendon R E, Parsons R, Friedman A H, Friedman H S, Bigner D D, Bigner S H. Cancer Res. 1997;57:4187–4190. [PubMed] [Google Scholar]
- 32.Watton S J, Downward J. Curr Biol. 1999;9:433–436. doi: 10.1016/s0960-9822(99)80192-4. [DOI] [PubMed] [Google Scholar]
- 33.Koh Y H, Popova E, Thomas U, Griffith L C, Budnik V. Cell. 1999;98:353–363. doi: 10.1016/s0092-8674(00)81964-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Adey N B, Huang L, Ormonde P A, Baumgard M L, Pero R, Byreddy D V, Tavtigian S V, Bartel P L. Cancer Res. 2000;60:35–37. [PubMed] [Google Scholar]
- 35.Macias M J, Hyvonen M, Baraldi E, Schultz J, Sudol M, Saraste M, Oschkinat H. Nature (London) 1996;382:646–649. doi: 10.1038/382646a0. [DOI] [PubMed] [Google Scholar]
- 36.Lu P J, Zhou X Z, Shen M, Lu K P. Science. 1999;283:1325–1328. doi: 10.1126/science.283.5406.1325. [DOI] [PubMed] [Google Scholar]
- 37.Tohma Y, Gratas C, Biernat W, Peraud A, Fukuda M, Yonekawa Y, Kleihues P, Ohgaki H. J Neuropathol Exp Neurol. 1998;57:684–689. doi: 10.1097/00005072-199807000-00005. [DOI] [PubMed] [Google Scholar]
- 38.Zhou X P, Li Y J, Hoang-Xuan K, Laurent-Puig P, Mokhtari K, Longy M, Sanson M, Delattre J Y, Thomas G, Hamelin R. Int J Cancer. 1999;84:150–154. doi: 10.1002/(sici)1097-0215(19990420)84:2<150::aid-ijc10>3.0.co;2-#. [DOI] [PubMed] [Google Scholar]
- 39.Schmidt E E, Ichimura K, Goike H M, Moshref A, Liu L, Collins V P. J Neuropathol Exp Neurol. 1999;58:1170–1183. doi: 10.1097/00005072-199911000-00007. [DOI] [PubMed] [Google Scholar]
- 40.Morimoto A M, Berson A E, Fujii G H, Teng D H, Tavtigian S V, Bookstein R, Steck P A, Bolen J B. Oncogene. 1999;18:1261–1266. doi: 10.1038/sj.onc.1202441. [DOI] [PubMed] [Google Scholar]
- 41.Georgescu M M, Kirsch K H, Akagi T, Shishido T, Hanafusa H. Proc Natl Acad Sci USA. 1999;96:10182–10187. doi: 10.1073/pnas.96.18.10182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lee J O, Yang H, Georgescu M M, Di Cristofano A, Maehama T, Shi Y, Dixon J E, Pandolfi P, Pavletich N P. Cell. 1999;99:323–334. doi: 10.1016/s0092-8674(00)81663-3. [DOI] [PubMed] [Google Scholar]
- 43.Pagano M, Tam S W, Theodoras A M, Beer-Romero P, Del Sal G, Chau V, Yew P R, Draetta G F, Rolfe M. Science. 1995;269:682–685. doi: 10.1126/science.7624798. [DOI] [PubMed] [Google Scholar]
- 44.Scheffner M, Werness B A, Huibregtse J M, Levine A J, Howley P M. Cell. 1990;63:1129–1136. doi: 10.1016/0092-8674(90)90409-8. [DOI] [PubMed] [Google Scholar]
- 45.Dahia P L, Aguiar R C, Alberta J, Kum J B, Caron S, Sill H, Marsh D J, Ritz J, Freedman A, Stiles C, et al. Hum Mol Genet. 1999;8:185–193. doi: 10.1093/hmg/8.2.185. [DOI] [PubMed] [Google Scholar]
- 46.Woods D F, Bryant P J. Cell. 1991;66:451–464. doi: 10.1016/0092-8674(81)90009-x. [DOI] [PubMed] [Google Scholar]
- 47.Ishwad C S, Ferrell R E, Davare J, Meloni A M, Sandberg A A, Surti U. Genes Chromosomes Cancer. 1995;14:51–55. doi: 10.1002/gcc.2870140109. [DOI] [PubMed] [Google Scholar]
- 48.Cui J, Deubler D A, Rohr L R, Zhu X L, Maxwell T M, Changus J E, Brothman A R. Cancer Genet Cytogenet. 1998;107:51–60. doi: 10.1016/s0165-4608(98)00074-0. [DOI] [PubMed] [Google Scholar]
- 49.Cunningham J M, Shan A, Wick M J, McDonnell S K, Schaid D J, Tester D J, Qian J, Takahashi S, Jenkins R B, Bostwick D G, et al. Cancer Res. 1996;56:4475–4482. [PubMed] [Google Scholar]
- 50.Kim D H, Mohapatra G, Bollen A, Waldman F M, Feuerstein B G. Int J Cancer. 1995;60:812–819. doi: 10.1002/ijc.2910600615. [DOI] [PubMed] [Google Scholar]
- 51.Shoji H, Tsuchida K, Kishi H, Yamakawa N, Matsuzaki T, Liu Z, Nakamura T, Sugino H. J Biol Chem. 2000;275:5485–5492. doi: 10.1074/jbc.275.8.5485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}