

Abstract
Hepatocellular carcinoma (HCC), one of the most common and malignant tumors worldwide, is unresponsive to any of the available therapies. Using intact HCC cells as therapeutic targets, we isolated a novel peptide, denoted HCC79 (KSLSRHDHIHHH), from a phage display peptide library. HCC79 can bind to hepatoma cell membranes with high affinity and specificity. Remarkably, competitive binding assays demonstrated that HCC79 competed with HAb25, a specific antibody for HCC, in binding to hepatoma cells. The corresponding synthetic peptide did not inhibit tumor proliferation directly, but repressed tumor invasion significantly in a cell migration assay. Moreover, we explored the potential of the selected peptide to deliver a superantigen (SAg) to cancer cells, to attain a significant cell-targeting effect. When the peptide is fused to the TSST-1 SAg, the resulting fusion protein could bind to hepatoma cells with high affinity in vitro and improved the tumor inhibition effect by activating T lymphocyte cells in vitro and in vivo, compared with TSST-1 alone. Taken together, our results indicate that this peptide and its future derivatives may have the potential to be developed into highly specific therapeutic agents against cancer.
INTRODUCTION
Hepatocellular carcinoma (HCC) is one of the most common and fatal malignancies worldwide, with a mortality rate of ≥94%. It exhibits a dangerous latency and an early translocation potential. In China, HCC comprises more than 2% of all disease types (1).
The main goal of cancer therapy currently is to eradicate cancer cells while sparing normal tissues. This process requires the selective targeting of cancer cells at the site of malignancy (2). Certain membrane proteins are expressed specifically in cancer cells, and likely house unique molecular cell surface markers that are useful as anticancer targets. Some studies have shown that generating specific monoclonal and genetically engineered antibodies can improve tumor targeting (3). An anti-HCC antibody–targeted agent has been used for clinical diagnoses in China and has shown favorable cellular carcinoma-targeting results (1,4–6). Additionally, a novel genetically engineered antibody from a single-chain disulfide-stabilized Fv (scdsFv) of the antihepatoma monoclonal antibody HAb25 was developed in our laboratory (7). Despite the exquisite targeting specificity of this antibody, however, its high molecular weight, low tissue penetration, and poor cellular uptake have impeded its clinical application.
Small peptides that recognize tumor cells selectively should overcome some of these antibody limitations (8). The effective tissue penetration of short synthetic peptides, in combination with their selective binding capacity for the targeted cancer cells, make these agents ideal candidates for delivering therapeutic molecules. Moreover, in contrast to antibody approaches, peptides are nearly invisible to the immune system and may cause few or no side effects (9). Therefore, small peptides represent more appropriate therapeutic delivery vehicles than larger molecules, such as antibodies.
Screening phage display peptide libraries is an effective and simple method for isolating cell-targeting peptides. The phage display technique involves producing libraries of peptides displayed on phage. These libraries may contain as many as 1010 different peptides. Peptide selection from these libraries can provide dozens to hundreds of potential cellular binders to many different sites on a target protein (10,11). Most cell-binding peptide screening is based on known targets, such as Her2 and ErbB-2; very few tumor-specific antigens have been identified on the HCC membrane. Several research groups have reported the use of intact cells and the selection of cell surface–binding peptides from phage display libraries (12–16). The panning of the phage-display libraries on intact cells is more likely to enrich for peptides that bind to cell surface receptors in their native conformation, and this method of selection requires no prior knowledge about the targeted receptors.
Toxic shock syndrome toxin 1 (TSST-1), a bacterial SAg produced by Staphylococcus aureus, has the smallest molecular weight in the SAg family. It stimulates a robust activation of T cells bearing certain TCR Vβ elements when it is bound as an unprocessed protein outside the antigenic groove of MHC II molecules to generate tumor-suppressive cytokines (for example, TNF-α and IFN-γ), even at picomolar concentrations (17,18). Therefore, TSST-1–based antitumor strategies may offer promising therapeutic options to HCC patients (19–21). SAgs preferentially direct cytotoxicity against MHC II–positive cells, however, so the in vivo administration of intact SAgs at the therapeutic dosage may induce cytotoxicity against normal cells. Therefore, successful employment of TSST-1 in tumor immunotherapy likely requires a targeting moiety. Selective delivery of TSST-1 to cancer cells would improve their efficacy and minimize any potential adverse side effects (22).
In this study, we developed a strategy for targeting TSST-1 to HCC tumor cells using a short peptide. Initially, we searched for peptides that bound specifically to intact hepatoma cells. A random 12-mer peptide library displayed on the surfaces of filamentous phages (M13) was screened by biopanning against the intact hepatoma cell line SMMC-7721. From these experiments, we isolated the peptide HCC79 (KSLSRHDHIHHH), which not only binds the hepatoma cell specifically, but also inhibits tumor invasion significantly. Subsequently, we explored the potential of HCC79 to deliver TSST-1 to cancer cells to estimate the peptide’s cell-targeting effect. When the peptide was fused to TSST-1, the resulting fusion protein bound to hepatoma cells with high affinity in vitro and achieved a better tumor inhibition effect by activating T lymphocytes than TSST-1 alone in vitro and in vivo.
MATERIAL AND METHODS
Biological Material and Reagents
Primers for preparing 12T encoding the HCC79/TSST-1 fusion gene were synthesized by Bioasia (Shanghai, China). The sequences are as follows: forward P1 5′-GCAGAATTCATGAAGTCTCTTAGTCGGCATGATCATATTCATCATCATGGCGGCTCTACAAACGATAATATAAAGGAT-3′ and reverse P2 5′-CACAAGCTTTTAATTAATTTCTGCTTCTATAG-3′. The S. aureus FRI1169 genome DNA used as a template for amplifying the TSST-1 gene was extracted according to the methods described in the manufacturer’s protocol (Takara, Dalian, China). The 12-mer phage-displayed library was purchased from New England BioLabs (Beverly, MA, USA). The peptide HCC79 (KSLSRHDHIHHH) was synthesized by GL Biochem (Shanghai, China). The hepatoma cell lines SMMC-7721 and BEL-7402, the stomach carcinoma cell line BGC-823, the oral squamous carcinoma cell line KB, the normal liver cell line HL-02, and H22 tumor tissue were obtained from the Cell Library of China Union Medical University (Beijing, China). Balb/c mice were provided by the Animal Center of the Academy of Military Medical Sciences (Beijing, China). TSST-1, anti–TSST-1 rabbit serum, and scFv25-TSST-1 (DT) were produced in our laboratory (20). The HAb25 antibody used for the competition ELISA assay was donated by Dr. Sun Zhiwei (1,4–6). The horseradish peroxidase (HRP)-conjugated anti-M13 phage antibody was purchased from Amersham Biosciences (Uppsala, Sweden).
Screening of a 12-mer Phage Display Library
The procedure for screening the phage-displayed library was modified from the manufacturer’s protocol (New England BioLabs). The human hepatoma cell line SMMC-7721 was grown in RPMI 1640 medium supplemented with 15% FBS. The normal liver cell line HL-02 was grown in Dulbecco’s modified Eagle’s medium (Gibco, Middlesex, England) supplemented with 15% FBS and insulin (1 unit/mL). The HL-02 or SMMC-7721 cells were seeded in 96-well plates at a density of 5 × 105 cells/well. After an overnight incubation at 37 °C, the cells were fixed with ice-cold methanol and acetone solution (1:1) for 10 min at room temperature. Blocking buffer (100 μL) (PBS supplemented with 0.3% H2O2) was added to each well, and the plates were incubated at 4 °C for 30 min. After the plates were washed 3 times with PBS, the phage library containing 1012 clones was added to the HL-02 cell wells for pre-absorption, and the plate was agitated gently at room temperature for 1 h. The pre-absorbed library was then applied to the SMMC-7721–coated wells for specific screening. After thorough washing, the plate-bound phage clones were eluted with elution buffer (0.22 M Glycine-HCl, pH 2.2) and neutralized immediately. Three rounds of random plaque selections were performed. After selection, the individual plaques were subjected to analysis by phage ELISA and DNA sequencing, following amplification in E. coli ER2738.
Phage ELISA
For the phage ELISA assays, the human cell lines SMMC-7721, BEL-7402, and HL-02 were seeded on 96-well plates and blocked as described above. After blocking, the amplified phages (5 × 1012 pfu/mL) were added to the wells via 3 rounds of screening. The plates were incubated for 2 h at room temperature and washed 4 times with Tris-buffered saline (50 mM Tris, pH 7.5, 0.1% Tween 20). The peptide-bound phages were detected by ELISA using a HRP-conjugated anti-M13 monoclonal antibody.
Competition Binding Assay
The phage clones with the relatively higher affinities, as determined by the phage ELISA, were selected for subsequent competition assays. The phage clones alone (at a concentration of 1013 pfu/mL) or together with the HAb25 antibody were incubated with the SMMC-7721 cell–coated plates for 1 h at room temperature. BSA (100 μg/mL) was used as a negative control. After washing 3 times with PBST, HRP-conjugated anti-M13 phage antibody was added to incubate for 45 min at 37 °C. After washing 3 times with PBST, the reactions were supplemented with tetramethyl benzidine (TMB) and incubated for 5 min, and the absorbance at 450 nm of each well was measured by SpectraMaxPlus spectrophotometer (Molecular Device Corp., Sunnyvale, CA, USA).
Cell Migration Assay
The cell migration assay was conducted according to the previously described protocol, with some modifications (23). The SMMC-7721 cells were resuspended in RPMI 1640 containing 10% FBS and counted with a hemacyto-mometer[Anno1]. The lower compartment of the modified Boyden chambers (Millipore, Billerica, MA, USA) was filled with 200 μL RPMI 1640 containing 10% FBS and the supernatants of the NIH3T3 cell culture. An polycarbonate membrane (8.0-μM pore) coated with 0.2% matrix gel was inserted into the chamber. Cell suspension (200 μL) containing 5 × 104 cells, in the presence or absence of synthetic peptides, was added to the upper compartment of each chamber and incubated at 37 °C for 18 h. At the end of the incubation period, the cells on the upper surface of the filters were removed by scraping twice with a cotton tip. The membranes were then fixed and stained with Diff-Quick, placed on a microscope slide and covered with a coverslip. The cells that migrated across the membranes were counted under a light microscope. The data are presented as means ± standard errors of 5 separate counts under the microscope.
Construction, Expression, and Purification of Superantigen TSST-1 (12T)
The gene encoding 12T was amplified by PCR using the S. aureus FRI1169 genome DNA as the template and the P1 and P2 primers. The PCR product was cloned into the PBV220 vector and sequenced. E. coli DH5α competent cells were transformed with PBV220-12T and cultured in Luria-Bertani (LB) medium supplemented with 100 μg/mL ampicillin at 37 °C. The expression of the fusion protein was induced by raising the temperature to 42 °C for 4 h. The bacterial pellets containing the expressed protein were ultrasonicated on ice in TE buffer (20 mM Tris-HCl, pH 8.0, 1 mM EDTA) and centrifuged at 17000g rpm for 10 min at 4 °C. The resulting precipitates and supernatants were analyzed with SDS-PAGE electrophoresis and identified with Western blotting techniques. The supernatant containing 12T was purified with CM-Sepharose FF (Amersham Biosciences, Uppsala, Sweden), as described previously (20).
Binding Assays of 12T
Binding of 12T to the SMMC-7721, BGC-823, KB, and HL-02 cells (cell-ELISA assay) was performed as described previously (7). Briefly, the cells were immobilized on 96-well plates and blocked as described above. Purified 12T protein was then added to the wells (PBS, TSST-1, and scFV25-TSST-1 were used as controls) and incubated for 1 h at 37 °C. After washing 3 times with PBST, rabbit anti–TSST-1 serum were added and incubated for 45 min at 37 °C, followed by incubation for 30 min at 37 °C with the HRP-labeled sheep antirabbit antibodies. TMB was then added for 5 min, and the absorbance at 450 nm was measured by SpectraMaxPlus spectrophotometers (Molecular Device Corp.).
Cytotoxicity Assays In Vitro
Redirected T-cell cytotoxicity (superantigen-dependent cellular cytotoxicity) was analyzed following the protocol of the cell titer 96AQueous Non-Radioactive cell proliferation assay (Promega, Madison, WI, USA), with some minor modifications (7,24). Briefly, 5 × 103 SMMC-7721 cells and 4 × 104 human T cells were added to each well, followed by the addition of diluted 12T in a total volume of 200 mL. The cells were cultured for 4 to 5 days at 37 °C in a humidified 5% CO2 atmosphere. Each well was washed 3 times with RPMI 1640 to remove the effector cells and the dead target cells. This was followed by the addition of 100 mL/well of culture medium and 20 mL/well of a fresh mixture of MTS/phenazine methosulfate solution (Promega). The absorbance at 490 nm was recorded using a SpectraMaxPlus spectrophotometer (Molecular Device Corp.) after incubating for 1 to 4 h at 37 °C. Redirected T-cell cytotoxicity of 12T to SMMC-7721 cells by a different effector:target ratio was also analyzed as described above.
Mouse HCC Tumor Growth Inhibition In Vivo
BABL/c mice (male, 18–22 g) were injected beneath the armpit with 2 × 106 H22 tumor cells suspended in PBS. The mice were then randomized into separate groups (8 mice per group). On the following day, 3 doses of 12T (2, 0.4, and 0.08 nM per mouse) or control agents (TSST-1 or PBS) were injected in the tail vein 3 times every other day. The experimental groups were designated as 12T high-dose, middle-dose, group and low-dose groups; the controls included the TSST-1 and the PBS groups. On the 9th day after cell injection, mice were killed by cervical dislocation. Nodules formed by implanted tumors at the injection sites were isolated and weighed. Statistical significance between the test and control groups was determined using the Student t test.
RESULTS
Selection of HCC-Specific Binding Peptides
Displaying peptides on the surfaces of bacteriophages provides an opportunity to screen peptides with desired binding specificities. We used the HCC cell line as a target to screen peptides from a 12-mer random peptide phage library. To eliminate phages with a binding capacity for common cellular receptors, the library was pre-absorbed with the human normal liver cell line HL-02. Three rounds of screening were then performed. The output of each round of biopanning was calculated to prove the validity of bacteriophage peptide library screening. The library was enriched to 152-fold after 3 rounds of biopanning (Figure 1A), from which we selected 5 positive clones. These clones were tested for their binding aptitudes for the hepatoma cell lines SMMC-7721 and BEL-7402. The normal liver cell line HL-02 was used as a binding control. The present experiment showed that all 5 clones exhibited high binding affinities for the hepatoma cell lines and low binding affinities for the normal liver cells (Figure 1B). These 5 positive phage clones were then selected for sequence analyses. Interestingly, all clones showed perfect sequence identity (KSLSRHDHIHHH). The screened peptide was designated as HCC79. Possessing high hydrophobicity and histidine richness, HCC79 is similar to the VEGF receptor binding peptide, K237 (HYMYYHHYQHHL) (23).
Figure 1.

Screening of HCC-specific peptides from 12-mer peptide libraries. (A) Output assay for each plaque selection round. The output ratio was calculated as output number/input number. (B) Binding assay of positive clones to the hepatoma cell lines (SMMC-7721 and BEL-7402) and to the normal liver cell line (HL-02). The binding capacity was quantified by phage ELISA assay. The data are presented as means of 5 positive phage clones, and the error bars indicate standard deviation. (C) Competitive binding of phage clones to SMMC-7721 with HAb25, which was diluted at the starting concentration (100 μg mL−1). The inhibition rate was calculated as the signal of phage binding with versus without HAb25.
The hepatoma cell line SMMC-7721 is recognized by HAb25, an antibody that binds specifically to hepatoma cell membrane receptors (1). An HAb25 antibody labeled with 131I has been used clinically for HCC diagnosis and is reported to be very specific for hepatoma cell labeling (4). To further identify the binding specificity of the selected positive phage clones, we performed competition ELISA assays to analyze its competitive binding ability toward hepatoma cells against a gradient diluted HAb25. These results showed that the increased HCC79 binding activity was accompanied by a decrease in HAb25 concentration (Figure 1C), whereas BSA did not compete against HAb25 to bind the hepatoma cells. Therefore, this peptide is specific for HCC cells and may share binding sites with the HAb25 antibody.
Synthetic Peptide HCC79 Inhibits Tumor Invasion of HCC
We synthesized the HCC79 peptide (KSLSRHDHIHHH) to examine its therapeutical potential as an anti-HCC agent. Because HCC79 is a cell-specific binding peptide, we tested whether HCC79 could inhibit tumor cell growth directly by blocking the targeted membrane receptor. Our results revealed that HCC79 did not suppress HCC cell proliferation, however, even at high concentrations (200 μg/mL) (data not shown).
The peptide K237-HYMYYHHYQHHL, screened previously for binding to the VEGF receptor, has an antitu-mor invasion activity. Similar to K237-(HYMYYHHYQHHL), HCC79-(KSLSRHDHIHHH) contains multiple histidines. Thus, we performed a cell migration assay to identify whether HCC79 inhibits cell migration. The results showed that migration of the SMMC-7721 cells was reduced in the presence of HCC-79 in a dose-dependent manner (Figure 2). The inhibitory rates of 200, 40, and 8 μg/mL HCC79 on SMMC-7721 cell migration were 70.3%, 60.1%, and 57.6%, respectively. These findings suggest that the synthetic peptide was capable of significantly inhibiting tumor invasion.
Figure 2.
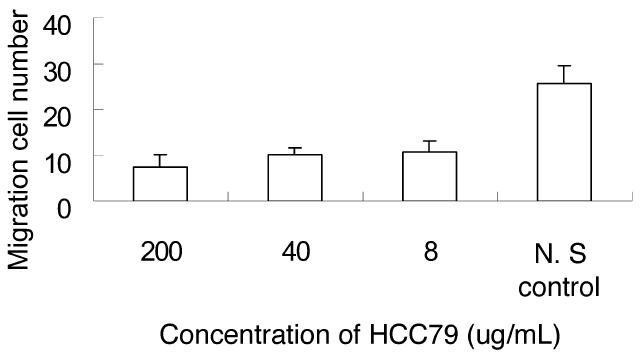
Inhibitory effect of peptide HCC79 on SMMC772 cell migration. The data presented are the means of 5 separate counts under a microscope, and the error bars indicate standard deviation.
12T Binds to Hepatoma Cells Efficiently and Specifically, Independent of Phage
All binding assays described above were performed using phages that formed the G-III protein and the 12-mer peptide fusion protein. To obtain the actual binding situation of HCC79 independent of the bound phage, we prepared the HCC79 and TSST-1 fusion protein with a Gly-Gly-Ser linker (12T). In the fusion protein, TSST-1 may serve as a binding detection tag and functions as an antitumor molecule to allow testing of the targeting effects of HCC79 in vitro and in vivo for its high potency to activate human T lymphocytes. 12T was expressed in bacterial cells, mostly as a soluble form, and was detected by Western blot procedures. The soluble protein in supernatant was purified by CM-Sepharose FF and analyzed by SDS-PAGE (Figure 3A). the purification process rendered a 90% pure fusion protein product.
Figure 3.

Characterization of the 12T fusion protein. (A) Purification of the 12T fusion protein. 12T was expressed in E. coli DH5α cells and analyzed by SDS-PAGE electrophoresis. Coomassie blue stain. Lanes 1–9 represent SDS-PAGE analysis of fusion protein expression. 1, 2: Total lysates of induced E. coli; 3: flowthrough of loaded proteins after purification; 4: proteins before purification; 5–9: various elutions of the 12T protein after purification. M indicates low molecular marker (Amersham). (B) Binding assay of 12T, scFv25-TSST-1 (DT), or TSST-1 alone to hepatoma cells under different concentrations (same starting concentration 3 μM, 5× serial dilution). (C) Binding assay of 12T to SMMC-7721, BGC823, KB versus normal liver cells by cell-ELISA assays under different concentrations (same starting concentration 3 μM, 5× serial dilution). The data are the means of triplicate samples, and the error bars indicate standard deviation.
We then examined the binding potential of 12T to SMMC-7721 hepatoma cells using cell-ELISA. scFv 25-TSST-1 is a fusion molecule produced in our laboratory that binds SMMC-7721 with high specificity and affinity (unpublished data). As shown in Figure 3B, the 12T protein bound to the SMMC-7721 cells with a binding capacity equivalent to that of scFv25-TSST-1, whereas TSST-1 demonstrated a low binding affinity for these cells. As shown in Figure 3C, 12T bound to the hepatoma cell SMMC-7721 with high affinity, but bound to the other tumor cells (BGC-823, KB, and normal liver cell HL-02) with poor affinity. This finding demonstrated that HCC79 may target HCC specifically, with high affinity and good specificity despite its low molecular weight (1.5 kDa).
12T Inhibits the Proliferation of Hepatoma Cells In Vitro and In Vivo
It is well known that the SAg TSST-1 can activate human T lymphocytes to kill hepatoma cells. Additionally, the 12T fusion protein can bind to hepatoma cells specifically. Thus, we tested whether the HCC79 peptide targets the TSST-1 SAg. Interestingly, our results showed that the effect of 12T on tumor inhibition was much stronger than that of native TSST-1 alone. As shown in Figure 4A, at an effector:target ratio of 8:1, the cytotoxic activity of 12T began at a concentration as low as 32 pM, whereas the native TSST-1 showed only minor cytotoxic activity at the same concentration. The protein concentrations of 12T and TSST-1 at which the tumor growth was inhibited by 50% (IC50) were 0.014 pM and 34.5 nM, respectively (Figure 4A). Considering the fact that HCC79 alone did not exert any tumor growth inhibition effect under the same conditions (data not shown), we hypothesize that this peptide may enhance the cytotoxic activity of TSST-1 in vitro by facilitating the targeting of tumor cells (Figure 4A). Our results also suggested that 12T inhibited hepatoma cell proliferation by activating a T-cell response (Figure 4B), possibly because of the higher effector:target ratio, which may enhance better tumor inhibitory effect.
Figure 4.

12T fusion protein inhibits tumor growth in vitro and in vivo. (A) Cytotoxic effects on the hepatoma cell line SMMC-7721 by 12T and TSST-1 in vitro. SMMC-7721 cells were incubated with human T cells, with either 12T or native TSST-1, which were diluted at the same starting concentration (10 nM) before the surviving tumor cells were measured by MTS assays. Effector:target ratio is 8:1. The data are the means of triplicate samples, and the error bars indicate standard deviation. (B) Cytotoxic effects of 12T on SMMC-7721 cells with different effector:target ratios. These were diluted at the same starting concentration (10 nM) before the surviving tumor cells were measured by MTS assays. The data are the means of triplicate samples, and the error bars indicate standard deviation. (C) 12T inhibits tumor growth in vivo. The tumor nodules were isolated and weighed after injecting different doses of 12T, TSST-1 alone, or a control. The inhibition rate of the experimental group was calculated versus the control. The data are the mean ± SD of 8 individual mice.
To determine the inhibitory effect of 12T on tumor growth in vivo, we established an animal tumor model by injecting H22 tumor cells into Balb/c mice. 12T and TSST-1 were administered, and tumor growth was evaluated by assessing tumor weights. Our results showed that 12T inhibited tumor growth effectively compared to TSST-1 alone (Figure 4C). At a 5-fold lower dosage (0.4 nM per mouse), the 12T protein inhibited tumor growth at a greater level than TSST-1 alone (2 nM per mouse) in vivo. Additionally, the 12T fusion protein displayed a lower toxicity compared with TSST-1. Two mice died at the medium dosage level in the TSST-1–treated group, whereas no mortality occurred in the 12T-treated groups, even at a dosage 5 times higher. These results indicate that 12T effectively inhibits tumor growth (Figure 4C).
DISCUSSION
The selective delivery of drugs to tumor cells provides an ideal approach to enhance therapeutic efficacy and minimize adverse side effects. To establish efficient and reliable therapeutic drug delivery into cancer cells, a number of delivery agents have been investigated in recent years (16). Among these, applying cell-targeting peptides has emerged as the most valuable nonimmunogenic approach (11). In the present study, we successfully used a cell-based selection system to isolate cell-targeting peptides and constructed a target drug molecule for HCC by fusing the peptide to TSST-1.
The hepatoma cell line SMMC-7721 serves as the panning cell in our system, in which we isolated a novel peptide, HCC79-(KSLSRHDHIHHH), that binds specifically to HCC cells. Surprisingly, this peptide can compete with a specific hepatoma cell membrane receptor antibody, HAb25, to bind to the hepatoma cells. Therefore, this peptide may recognize the same site as HAb25, which implicates its high targeting specificity. Our cell invasion assay indicates that HCC79 can significantly reduce the invasion of SMMC-7721 cells. Along with its binding capability and specificity toward the human hepatoma cell line, the peptide HCC79 demonstrates binding affinity toward murine HCC tumor H22. A biotin-labeled HCC79 was shown to bind to H22 cells by flow cytometry analysis, but a biotin-labeled unrelated 12-mer peptide cannot bind to H22 cells (Ning baoan, unpublished observations). This suggests that the peptide might interact with the conserved cell surface antigens that exist in both human and murine tumor cell lines. Thus we can use the mouse H22 model in experiments to evaluate the targeting efficacy of HCC79. The membrane receptor for HCC79 in the hepatoma cell remains unclear, however. Research in this area is ongoing, geared toward understanding the molecular mechanism of peptide-induced tumor suppression.
We produced a 12mer cell-targeting peptide fused with TSST-1 (12T) and evaluated its efficacy as a cancer cell–targeting drug. The fusion protein has some unique features, enabling ready detection using an anti–TSST-1 antibody, and possessing a low molecular weight and a strong immune-activating capability among the SAg family. Thus, 12T is an ideal effector for targeted anticancer therapy. These features allowed us to study the targeting effects of 12T in vitro and in vivo.
Our findings suggest that the peptide HCC79 has a high HCC cell–targeting capability, similar to that of the engineered antibody HAb25. Moreover, its molecular size is much smaller than that of HAb25. The fusion protein provides a better targeting and tumor inhibitory effect, both in vitro and in vivo, than TSST-1 alone. Furthermore, the fusion protein exhibits an improved therapeutic efficacy and reduced toxicity in animal models. These results suggest that the fusion protein is a promising agent for the targeted therapy of HCC.
In summary, the peptide that we identified by screening the phage libraries on HCC cells may be useful agent for targeting HCC. The isolated peptide fused to TSST-1 can bind specifically to hepatoma cells, suppress tumor invasion, and inhibit tumor invasion growth in vitro and in vivo. The results suggest that this peptide and its derivative have properties that merit further evaluation for treatment of human tumor diseases.
ACKNOWLEDGMENTS
We thank Dr. Cheng Li for the critical reading and helpful proposal for this manuscript. This work was supported by grants from the National Natural Science Foundation of China (no. 30171091, no. 30271478).
Footnotes
Online address: http://www.molmed.org
REFERENCES
- 1.Hu CM, Liu YF, Chen ZN, Sui YF, Chou K, Xu LQ. Preparation and characterization of two clones of monoclonal antibodies against hepatocellular carcinoma. J Monoclonal Antibody. 1994;10:16–20. [Google Scholar]
- 2.Shadidi M, Sioud M. Selective targeting of cancer cells using synthetic peptides. Drug Resistance Updates. 2003;6:363–71. doi: 10.1016/j.drup.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Nielsen UB, Marks JD. Internalizing antibodies and targeted cancer therapy: direct selection from phage display libraries. Pharm Sci Technol Today. 2000;3:282–91. doi: 10.1016/s1461-5347(00)00280-7. [DOI] [PubMed] [Google Scholar]
- 4.Hu CM, Liu YF, Ye HG, Yang SQ, Sui YF, Liu CG. Targeting location and pharmacokinetics Hab25 monoclonal antibody against primary hepatocellular carcinoma in human body. J Monoclonal Antibody. 1995;11:1–4. [Google Scholar]
- 5.Ma LH, Liu YF, Sun ZW. Expression, purification and targeting therapy of mscFv25-TNF against hepatocellular carcinoma. J Med Coll PLA. 2000;15:121–4. [Google Scholar]
- 6.Hu CM, Liu YF, Gao L. Cloning and sequencing of variable region genes of Hab25 McAb against hepatocellular carcinoma. Zhonghua Gan Zang Bing Za Zhi. 1999;7:101–3. [PubMed] [Google Scholar]
- 7.Hao HJ, Jiang YQ, Zheng YL, Ma R, Yu DW. Improved stability and yield of Fv targeted superantigen by introducing both linker and disulfide bond into the targeting moiety. Biochimie. 2005;87:661–7. doi: 10.1016/j.biochi.2005.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Graff CP, Wittrup KD. Theoretical analysis of antibody targeting of tumor spheroids: importance of dosage for penetration and affinity for retention. Cancer Res. 2003;63:1288–96. [PubMed] [Google Scholar]
- 9.Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. In vivo protein transduction: delivery of a biologically active protein into the mouse. Science. 1999;285:1569–72. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]
- 10.Ladner RC, Sato AK, Gorzelany J, de Souza M. Phage display-derived peptides as therapeutic alternatives to antibodies. Drug Discov Today. 2004;9:525–9. doi: 10.1016/S1359-6446(04)03104-6. [DOI] [PubMed] [Google Scholar]
- 11.Aina OH, Sroka TC, Chen ML, Lam KS. Therapeutic cancer targeting peptides. Biopolymers. 2002;66:184–99. doi: 10.1002/bip.10257. [DOI] [PubMed] [Google Scholar]
- 12.Hong FD, Clayman GL. Isolation of a peptide for targeted drug delivery into human head and neck solid tumors. Cancer Res. 2000;60:6551–6. [PubMed] [Google Scholar]
- 13.Rasmussen UB, Schreiber V, Schultz H, Mischler F, Schughart K. Tumor cell-targeting by phage-displayed peptides. Cancer Gene Ther. 2000;9:606–12. doi: 10.1038/sj.cgt.7700476. [DOI] [PubMed] [Google Scholar]
- 14.Shadidi M, Sioud M. Identification of novel carrier peptides for the specific delivery of therapeutics into cancer cells. FASEB J. 2003;17:256–8. doi: 10.1096/fj.02-0280fje. [DOI] [PubMed] [Google Scholar]
- 15.Michon IN, Hauer AD, von der Thusen JH, Molenaar TJ, van Berkel TJ, Biessen EA, Kuiper J. Targeting of peptides to restenotic vascular smooth muscle cells using phage display in vitro and in vivo. Biochim Biophys Acta. 2002;1591:87–97. doi: 10.1016/s0167-4889(02)00254-9. [DOI] [PubMed] [Google Scholar]
- 16.Torchilin VP, Lukyanov AN. Peptide and protein drug delivery to and into tumors: challenges and solutions. Drug Discov Today. 2003;8:259–65. doi: 10.1016/s1359-6446(03)02623-0. [DOI] [PubMed] [Google Scholar]
- 17.Marrack P, Kappler J. The staphylococcal enterotoxins and their relatives. Science. 1990;248:705–11. doi: 10.1126/science.2185544. [DOI] [PubMed] [Google Scholar]
- 18.Dohlsten M, Sundstedt A, Bjorklund M, Hedlund G, Kalland T. Superantigen-induced cytokines suppress growth of human colon-carcinoma cells. Int J Cancer. 1993;54:482–8. doi: 10.1002/ijc.2910540321. [DOI] [PubMed] [Google Scholar]
- 19.Wahlsten JL, Mills CD, Ramakrishnan S. Antitumor response elicited by a superantigen-transmembrane sequence fusion protein anchored onto tumor cells. J Immunol. 1998;161:6761–6. [PubMed] [Google Scholar]
- 20.Jiang YQ, Zheng YL, Ning BA, Ma R, Wang ZZ, Li HP. Purification and in vitro anti-hepatocellular carcinoma activity of toxic shock syndrome toxin–1. China Life Sci Res. 2002;6:347–50. [Google Scholar]
- 21.Litton MJ, Dohlsten M, Lando PA, Kalland T, Ohlsson L, Andersson J, Andersson U. Antibody-targeted superantigen therapy induces tumor infiltrating lymphocytes, excessive cytokine production, and apoptosis in human colon carcinoma. Eur J Immunol. 1996;26:1–9. doi: 10.1002/eji.1830260102. [DOI] [PubMed] [Google Scholar]
- 22.Dohlsten M, Hansson J, Ohlsson L, Litton M, Kalland T. Antibody-targeted superantigens are potent inducers of tumor-infiltrating T lymphocytes in vivo. Proc Natl Acad Sci U S A. 1995;92:9791–5. doi: 10.1073/pnas.92.21.9791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hetian L, et al. A novel peptide isolated from a phage display library inhibits tumor growth and metastasis by blocking the binding of vascular endothelial growth factor to its kinase domain receptor. J Biol Chem. 2002;277:43137–42. doi: 10.1074/jbc.M203103200. [DOI] [PubMed] [Google Scholar]
- 24.Dohlsten M, et al. Monoclonal antibody-superantigen fusion proteins: tumor-specific agents for T-cell-based tumor therapy. Proc Natl Acad Sci U S A. 1994;91:8945–9. doi: 10.1073/pnas.91.19.8945. [DOI] [PMC free article] [PubMed] [Google Scholar]