

Abstract
Nearly 50 individual DNAs with polynucleotide kinase-like activity were isolated from a random-sequence pool by using in vitro selection. Each self-phosphorylating deoxyribozyme makes use of one or more of the eight standard NTPs or dNTPs as a source of activated phosphate. Although most prototypic deoxyribozymes poorly differentiate between the ribose and deoxyribose moieties, further optimization by in vitro selection produced variants that display up to 100-fold discrimination between related NTP and dNTP substrates. An optimized ATP-dependent deoxyribozyme uses ATP >40,000-fold more efficiently than CTP, GTP, or UTP. This enzyme operates with a rate enhancement of nearly one billion-fold over the uncatalyzed rate of ATP hydrolysis. A bimolecular version of the ATP-dependent deoxyribozyme was further engineered to phosphorylate specific target DNAs with multiple turnover. The substrate-recognition patterns and rate enhancements intrinsic to these DNAs are characteristic of naturally occurring RNA and protein enzymes, supporting the hypothesis that DNA has sufficient catalytic potential to function as an enzyme in biological systems.
RNA has many well defined functions in cells, which span the range from the transfer of genetic information to molecular recognition and even biocatalysis. In stark contrast, DNA primarily serves a single purpose—the passive storage of genetic information. The distinctive biochemical properties of these two polymers have been ascribed exclusively to their differences in chemical composition. The presence of the 2′-hydroxyl group, for example, renders the phosphodiester linkage of RNA much more susceptible to strand scission under biological conditions compared with DNA (1). As a consequence, DNA is far superior to RNA as a format for the long-term storage of genetic information. Conversely, the 2′-hydroxyl group unquestionably contributes positively to the structural and functional complexity of RNA (2, 3). Considering the many natural (4–7) and artificial (8, 9) ribozymes that have been identified to date, it is evident that RNA has an enormous capacity for enzyme-like function. The extra chemical complexity provided by the 2′-hydroxyl group often is used to rationalize the biochemical functions that natural RNA maintains over DNA.
The fact that 2′-hydroxyl groups make important contributions to RNA folding supports the belief (10) that DNA may have great difficulty in forming highly complex and efficient active sites for substrate binding and enzymatic function. Nevertheless, the chemical compositions of RNA and DNA are otherwise very similar, and it seems reasonable to speculate that DNA may have a large untapped potential for catalytic function (11). The many examples of artificial deoxyribozymes, which have been isolated by using in vitro selection (9, 12), support this hypothesis. The rapidly expanding list of catalytic activities that deoxyribozymes exhibit includes DNA cleavage (13, 14), DNA ligation (15), and metalation of porphyrins (16). A variety of deoxyribozymes that catalyze RNA cleavage also have been isolated (17, 18), each displaying characteristics that are indicative of structural and functional complexity. For example, RNA-cleaving deoxyribozymes have been isolated that require metal ion (17–20) or amino acid cofactors (21), or that require no cofactor at all (22, 23). A representative Mg2+-dependent deoxyribozyme cleaves RNA with a kcat of 10 min−1 (20), which is comparable to the rates achieved by natural RNA-cleaving ribozymes (4). Such artificial deoxyribozymes may offer distinct advantages for certain practical applications where chemical stability and ease of enzyme synthesis are required. If further studies reveal that the catalytic function of DNA can approach the proficiency seen with ribozymes, then perhaps the existence of DNA enzymes in nature is not a biochemical impossibility.
In an effort to understand more about the catalytic capabilities of DNA, we sought to create deoxyribozymes with polynucleotide kinase activity. The analogous protein enzyme, T4 polynucleotide kinase (T4 PNK), catalyzes the transfer of the γ-phosphate from ATP to the 5′ hydroxyl of an RNA or DNA substrate in a sequence-independent fashion (24). A number of artificial ribozymes also have been isolated by in vitro selection that transfer the γ-thiophosphate moiety of adenosine 5′-[γ-thio]triphosphate (ATP[γS]) to the 5′ hydroxyl of RNA (25), establishing a precedent for self-phosphorylation by nucleic acids. Expansion of the catalytic repertoire of DNA to include similar phosphoryl transfer activity would allow direct comparisons to be made among the three enzyme formats and would also provide a new tool for the sequence-specific phosphorylation of oligonucleotides.
MATERIALS AND METHODS
Nucleotides and Oligonucleotides.
Synthetic DNAs were prepared by automated synthesis (21) and were purified by denaturing (8 M urea) PAGE. Single-stranded DNAs were internally 32P-labeled either during synthesis by PCR or by reverse transcriptase- (RT) catalyzed primer extension by using standard protocols. Nucleoside triphosphates and [α-32P]dGTP and [γ-32P]ATP were purchased from Amersham Pharmacia.
In Vitro Selection.
Each round was adapted from the general scheme depicted in Fig. 1A. For the first round, 1,000 pmol of the initial DNA population (G0) was heated in 400 μl of deionized water at 90°C for 1 min, cooled to room temperature, and combined with 500 μl of 2× selection buffer (100 mM Hepes [pH 7.0 at 23°C], 800 mM NaCl, 200 mM KCl, 20 mM MgCl2, 10 mM CaCl2, 2 mM MnCl2, and 100 μM CuCl2). One hundred microliters of a solution containing the four NTPs or dNTPs (10 mM each) was added to initiate either the NTP or dNTP selection reactions, respectively. Mixtures were incubated at 23°C for 48 hr and combined with an equal volume of 20 mM EDTA (pH 8.0). DNA was recovered by precipitation with ethanol and redissolved in 4.5 ml of 20 mM NaCl containing 1 μM template and 1.3 μM acceptor. Mixtures were incubated at 90°C for 1 min, cooled to room temperature, and combined with 500 μl of 10× T4 ligase buffer (NEB, Beverly, MA) and 10 μl of 400 units/μl T4 DNA ligase. After incubation at 16°C for 24 hr, DNA was precipitated with ethanol, and ligated products (≈123 nt in length) were isolated by 10% denaturing PAGE. DNA eluted from the gel was amplified by PCR (21) using 40 pmol each of primer 1 and primer 2 (5′-GAATTCTAATACGACTCACTATrA). DNA products were precipitated with ethanol, resuspended in 90 μl of 0.25 M NaOH, heated at 90°C for 10 min to cleave the single RNA phosphodiester link (18, 26, 27), and neutralized with 10 μl of 3 M NaOAc (pH 5.2 at 23°C). DNA was again precipitated with ethanol, purified by 10% denaturing PAGE, and DNA corresponding to the original pool size was recovered and used to initiate the next round of selection.
Figure 1.

Selection of self-phosphorylating deoxyribozymes. (A) Selection is initiated by incubating a population of synthetic 100-nt DNAs that carry a random-sequence domain of 70 nt. DNAs are (I) incubated with a mixture of either the four standard NTPs or the four standard dNTPs. After incubation, the DNA pool is (II) combined with excess acceptor and template oligonucleotides and treated with T4 DNA ligase in the presence of ATP. The ligated DNAs (≈123 nt) are (III) isolated by PAGE, and the recovered DNAs are selectively amplified by PCR using primer 1 and ribo-terminated primer 2. The resulting double-stranded PCR products are (IV) treated with NaOH to cleave the isolated RNA linkage, and the subsequent single-stranded DNAs are purified by PAGE and used (V) to initiate the next round. DNA populations from the final rounds of selection were (VI) PCR-amplified with primer 1 and the “all-DNA” version of primer 2 to facilitate cloning. N70 represents the random-sequence domain. (B) Sequences of the DNA population complexed with the acceptor and template strands. (C) Response of the NTP (open bars) and dNTP (filled bars) populations over the first six rounds of selection.
Subsequent rounds of selection were performed at 1/50th scale of the initial reaction. DNA was pretreated with alkaline phosphatase to prevent the emergence of selfish DNAs that acquire a 5′-terminal phosphate during PCR amplification (unpublished data). In addition, the selection time was reduced to 20 hr to favor the isolation of the most efficient deoxyribozymes, and the ligation time was reduced to a minimum of 10 min. Final populations were cloned by using a TOPO-TA cloning kit (Invitrogen) and sequenced by using a ThermoSequenase kit (Amersham Pharmacia).
Catalytic Assays and Kinetic Analyses.
Internally 32P-labeled DNAs (≈100 nM) were tested for self-phosphorylation activity individually in the presence of NTP or dNTP substrates by using the phosphorylation-ligation and PAGE methods employed for selection. In most cases, initial rate constants for DNA phosphorylation were determined by plotting the fraction of precursor converted to ligated product versus deoxyribozyme reaction time. The slope of the line that represents the initial velocity of the reaction (<10% reacted) was determined by a least-squares fit to the data. Although the extent of DNA phosphorylation was measured indirectly through analysis of ligation products, we observed that a 1-hr ligation reaction was sufficient to produce quantitative conversion of phosphorylated DNAs to ligated DNAs, indicating that this indirect analytical approach accurately reflects the true yield for the deoxyribozyme reaction.
RESULTS AND DISCUSSION
In Vitro Selection of Self-Phosphorylating Deoxyribozymes.
To isolate DNAs with polynucleotide kinase activity, we employed the in vitro selection strategy depicted in Fig. 1A. This approach relies on the ability of T4 DNA ligase to join an “acceptor” DNA to a 5′-phosphorylated “donor” DNA when the two are properly juxtaposed within a duplex (28). Therefore, only those DNAs that acquire a phosphate at any step preceding the ligation reaction will be joined to a DNA acceptor molecule (Fig. 1B). This coupling event distinguishes the reacted DNAs both during PAGE and during selective PCR amplification.
Two parallel selection efforts were undertaken, each with a DNA population comprised of ≈6 × 1014 different molecules. The populations were supplied with the four standard ribonucleoside 5′-triphosphates (NTP selection) or the four standard deoxyribonucleoside 5′-triphosphates (dNTP selection) during the reaction phase of the selection process (Fig. 1A, stage I or stage V). This provides a choice of phosphorylating agents that can be used by individual self-phosphorylating deoxyribozymes. Both populations responded positively to the selection constraints, such that ≈35% (NTP) and ≈45% (dNTP) of the DNA populations undergo ligation after six rounds (G6) (Fig. 1C). Further characterization of the active populations and of individual deoxyribozymes provided evidence that catalysis results in the transfer of the (d)NTP γ phosphate to the 5′-terminal oxygen of DNA (see below).
Deoxyribozymes with Diverse Substrate Specificities.
The G7 populations for the NTP and dNTP selections were cloned, and ≈20 representative clones from each pool were sequenced and assayed for catalytic activity. Analysis revealed surprising sequence diversity, suggesting that a variety of unique structural motifs are able to promote self-phosphorylation (data not shown). However, among five distinct sequence classes identified from the NTP pool (termed “NTP-G1” through “NTP-G5”), all require GTP for deoxyribozyme function. Likewise, dGTP-utilizing deoxyribozymes comprise three (dNTP-G1 through dNTP-G3) of the four distinct classes isolated from the G7 dNTP pool. The remaining class (dNTP-N1) is a “universal” kinase that can use any of the four dNTPs with similar efficiency.
Despite the fact that the G7 populations were dominated by GTP- or dGTP-utilizing deoxyribozymes, we speculated that individuals exhibiting specificities for the remaining nucleotide substrates did exist, albeit in less abundance. To confirm this, we repeated the in vitro selection process in parallel beginning with G2 DNA, in each case making available only the NTPs or dNTPs with base identities A, C, U, or T for use as substrates by each corresponding population. In this manner, representative deoxyribozymes were recovered that display distinct preferences for each of the four standard NTPs or dNTPs (Fig. 2).
Figure 2.

Self-phosphorylating DNAs with distinct substrate specificities. (A) Sequences of nine representative classes of deoxyribozymes. Nucleotides corresponding to the original random-sequence domain are numbered. Underlined nucleotides in dNTP-N1 identify BstN I, EagI and BsmAI sites from left to right. (B) Substrate specificity of individual deoxyribozymes. Filled arrowheads identify internally 32P-labeled precursor DNAs, and open arrowheads identify the positions of the ligated products that are indicative of deoxyribozyme activity. Reaction conditions were as follows: Nucleoside triphosphate concentrations: NTP-A1, U1, C1, N1, and dNTP-C1 and N1 (1 mM); NTP-G1, dNTP-G1, and dNTP-A1 (50 μM). Incubation times: NTP-G1, NTP-A1, and dNTP-G1, A1, and C1 (20 hr); NTP-C1 (46.5 hr), dNTP-N1 (60 hr), NTP-U1 (92.5 hr); NTP-N1 (94 hr).
Two major (NTP-A1 and NTP-A2) and four minor classes of ATP-dependent deoxyribozymes were isolated from the NTP G7 population produced by providing ATP exclusively to the selection reaction. Similarly, a single CTP-dependent deoxyribozyme (NTP-C1) was isolated from the corresponding G8 population produced by supplying only CTP to the reaction mixture. Finally, two classes of deoxyribozymes were isolated from the G10 population generated by the exclusive use of UTP. Although one class (NTP-U1) requires UTP for activity, the other forms a motif with universal substrate specificity (NTP-N1) that is distinct from the class represented by dNTP-N1 (Fig. 2A).
Successfully completing the analogous selections for different dNTP specificities proved more challenging. The G7 population of the dNTP selection is highly populated with the universal kinase dNTP-N1. With its unrestricted substrate specificity, this deoxyribozyme likely would have dominated the independent selections for DNAs that use dATP, dCTP, and dTTP, thereby precluding the enrichment of base-specific kinases. We adopted a strategy to prevent dNTP-N1 domination by destroying the deoxyribozyme in its double-stranded form by using the restriction enzyme EagI. Although dNTP-N1 quickly “evolved” to evade cleavage through mutation of the corresponding restriction site, we adopted a more vigorous strategy that included the simultaneous digestion of the population with three restriction enzymes (EagI, BstN I, and BsmA I). In addition, the surviving single-stranded population was preincubated with a mixture of the remaining three dNTPs that were not used for positive selection. DNAs that remained unreacted during this “negative” selection were separated from phosphorylated DNAs and subjected to positive selection in the presence of the target dNTP. In this manner, four classes of dATP-dependent deoxyribozymes and two classes of dCTP-dependent deoxyribozymes were isolated from the corresponding G8 pools. We were unsuccessful in our attempt to recover dTTP-dependent deoxyribozymes even after conducting a total of 14 rounds of selection and despite the fact that NTP-U1 also accepts dTTP (Fig. 2B). Although G14 was populated with self-phosphorylating DNAs, all proved to belong to one of a number of new classes with universal substrate specificities (data not shown). The emergence of so many new classes of universal kinases rendered impractical our continued pursuit of dTTP-specific deoxyribozymes.
Discrimination Between Ribo- and Deoxyribonucleotides.
A cursory survey of the accumulated sequence data has revealed many distinct sequence classes of self-phosphorylating deoxyribozymes (data not shown). Each class can be further organized into one of several groups based on NTP or dNTP substrate requirements. In addition to the specific groups described above (Fig. 2B), we have observed several individuals that function as “semiuniversal” kinases that, for example, use only (d)GTP and (d)CTP or that use (d)GTP and (d)ATP.
Interestingly, of the nine classes of deoxyribozymes depicted in Fig. 2A, only dNTP-N1 displays significant nucleotide discrimination based on the 2′-hydroxyl group. This finding brings into question the structural diversity of DNA and whether the limitations set by this polymer’s chemical simplicity preclude it from distinguishing between closely related organic compounds. To examine this issue in greater detail, we conducted two parallel selections to isolate deoxyribozymes that use GTP to the exclusion of dGTP, and vice versa. After six rounds of selection beginning with the G5 populations of the NTP and dNTP lineages, both resulting populations displayed significant discrimination between GTP and dGTP. The function of two individuals, NTP-G6 and dNTP-G4 (Fig. 3A), were examined in greater detail. NTP-G6 is >100-fold more active with GTP compared with dGTP, whereas dNTP-G4 is ≈25-fold more active with dGTP versus GTP (Fig. 3B; Table 1). For both deoxyribozymes, the differences in Km values between GTP and dGTP are negligible, however the maximum rate constants obtained under saturating conditions are unequal. Surprisingly, specificity in each case is not due to differential binding affinity but is a result of the differences in reactivity of the nucleotides when bound to the deoxyribozyme. This conclusion is supported by the observation that dGTP serves as a competitive inhibitor of NTP-G6 (data not shown). Although additional investigations are needed to understand the mechanism of discrimination, these and earlier findings (12, 21) support the view that DNA has sufficient structural complexity to recognize subtle chemical differences among substrate, cofactor, and effector molecules.
Figure 3.

Precise discrimination between GTP and dGTP by DNA. (A) Sequences for the GTP-specific deoxyribozyme NTP-G6 and for the dGTP-specific deoxyribozyme dNTP-G4. (B) Plots of the rate constants for NTP-G6 and dNTP-G4 with various concentrations of GTP (filled symbols) and dGTP (open symbols). Circles and diamonds or squares and triangles represent two replicate experiments conducted with GTP or dGTP, respectively. For selection, the G5 pool from the NTP selection was incubated with a mixture of the four dNTPs (1 mM each) for 50–100 hr, and the DNAs that remained unreacted after ligation were isolated by PAGE. The remaining DNAs were incubated with 100 μM GTP for 2 hr, and the phosphorylated DNAs were recovered and amplified as depicted in Fig. 1A. Likewise, the G5 pool from the dNTP lineage was preselected against function with the NTPs and selected for activity in the presence of 100 μM dGTP. Kinetic parameters are summarized in Table 1.
Table 1.
kcat (min−1) and Km (μM) values for NTP-G6 and dNTP-G4 with GTP and dGTP
| Species |
kcat
|
Km
|
Selectivity | ||
|---|---|---|---|---|---|
| dGTP | GTP | dGTP | GTP | ||
| NTP-G6 | 2.74 × 10−5 | 1.46 × 10−3 | 918 | 425 | 115 |
| dNTP-G4 | 4.43 × 10−4 | 1.90 × 10−5 | 98 | 106 | 26 |
Values were calculated with the Michaelis-Menten equation by using the data presented in Fig. 3, and each is the average of two replicate experiments. Selectivity is the ratio of the kcat/Km values with GTP and dGTP.
A Distinct Class of ATP-Dependent Deoxyribozymes.
All prototypic deoxyribozymes examined operate with kcat values of 10−4⋅min−1 or lower and Km values near 1 mM. However, we expected that variants of each deoxyribozyme would show improved kinetic characteristics if further optimized. A mutagenized pool based on the ATP-dependent deoxyribozyme NTP-A2 (d = 0.21 over 70 positions) was chemically synthesized (29). After five rounds of selection by using progressively lower concentrations of ATP and shorter reaction times, a population of variant deoxyribozymes was recovered (Fig. 4). Improvements in both catalytic rate and ATP-binding affinity were evident on examination of NTP-A2.1, a representative clone from the G5 population. NTP-A2 has a kcat of 1.7 × 10−4⋅min−1 and a Km of ≈1 mM, whereas NTP-A2.1 has a kcat of 5.5 × 10−3⋅min−1 and a Km of 3.3 μM, corresponding to an improvement in catalytic efficiency (kcat/Km ) of ≈10,000-fold. We were unable to detect the background reaction rate of DNA phosphorylation by ATP to report a true rate enhancement over uncatalyzed phosphorylation. However, the rate enhancement can be estimated by ATP hydrolysis in the presence of Ca2+ (30), which has a reported second-order rate constant of ≈25 × 10−7 M−1⋅min−1 at 23°C. Therefore, the rate enhancement (kcat/Km )/khydrolysis for NTP-A2.1 is ≈7 × 108, indicating that this deoxyribozyme transfers the γ-phosphate from ATP to its 5′-hydroxyl group nearly one billion times faster than the spontaneous hydrolysis of ATP.
Figure 4.

Artificial phylogeny comprised of deoxyribozyme variants isolated after five rounds of selection beginning with mutagenized NTP-A2. Nucleotides depicted correspond to the original random-sequence domain. The donor domain (not shown) was found to vary, and each conforms to one of the following states represented by zero through four asterisks, respectively: GGAAAGATGCGAC (original), GGAAAGATG-GAC, GGAAAGATGTGAC, GGAAAGATGGGAC, and GGAAAGAAGCGAC. Overlined nucleotides identify four regions (I–IV) of conserved tracts of guanosine residues. (†) identifies variant “NTP-A2.1,” which was used in further analyses. Reactions were performed for 1 hr by using 100 μM ATP for first two rounds and for 5 min by using 10 μM ATP for the final three rounds.
NTP-A2.1 is highly selective for ATP versus CTP, GTP and UTP. The apparent Kd for ATP, interpreted as the concentration required for half-maximal deoxyribozyme activity, corresponds with the Km value determined for the same deoxyribozyme (Fig. 5A). In contrast, the binding constants for the noncognate NTPs appear to be more than four orders of magnitude lower. As expected, the kcat/Km value for ATP is >10,000-fold larger than the kcat/Km values determined for the remaining NTPs (data not shown). These findings are indicative of a polymer that is of sufficient structural sophistication to exploit differences in molecular composition to achieve precise substrate recognition.
Figure 5.

Kinetic analysis of an improved ATP-dependent deoxyribozyme. (A) Rate constants exhibited by NTP-A2.1 with different concentrations and forms of NTPs. The left axis provides the scale for ATP (●), and the right axis provides the scale for GTP (□), CTP (▵) and UTP (⋄). (B) Self-phosphorylation of NTP-A2.1 with [γ-32P]ATP. Unlabeled deoxyribozyme (0.25 μM) was incubated with selection buffer containing 10 μM ATP (spiked with a trace of [γ-32P]ATP) for 0, 5, 10, 20, 40, 60, and 90 min (lanes 1–7, respectively) at 23°C and examined by PAGE (see Inset). A plot of the natural logarithm of the fraction unreacted (determined by using the maximum extent of self- and T4 PNK-mediated phosphorylation) versus time provides a negative slope of 4.0 × 10−3⋅min−1, which reflects the rate constant of DNA self-phosphorylation. This value corresponds well with the rate constant determined by using the DNA ligation method (5.5 × 10−3⋅min−1; Fig. 5A).
NTP-A2 requires both Ca2+ and K+ for activity. However, the improved variant NTP-A2.1 functions at a maximum rate (kobs = 0.012 min−1) in the presence of 20 mM Ca2+ (apparent Kd for Ca2+ ≈5 mM), and remains active in the absence of K+. Among the different sequence classes of deoxyribozymes examined, only Ca2+ and Mn2+ serve as divalent metal ion cofactors. Ca2+ dependence also has been demonstrated for a recently described capping ribozyme (31), indicating that this divalent ion may be well suited for catalytic roles in certain phosphate-transfer reactions. In contrast, the self-phosphorylating ribozymes previously isolated (25) require Mg2+ as a cofactor. However, these ribozymes were selected to perform in a reaction mixture that contained Mg2+ alone. Although Ca2+ is only 5-fold more effective than Mg2+ at promoting ATP hydrolysis (30), RNA may also favor Ca2+ as a cofactor for self-phosphorylation if presented with the option.
Verification of Self-Phosphorylation by DNA.
The selection protocol was designed to isolate DNAs that acquire a 5′-phosphate group when incubated with nucleoside 5′-triphosphates and that serve as donor oligonucleotides during the subsequent ligation reaction. NTP-A2 and dNTP-G4 show patterns of DNA ligation that are consistent with the acquisition of a 5′-phosphate group at their 5′ terminus in an ATP- or dGTP-dependent manner, respectively (data not shown). Likewise, successful DNA ligation requires the addition of DNA ligase, template and acceptor oligonucleotides, and ATP, requirements that are in agreement with the selection scheme proposed in Fig. 1A.
To further investigate the nature of the deoxyribozyme-mediated reaction, unlabeled NTP-A2.1 was incubated with [γ-32P]ATP under permissive reaction conditions. The deoxyribozyme becomes radiolabeled in a time-dependent fashion (Fig. 5B Inset), and forms a product that comigrates on a polyacrylamide gel with an authentic 5′-phosphorylated DNA prepared with T4 PNK. These findings are consistent with the transfer of the γ-phosphate of ATP to the 5′-hydroxyl group of DNA. Moreover, the rate constant derived for the increase in 32P-labeled DNA (kobs = 4.0 × 10−3⋅min−1) matches the rate constant obtained by indirectly measuring phosphorylation via DNA ligation (Fig. 5A).
A Structural Model for NTP-A2.
Examination of an artificial phylogeny for NTP-A2 (Fig. 4) reveals several domains of conserved sequence interspersed with domains that are highly variable. Most obvious are the four strictly conserved spans of three consecutive G residues. The presence of these G-rich domains suggests the formation of a three-tiered guanine quartet. Similar “G-quartet” domains have been identified in a number of other structured DNAs (11, 12) and are the basic structural element of telomeric DNA (32). We also recognized that the last two nucleotides of the donor domain and the first three nucleotides of the original random-sequence domain are complementary to nucleotides spanning positions 47–52 and that covariation in these regions is common (Fig. 4).
A structural model that is consistent with the phylogenetic data is depicted in Fig. 6A. The primary feature of this model is a single base-paired structure that constrains a large hairpin loop. We speculate that the loop folds back on itself, perhaps stabilized by a three-tiered G quartet, thereby closely positioning three otherwise distal conserved domains. Some G-quartet structures are stabilized by K+ (32), which may explain why NTP-A2 requires K+ for function. Additional support for the formation of G quartets was obtained by chemical modification experiments with NTP-A2.1. The guanosine residues of domains I–IV are efficiently methylated by dimethyl sulfate (DMS) in the absence of divalent metal ions but become fully protected on the addition 20 mM Ca2+ (data not shown). This indicates that the N7 position of each residue may be involved in tertiary contacts like those involved in the formation of G quartets.
Figure 6.

Engineering an ATP-dependent deoxyribozyme. (A) A secondary-structure model for NTP-A2. Encircled regions identify highly conserved nucleotides. The 3′-terminal domain (∗) can be deleted with minor loss of activity. Filled and open arrowheads designate cleaved sites for the construction of bimolecular complexes. (B) Bimolecular constructs composed of separate NTP-A2.1 enzyme (E) and substrate (S) domains used to examine determinants of oligonucleotide substrate specificity. (C) PAGE analysis of the activity of E1 and E2 with the matched or mismatched substrates S1 and S2 that were internally 32P-labeled. Upper bands indicate deoxyribozyme activity. Substrate DNA (10 nM) was incubated with 100-fold excess of enzyme in the presence of 1 mM ATP for 5 hr before treatment with T4 DNA ligase.
Sequence-Specific Phosphorylation of DNA by DNA.
The structural model for NTP-A2 was examined by testing several reorganized DNA constructs based on the sequence of NTP-A2.1. Nucleotides comprising the highly variable positions 50–70 of the original random-sequence domain and the 3′ primer binding site can be deleted, resulting in a truncated enzyme with ≈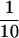 catalytic activity. However, further deletion at the 3′ terminus results in complete loss of activity. The latter observation is consistent with the hypothesis that formation of the putative stem element is necessary for catalytic function.
catalytic activity. However, further deletion at the 3′ terminus results in complete loss of activity. The latter observation is consistent with the hypothesis that formation of the putative stem element is necessary for catalytic function.
We created bimolecular constructs wherein the DNA strand was severed either before the proposed stem or immediately after the first G-rich domain (I; Fig. 6A). A combination of the two DNAs split before the duplex is almost completely inactive, indicating that this constraint is necessary for maximum catalytic activity (data not shown). In contrast, the bimolecular system formed by a split after domain I (Fig. 6B) remains fully active. This creates the possibility of phosphorylating target DNAs with specific sequences, if indeed the stem serves as a recognition element for substrate binding. To examine the importance of the proposed duplex, two synthetic deoxyribozymes (E1 and E2) were synthesized and tested for activity with two DNA substrates (S1 and S2). Each enzyme was engineered to target the phosphorylation of its complementary substrate via the putative Watson-Crick base pairing within the stem (Fig. 6B). As predicted, E1 functions only with its corresponding substrate S1, whereas E2 only accepts S2 as a substrate. Moreover, the bimolecular complex formed between S1 and E1 displays multiple-turnover kinetics with a rate that approximates the rate for self-phosphorylation. However, to achieve this effect, the reaction must occasionally be thermocycled, which presumably denatures the enzyme–product complex and allows another substrate to bind.
T4 PNK has no significant sequence restrictions, and therefore has no selectivity for the phosphorylation of individual DNAs within a complex mixture. In contrast, the NTP-A2 class of DNA kinases maintains a significant sequence requirement for substrate recognition in addition to the above described stem element. For example, deletion of the 5′-terminal guanosine of the deoxyribozyme, or replacement of this residue with A, C, or T eliminates catalytic activity. Moreover, the conserved G residues that reside within the substrate domain are expected to be a necessary sequence element for this class of deoxyribozymes.
Comparisons of Kinases Made of Protein, RNA, and DNA.
Previously, Lorsch and Szostak (25) described the in vitro selection of several self-phosphorylating ribozymes that use ATP[γS], a thiophosphate-containing analog of ATP. Although the pool designs and selection strategies used were fundamentally different between these two studies, both efforts produced a number of distinct motifs that catalyze the phosphorylation of nucleic acids. In our study, we have targeted the remaining natural NTP and dNTP substrates with almost equal effectiveness. It is clear from these results that DNA, like RNA, is capable of forming a variety of sophisticated tertiary structures that effectively promote chemical catalysis.
Perhaps the most important issue is that of catalytic rates and efficiency. The kcat for our optimized NTP-A2.1 deoxyribozyme is approximately 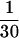 that of the kcat for the most active ATP[γS]-dependent ribozyme isolated (25, 33). The uncatalyzed rate of ATP[γS] hydrolysis is up to 10-fold faster than ATP (34, 35), therefore the differences in chemical rate enhancements between the two polymers are negligible. Interestingly, the Km for ATP exhibited by NTP-A2.1 is more than 10-fold improved from that of the best kinase ribozymes. As a result, the catalytic efficiencies of the DNA and RNA enzymes are nearly equivalent. The kcat/Km−1 values for NTP-A2.1 and “Kin.25” (25) are 3,600 M−1⋅min−1 and 7,900 M−1⋅min−1, respectively. In addition, the Km of the deoxyribozyme is more than 10-fold improved compared with the Km for ATP exhibited by T4 PNK (36). However, T4 PNK has a kcat of ≈25,000 min−1, which equates to a catalytic efficiency (≈6 × 108 M−1⋅min−1) that is approximately five orders of magnitude greater than NTP-A2.1. It is conceivable that ribozymes and deoxyribozymes with higher activities could be created by exploring DNA libraries of larger size or by performing additional rounds of mutagenesis and selection with existing catalysts. Such efficient DNA enzymes could be very useful for promoting the sequence-specific phosphorylation of DNA or RNA sequences.
that of the kcat for the most active ATP[γS]-dependent ribozyme isolated (25, 33). The uncatalyzed rate of ATP[γS] hydrolysis is up to 10-fold faster than ATP (34, 35), therefore the differences in chemical rate enhancements between the two polymers are negligible. Interestingly, the Km for ATP exhibited by NTP-A2.1 is more than 10-fold improved from that of the best kinase ribozymes. As a result, the catalytic efficiencies of the DNA and RNA enzymes are nearly equivalent. The kcat/Km−1 values for NTP-A2.1 and “Kin.25” (25) are 3,600 M−1⋅min−1 and 7,900 M−1⋅min−1, respectively. In addition, the Km of the deoxyribozyme is more than 10-fold improved compared with the Km for ATP exhibited by T4 PNK (36). However, T4 PNK has a kcat of ≈25,000 min−1, which equates to a catalytic efficiency (≈6 × 108 M−1⋅min−1) that is approximately five orders of magnitude greater than NTP-A2.1. It is conceivable that ribozymes and deoxyribozymes with higher activities could be created by exploring DNA libraries of larger size or by performing additional rounds of mutagenesis and selection with existing catalysts. Such efficient DNA enzymes could be very useful for promoting the sequence-specific phosphorylation of DNA or RNA sequences.
CONCLUSIONS
Nearly 50 distinct sequence classes of self-phosphorylating deoxyribozymes were isolated from a population of ≈1015 single-stranded DNA molecules. Representative deoxyribozymes achieve substantial catalytic rate enhancements, tight substrate binding, and precise recognition of nucleotides and oligonucleotides. These results provide compelling evidence that DNA, like RNA and protein, has sufficient structural diversity to form diverse and sophisticated active sites (11). The untapped potential for DNA catalysis could be exploited for various applications. For example, the optimization of a small deoxyribozyme that can be engineered to precisely phosphorylate any DNA or RNA sequence would be of significant utility.
Catalysis by DNA is not known to occur in nature. However, the kinase deoxyribozymes described herein provide additional examples of DNA self-processing reactions that are of biological relevance. Along with self-phosphorylation, DNA-mediated DNA cleavage (13, 14) and ligation (14) activities form an important collection of catalytic functions that are fundamental to the maintenance of all DNA genomes. Moreover, the proven utility of biologically relevant cofactors, such as Mg2+, Ca2+, Zn2+, and histidine (12, 19, 21), that augment chemical diversity in certain deoxyribozymes is consistent with the view that DNA could play a role in biological catalysis.
Acknowledgments
We thank Krishan Soni for compiling the data presented in Fig. 2B. We also thank members of the Breaker laboratory for helpful discussions. This work was supported by a grant from the National Institutes of Health (GM57500) and by a Hellman Family Fellowship to R.R.B.; Y.L. holds a postdoctoral fellowship from the Medical Research Council of Canada.
ABBREVIATIONS
- T4 PNK
T4 polynucleotide kinase
- ATP[γS]
adenosine 5′-[γ-thio]triphosphate
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
References
- 1.Oivanen M, Kuusela S, Lönnberg H. Chem Rev. 1998;98:961–990. doi: 10.1021/cr960425x. [DOI] [PubMed] [Google Scholar]
- 2.Draper D E. Trends Biol Sci. 1996;21:145–149. [PubMed] [Google Scholar]
- 3.Pyle A M, Green J B. Curr Opin Struct Biol. 1995;5:303–310. doi: 10.1016/0959-440x(95)80091-3. [DOI] [PubMed] [Google Scholar]
- 4.Cech T R. Annu Rev Biochem. 1990;59:543–568. doi: 10.1146/annurev.bi.59.070190.002551. [DOI] [PubMed] [Google Scholar]
- 5.Symons R H. Annu Rev Biochem. 1992;61:641–671. doi: 10.1146/annurev.bi.61.070192.003233. [DOI] [PubMed] [Google Scholar]
- 6.Michel F, Ferat J-L. Annu Rev Biochem. 1995;64:435–461. doi: 10.1146/annurev.bi.64.070195.002251. [DOI] [PubMed] [Google Scholar]
- 7.Frank D N, Pace N R. Annu Rev Biochem. 1998;67:153–180. doi: 10.1146/annurev.biochem.67.1.153. [DOI] [PubMed] [Google Scholar]
- 8.Williams K P, Bartel D P. Nucleic Acids Molec Biol. 1996;10:367–381. [Google Scholar]
- 9.Breaker R R. Chem Rev. 1997;97:371–390. doi: 10.1021/cr960008k. [DOI] [PubMed] [Google Scholar]
- 10.Cate J H, Gooding A R, Podell E, Zhou K, Golden B L, Kundrot C E, Cech T R, Doudna J A. Science. 1996;273:1678–1685. doi: 10.1126/science.273.5282.1678. [DOI] [PubMed] [Google Scholar]
- 11.Breaker R R. Nat Biotechnol. 1997;15:427–431. doi: 10.1038/nbt0597-427. [DOI] [PubMed] [Google Scholar]
- 12.Breaker R R. Curr Opin Chem Biol. 1997;1:26–31. doi: 10.1016/s1367-5931(97)80105-6. [DOI] [PubMed] [Google Scholar]
- 13.Carmi N, Shultz L A, Breaker R R. Chem Biol. 1996;3:1039–1046. doi: 10.1016/s1074-5521(96)90170-2. [DOI] [PubMed] [Google Scholar]
- 14.Carmi N, Balkhi S R, Breaker R R. Proc Natl Acad Sci USA. 1997;95:2233–2237. doi: 10.1073/pnas.95.5.2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cuenoud B, Szostak J W. Nature (London) 1995;375:611–614. doi: 10.1038/375611a0. [DOI] [PubMed] [Google Scholar]
- 16.Li Y, Sen D. Nat Struct Biol. 1996;3:743–747. doi: 10.1038/nsb0996-743. [DOI] [PubMed] [Google Scholar]
- 17.Breaker R R, Joyce G F. Chem Biol. 1994;1:223–229. doi: 10.1016/1074-5521(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 18.Breaker R R, Joyce G F. Chem Biol. 1995;2:655–660. doi: 10.1016/1074-5521(95)90028-4. [DOI] [PubMed] [Google Scholar]
- 19.Santoro S W, Joyce G F. Proc Natl Acad Sci USA. 1997;94:4262–4266. doi: 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santoro S W, Joyce G F. Biochemistry. 1998;37:13330–13342. doi: 10.1021/bi9812221. [DOI] [PubMed] [Google Scholar]
- 21.Roth A, Breaker R R. Proc Natl Acad Sci USA. 1998;95:6027–6031. doi: 10.1073/pnas.95.11.6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faulhammer D, Famulok M. Angew Chem Int Ed Engl. 1996;35:2837–2841. [Google Scholar]
- 23.Geyer C R, Sen D. Chem Biol. 1997;4:579–593. doi: 10.1016/s1074-5521(97)90244-1. [DOI] [PubMed] [Google Scholar]
- 24.Richardson C C. Proc Natl Acad Sci USA. 1965;54:158–165. doi: 10.1073/pnas.54.1.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lorsch J R, Szostak J W. Nature (London) 1994;371:31–36. doi: 10.1038/371031a0. [DOI] [PubMed] [Google Scholar]
- 26.Walder R Y, Hayes J R, Walder J A. Nucleic Acids Res. 1993;21:4339–4343. doi: 10.1093/nar/21.18.4339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Silveira M H, Orgel L E. Nucleic Acids Res. 1995;23:1083–1084. doi: 10.1093/nar/23.6.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Weiss B, Jacquemin-Sablon A, Live T R, Fareed G C, Richardson C C. J Biol Chem. 1968;243:4543–4555. [PubMed] [Google Scholar]
- 29.Breaker R R, Joyce G F. Trends Biotechnol. 1994;12:268–275. doi: 10.1016/0167-7799(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 30.Ramirez F, Marecek J F, Szamosi J. J Org Chem. 1980;45:4748–4752. [Google Scholar]
- 31.Huang F, Yarus M. Proc Natl Acad Sci USA. 1997;94:8965–8969. doi: 10.1073/pnas.94.17.8965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Williamson J R. Annu Rev Biophys Biomol Struct. 1994;23:703–730. doi: 10.1146/annurev.bb.23.060194.003415. [DOI] [PubMed] [Google Scholar]
- 33.Lorsch J R, Szostak J W. Biochemistry. 1995;34:15315–15327. doi: 10.1021/bi00046a041. [DOI] [PubMed] [Google Scholar]
- 34.Herschlag D, Piccirilli J A, Cech T R. Biochemistry. 1991;30:4844–4854. doi: 10.1021/bi00234a003. [DOI] [PubMed] [Google Scholar]
- 35.Breslow R, Katz I. J Am Chem Soc. 1968;90:7367–7377. [Google Scholar]
- 36.Lillehaug J R, Kleppe K. Biochemistry. 1975;14:1221–1225. doi: 10.1021/bi00677a020. [DOI] [PubMed] [Google Scholar]