

Abstract
The site-specific recombination pathway by which the bacteriophage λ chromosome is excised from its Escherichia coli host chromosome is a tightly regulated, highly directional, multistep reaction that is executed by a series of multiprotein complexes. Until now, it has been difficult to study the individual steps of such reactions in the context of the entire pathway. Using single-molecule light microscopy, we have examined this process from start to finish. Stable bent-DNA complexes containing integrase and the accessory proteins IHF (integration host factor) and Xis form rapidly on attL and attR recombination partners, and synapsis of partner complexes follows rapidly after their formation. Integrase-mediated DNA cleavage before or immediately after synapsis is required to stabilize the synaptic assemblies. Those complexes that synapsed (∼50% of the total) yield recombinant product with a remarkable ∼100% efficiency. The rate-limiting step of excision occurs after synapsis, but closely precedes or is concomitant with the appearance of a stable Holliday junction. Our kinetic analysis shows that directionality of this recombination reaction is conferred by the irreversibility of multiple reaction steps.
Keywords: bacteriophage λ, DNA recombination, Holliday junction, synapsis, tyrosine recombinase
Introduction
The tyrosine recombinase family of site-specific recombinases is responsible for many important biological processes including regulation of gene expression and DNA replication, plasmid copy number maintenance, conjugative transposition, catenated circle resolution, daughter chromosome segregation, and prokaryotic telomere processing. The hallmark of this large family is a multistep reaction pathway in which a recombinase tetramer executes a sequential pair of strand exchanges that first generate and then resolve a four-way DNA junction (Holliday intermediate). This is accomplished via two pairs of staggered DNA cleavage and ligation reactions mediated by covalent 3′ phosphotyrosine linkages in the absence of high-energy cofactors (Figure 1A; for review see Sadowski, 1993; Grainge and Jayaram, 1999; Voziyanov et al, 1999; Azaro and Landy, 2002; Van Duyne, 2002).
Figure 1.
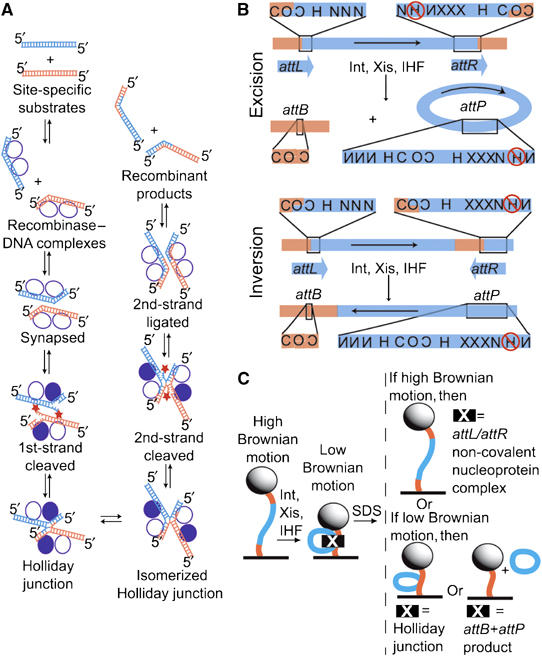
Intramolecular recombination substrates, generated products, and experimental design. (A) General scheme of the site-specific recombination reaction, in which parental DNAs (orange and blue) are recombined to form two hybrid DNA products (see text for details). The recombinase subunits (circles) that catalyze these reactions form a tetramer in which only one pair (filled) is DNA cleavage active. The reactivity of the Int pairs is reversed for the second pair of strand exchanges. All strand exchanges proceed through two successive transesterification reactions: a strand cleavage reaction in which a tyrosine side chain attacks the DNA backbone and a ligation in which the resulting phosphotyrosine linkage is attacked by a free DNA 5′ hydroxyl (star). (B) Maps of the recombination substrates and the products generated from their recombination. Top (excision): when the phage λ attachment sites are in direct repeat (attL+attR substrate), recombination generates separated linear attB and circular attP products. Bottom (inversion): the indirect repeat substrate (attLinvattR) recombines to form a single linear product in which the DNA between attB and attP attachment sites has been inverted. The presence of Xis prevents further recombination of both the excision and inversion products (Abremski and Gottesman, 1982). att sites contain 7 bp homologous DNA sequences (overlap region, O) between cleavage sites and multiple sites for binding of protein domains: N, Int N-terminal domain; C, Int catalytic domain; H, IHF; and X, Xis. Reversed lettering indicates inverted site orientation. IHF binding to the circled site inhibits excision; to avoid this, this site was inactivated by mutations (Thompson et al, 1986). The binding site for Fis protein, which overlaps with one of the Xis sites, is not shown because this host-encoded DNA-bending protein is not required for excision in the presence of sufficient Xis (Thompson et al, 1987a, 1987b) and was not included in these experiments. Both substrates are 1943 bp long and have att site overlap regions positioned 301 and 290 bp from the closest end. (C) Experimental design with attL+attR DNA. Each 200 nm polystyrene bead (sphere) is tethered to the coverslip surface of a flow chamber (black line) by a single substrate DNA molecule (orange and blue curve; Finzi and Gelles, 1995). Formation of non-covalent protein–DNA complexes that bend or loop DNA (e.g., att-site complexes or synaptic complexes) reduce the effective length of the tether and can be detected as a decrease in bead Brownian motion. Formation of covalent links between two DNA segments (in a Holliday junction) or excision of a DNA segment will have a similar effect. Effective tether length changes resulting from non-covalent protein–DNA interactions and those from covalent DNA backbone modifications can be distinguished by challenge with the protein denaturant sodium dodecyl sulfate (SDS).
The site-specific recombination system of bacteriophage λ, which directs integration and excision of the viral chromosome into and out of the Escherichia coli host chromosome (Campbell, 1962), adds several levels of complexity to this basic mechanistic framework (for review see Nash, 1996; Azaro and Landy, 2002). Like other well-studied recombinase systems from this subfamily, including Tn916 (Rudy et al, 1997a, 1997b), HP1 (Hickman et al, 1997), and L5 (Lewis and Hatfull, 2001, 2003), the virally encoded recombinase (Int) is a heterobivalent DNA binding protein. It consists of a large carboxy-terminal domain that binds to ‘core-type' DNA sites (C in Figure 1B) and carries out the enzymatic steps of recombination. This domain is analogous to the well-studied monovalent tyrosine recombinase proteins Cre and Flp (Esposito and Scocca, 1997; Nunes-Düby et al, 1998). Additionally, Int contains a small amino-terminal domain that binds with high affinity to ‘arm-type' sites (N in Figure 1B) that are distant from the sites of DNA strand exchange. The ability of single Int protomers to bind distinct and widely separated DNA sites depends upon the presence of accessory proteins, which introduce sharp site-specific DNA bends and thereby bring the two classes of Int sites sufficiently close for the formation of specific Int bridges between the sites. Three site-specific accessory DNA-bending proteins and 16 protein binding sites are used to assemble large multiprotein recombinogenic complexes at specific loci, called att sites. Integrative recombination between attP and attB, on the phage and bacterial chromosomes, respectively, generates attL and attR sites at the junctions between the integrated prophage and the bacterial chromosome. During excisive recombination, complexes formed at the attL and attR sites recombine to regenerate attP on the excised viral DNA and attB on the restored E. coli chromosome. Excision is not the simple reverse of integration—the two reactions use different, partially overlapping subsets of protein binding sites (Figure 1) and different combinations of accessory DNA-bending proteins.
In this report, we focus on excisive recombination, which is distinguished by its requirement for the virally encoded DNA-bending protein, Xis, (or its use of the host-encoded Fis protein when Xis is limiting). The third DNA-bending protein, host-encoded IHF (integration host factor), is required for both integrative and excisive recombination reactions but binds a different subset of loci in each. The heterobivalent feature of Int in conjunction with the accessory DNA-bending proteins confers a sophisticated regulation that is sensitive to bacterial host physiology (Bushman et al, 1985; Gardner and Nash, 1986; Thompson et al, 1987a; Ball and Johnson, 1991; Ball et al, 1992; Ditto et al, 1994). These features, which are absent in simpler DNA recombination reactions, are fundamental to lambda's environment-sensitive modulation of the viral life cycle.
Although the λ Int pathway has been the subject of extensive genetic and biochemical analyses, it has been difficult with these traditional approaches to distinguish kinetically relevant from off-pathway species (a generic problem in the analysis of recombination pathways) and to measure the rates at which functionally important intermediates are processed in both the forward and reverse directions. Elucidating these mechanistic features is essential when determining how binding energy from the multiple protein–DNA interactions is used to achieve efficiency and directionality in the overall reaction. We addressed these issues by performing experiments in which recombination of individual isolated DNA molecules was observed from start to finish using light microscopy.
We visualized the behavior of single DNA molecules by using them to tether a microscopic bead to a flow chamber bed. The DNA-tethered bead exhibits Brownian motion restricted to a hemisphere of radius equal to the DNA contour length. Using light microscopy techniques to visualize and record this bead motion, any change in the effective length of the DNA tether can be determined in real time. The effective tether length changes when the mean end-to-end distance of the DNA is decreased by protein-mediated DNA looping or bending (Figure 1C; Finzi and Gelles, 1995), or when a portion of the DNA is excised.
Results
Complex assembly on att site DNA
To help elucidate the initial steps of the overall excision mechanism, we first determined the rates of formation and stability of the protein complex formed on a 1943 bp DNA containing a single attL (with its overlap region centered 301 bp from one end of the DNA). When Int, IHF, and Xis were added to a DNA tether containing attL, an abrupt decrease in effective tether length was detected in 100% of the molecules analyzed (Figure 2A and D, compare black and blue curves). The decrease (by 443±16 bp (mean±s.e.)) from the 1943 bp tether length of the naked DNA) is consistent with the formation of a bent nucleoprotein complex. In control experiments that used DNA without any att sites, addition of the three proteins had no discernable effect (Figure 2B and E, compare black and blue curves). Similarly, addition of Xis alone, which has no binding sites on attL, had no effect on the attL-containing DNA (data not shown). In contrast, IHF, which does have a single binding site on attL, was sufficient by itself to reduce the effective tether length by 284±15 bp (Figure 2D, compare black and red curves). Shortening by IHF alone is predicted from the >160° DNA bend it induces upon binding to its cognate site in attL (Rice et al, 1996). In accordance with previously published results (Moitoso de Vargas et al, 1989; Kim et al, 1990; Segall and Nash, 1996; MacWilliams et al, 1997), the significant (P=0.05) additional shortening observed with all three proteins over that seen with IHF alone is consistent with the attL complexes having at least one Int protomer simultaneously bound to and bridging N- and C-type DNA sites, thereby closing a DNA loop between these sites (Figure 1B). Behavior of cleavage-incompetent mutants K235A and H308L on attL DNA is indistinguishable from that of wild-type Int, demonstrating that formation of a stable attL complex does not require a covalent Int-DNA linkage (data not shown). This interpretation is consistent with the observation that in all experiments, addition of 0.2% SDS returned the DNA to its original length within experimental uncertainty. Taken together, the data suggest that the attL complexes observed here by single-molecule microscopy are identical, or very similar, to those characterized previously by genetics and solution biochemistry (Moitoso de Vargas et al, 1989; Kim et al, 1990; Segall and Nash, 1996; MacWilliams et al, 1997).
Figure 2.
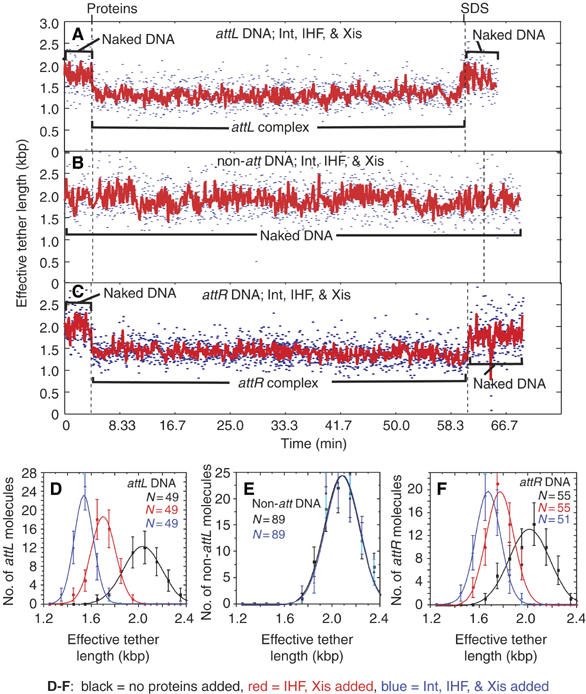
Specific complex formation on DNA molecules with only one att site. (A–C) Changes in the effective lengths of single DNA molecules in response to the addition of recombination proteins Int, Xis, and IHF. The three proteins or the SDS challenge were added at the times indicated by dashed lines. Both unfiltered (blue; 2.13 s time resolution) and filtered (red) data are shown. Noise in tethered-particle motion (TPM) measurements increases with the increasing tether length (Yin et al, 1994). Wild-type proteins were added to single DNA molecules containing a single attL (A), no att sites (B), or a single attR (C). (D–F) The distributions (±s.e.) of effective tether lengths (and Gaussian fits) compiled from all experiments for attL-containing molecules (D), non-att DNA molecules (E), or attR-containing DNA molecules (F). The measurement for each type of molecule was calculated as the mean of the 10.5 s of tether length data: commencing at t=0 min (before protein addition; black squares), commencing at t=7.5 min (after IHF and Xis addition; red circles), or commencing at t=7.5 min (after wild-type Int, Xis, and IHF addition; blue triangles).
Every attL DNA molecule observed adopted its final, shortened tether length within the 20 s mixing time in which no measurements could be made owing to the solution flow used to introduce the proteins. Every molecule remained at its final length for the duration of recording (20–60 min), even in experiments in which free IHF, Int, and Xis were removed from the chamber by buffer flow. These results show that complex formation is rapid (with an apparent first-order rate constant of >3.0 min−1) and is essentially irreversible under these reaction conditions.
In a similar manner, we looked for the formation of specific complexes on a DNA containing only a single attR site (overlap region centered 290 bp from one end). Like attL, the attR sequence used here has a single IHF binding site (Figure 1B) and behavior of this DNA in the presence of IHF and Xis was identical to that seen with attL (data not shown): apparent tether lengths of all molecules decreased by 241±7 bp during the mixing time (Figure 2F, compare black and red curves). The addition of Xis alone or in conjunction with IHF did not result in any additional apparent attR shortening (data not shown). When all three proteins were added, the attR DNA shortened by an additional 95 bp, to a final effective tether length 336±9 bp shorter than the original 1943 bp length (Figure 2C and F; compare black and blue curves in Figure 2F). Other characteristics of the attR complexes were identical to attL complexes: they formed rapidly (>3.0 min−1), their dissociation was never observed within the 60 min time of measurement (dissociation rate constant <0.017 min−1), they formed equally well with cleavage-incompetent IntK235A or IntH308L (data not shown), and they were disrupted by SDS. For both attL and attR, full complex formation depended upon the presence of IHF; neither Int nor Xis, individually or together, produced detectable shortening.
Complex assembly on attL+attR DNA
Having established that nucleoprotein complexes form independently on attL and attR, we examined complex formation on DNA molecules containing both attL and attR in their canonical orientation for excisive recombination and separated by 1353 bp (Figure 1B, top). Examples of single-molecule traces with this substrate upon addition of Int, IHF, and Xis are shown in Figure 3A and B. In 89 of the 94 molecules analyzed, the effective length decreased by 873±12 bp during the mixing time (Figure 3C). This reduction corresponds roughly to the sum of those measured with attL and attR individually (779±18 bp). Like those complexes seen with a single attL or a single attR, the tether remained shortened unless challenged with SDS and shortening occurred even with cleavage-deficient Int mutant K235A or H308L. This decrease is too small to be caused by DNA looping (Finzi and Gelles, 1995) between attL and attR; it likely reflects independent formation of the Int-containing attL and attR complexes.
Figure 3.
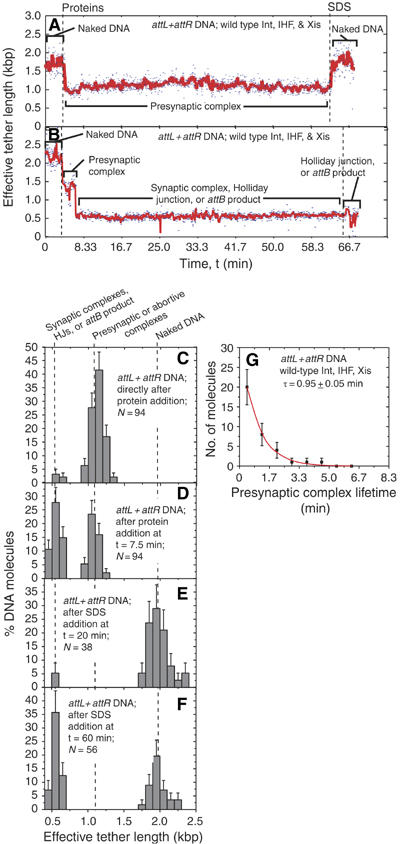
Complex formation and recombination of single attL+attR-containing molecules. (A, B) Changes in the effective lengths of single attL+attR DNA molecules in response to the addition of recombination proteins Int, Xis, and IHF. (A) Example of a molecule that failed to synapse within the duration of observation. (B) Example of a molecule that synapsed and went on to recombine. The three proteins or the SDS challenge were added at the times indicated by dashed lines. (C–F) Distributions (±s.e.) of effective tether lengths of attL+attR molecules. In (D), t=7.5 min is used because no additional synaptic complexes form after that time. In (E) and (F), the measurement was immediately preceded by addition of SDS at t=20 or 60 min, respectively. (G) Distribution of presynaptic complex lifetimes for attL+attR molecules that went on to form recombinant product. The lifetime τ was determined by fitting with a scaled exponential probability function (see Materials and methods) and is the inverse of the synapsis rate constant.
Synapsis of att-site complexes
If the DNA between the attL and attR sites is looped out or excised, the effective tether length is expected to be ⩽590 bp (Figure 1B). (If the DNA arms exit from a looped complex at an angle <180°, the effective tether length will be shorter than the summed length of the flanking DNA.) A further reduction in effective length (to 555±12 bp) was observed in 53% of the attL+attR molecules after the formation of att-site complexes (Figure 3B and D). (For the remaining 47%, the trace looked like that shown in Figure 3A; the molecules failed to synapse and their length distributions were similar at 7.5 min (Figure 3F) and just before SDS addition at 60 min (data not shown). The second shortening is likely caused by one or more of three key events that occur sequentially in recombination (Figure 1A): (a) association of the independent attL and attR complexes to form a non-covalent synaptic complex, (b) exchange of the first pair of DNA strands to generate a covalent four-way (Holliday) junction, or (c) resolution of the Holliday junction by a second pair of DNA strand exchanges to generate completed recombinant molecules. To help determine which of these events had occurred, buffer containing 0.2% SDS was added to one set of 38 attL+attR molecules at 20 min after protein addition. All but two of the 19 molecules that had shortened to ∼550 bp returned to their original effective length of 1943 bp (Figure 3E). Thus, after 20 min reaction, 95% of the molecules that had synapsed had not yet formed stable Holliday junction or recombinant products, neither of which would be disrupted by SDS (Figure 1C). In contrast, when SDS was added to the second set of samples 60 min after addition of proteins, 100% of the 31 attL+attR DNAs that had synapsed retained their ∼550 bp length, indicating that by this time all of these molecules were either Holliday junctions or recombinant products (Figure 3B and F).
In the molecules that synapsed, the time intervals between addition of proteins and the shortening to ∼550 bp were exponentially distributed (Figure 3G), consistent with a single rate-limiting step (rate constant 1.0 min−1) for synapsis. Thus, the attL and attR nucleoprotein complexes that become competent for synapsis do so faster than we can measure, probably within the 20 s period during which the proteins are introduced.
Recombination
To distinguish between Holliday junctions and recombinant products, we performed additional experiments using DNA in which the attR orientation was inverted with respect to attL. Recombination of this attLinvattR substrate inverts (rather than deletes) the intervening 1353 bp DNA segment leaving the overall DNA length unaltered (Figure 1B). This allows differentiation of long tether length recombinant products from short tether length Holliday junction. The presence of Xis inhibits any further recombination of the attP- and attB-containing inversion product (Abremski and Gottesman, 1982).
Recombination reactions with inverted or directly repeated att sites are expected to proceed with the same kinetics (Craig and Nash, 1983) and through the same intermediates (Nash and Pollock, 1983; Pollock and Nash, 1983). Consistent with this expectation, attLinvattR molecules to which Int, IHF, and Xis were added appeared similar to presynaptic and synaptic complexes observed on attL+attR molecules (see Figure 4A). That is to say, presynaptic and synaptic complexes form with the same effective tether lengths and kinetics on attLinvattR and attL+attR DNA (compare Figure 4B and C with 3G and D). When SDS was added to attLinvattR samples after 60 min reaction time, all 23 of the total attLinvattR molecules that had synapsed returned to their original 1943 bp length (Figure 4D). This implies that none of these molecules exist as Holliday junction (effective tether length ⩽590 bp) after 60 min reaction. Taken together with the results from attL+attR substrates, these data suggest that all synapsed complexes complete recombination within 60 min. Consistent with these interpretations, the fraction of single molecules completing excision at 20 and 60 min is similar to that measured by gel electrophoresis for DNA free in solution under similar conditions (data not shown).
Figure 4.
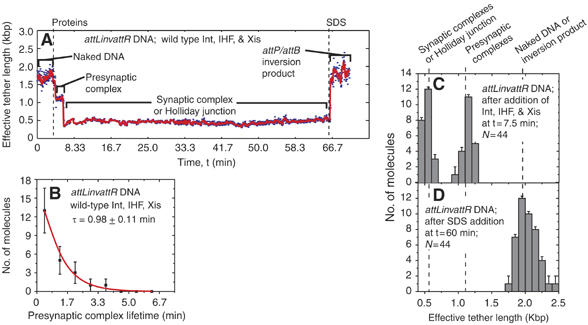
Kinetics of recombination examined with an inverted repeat substrate. (A) Changes in the effective lengths of a single DNA molecule with the attL and attR orientated as inverted repeats (attLinvattR) in response to the addition of recombination proteins Int, Xis, and IHF. The three proteins or the SDS challenge were added at the times indicated by dashed lines. SDS addition to this substrate at 60 min yields a recombination product identical in size to the substrate (but with the inverted DNA between att sites) rather than an ∼590 bp Holliday junction. (B) Distribution of presynaptic complex lifetimes for attLinvattR molecules that went on to synapse. The lifetime τ was determined by fitting with a scaled exponential probability function (see Materials and methods) and is the inverse of the synapsis rate constant. (C, D) The distributions of effective tether lengths of all attLinvattR molecules at t=7.5 min (after the addition of Int, IHF, and Xis; C) or t=60 min (after SDS addition; D).
DNA cleavage
To measure the forward and reverse rates of synaptic complex formation without the complication of covalently trapped intermediates, we performed experiments with cleavage-incompetent Int mutants. When IntK235A (Figure 5A) or IntH308L (data not shown) was added with Xis and IHF to attL+attR substrate, synaptic complexes formed with the same efficiency (∼50%), rate (compare Figure 5B and 3G), and apparent tether length as those formed with wild-type Int. However, unlike the wild-type complexes, formation of mutant synaptic complexes was reversible: the complexes dissociate with an apparent rate constant of 0.9 min−1 (Figure 5C), compared to wild-type synaptic complex dissociation, which was never observed within 60 min (k<0.017 min−1). We conclude that although synaptic complexes form rapidly, they are kinetically unstable unless trapped by a subsequent DNA cleavage step.
Figure 5.
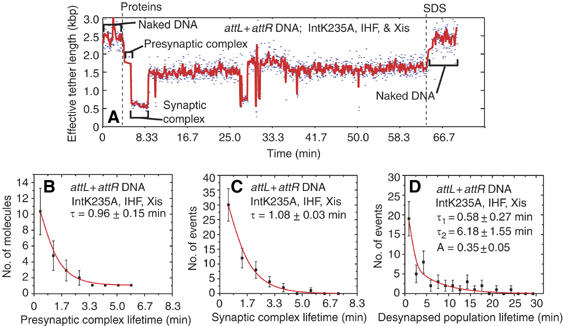
Unstable synaptic complexes form with cleavage-deficient Int mutants. (A) Changes in the effective lengths of single attL+attR molecules in response to the addition of recombination proteins IntK235A, Xis, and IHF. (B, C) Lifetime distributions from all single-molecule records after the addition of IntK235A, Xis, and IHF to attL+attR molecules: lifetimes of (B) initial presynaptic and (C) synaptic complexes formed with IntK235A. The lifetime τ for each distribution was determined by fitting to a scaled exponential probability function. The reciprocal of τ is the apparent rate constant for synapsis of IntK235A-containing presynaptic complexes (B) or the apparent rate constant for desynapsis of IntK235A-containing synaptic complexes (C). (D) Lifetimes of the desynapsed population formed after IntK235A-mediated synapsis (e.g., 11–26 min in panel A). The parameters τ1, τ2, and A were determined by fitting to a scaled biexponential probability function and determine the rate constants for resynapsis (see Discussion).
Comprehensive reaction pathway
The single-molecule experiments reported here delineate the kinetics and pathway of the excision reaction from start to finish (Figure 6). Nucleoprotein complex formation, synapsis, and DNA cleavage are rapid relative to the formation of a stable Holliday junction. Although DNA cleavage is not strictly required for synaptic complex formation, it stabilizes the synaptic complex. It is striking that virtually every observed cleavage-stabilized synaptic complex infallibly produced recombinant products.
Figure 6.
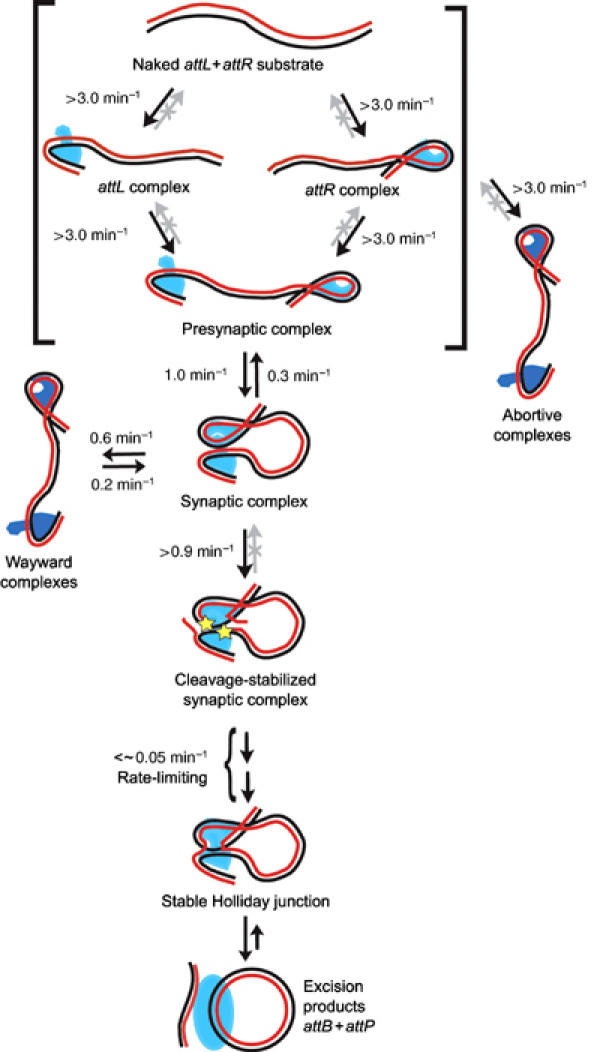
Mechanism of bacteriophage λ excision. The specified apparent first-order rate constants are the lower limits for reactions at 60 nM Int, 6 nM IHF, and 250 nM Xis. All reactions within brackets are completed within the mixing time of 20 s and, therefore, have lower rate limits of 1/20 s=3.0 min−1. Gray arrows denote processes that were not observed over the 60 min recording time and, therefore, have rate constants <∼0.017 min−1. Abortive complexes could arise from any of the species within brackets. Covalent phosphotyrosine linkages (stars) resulting from DNA cleavage are likely to occur in attL before synapsis (not shown) and in attR following synapsis (Nash and Robertson, 1989).
One implication of our kinetic model is that DNA cleavage intermediates covalently bound to Int (e.g., cleavage-stabilized synaptic complex; Figure 6) should be trapped in ensemble biochemical experiments by quenching the reaction with SDS between ∼2 and 20 min after protein addition. However, this was not observed in earlier studies; it was only through the use of suicide substrates or peptide inhibitors that significant amounts of covalent intermediate could be detected (Kitts and Nash, 1987, 1988; Nunes-Düby et al, 1987; Pargellis et al, 1988; Burgin Jr et al, 1995; Cassell et al, 2000). One explanation for the inability to detect covalent complexes is that SDS fails to quench Int ligation activity fast enough to prevent reformation of the initial phosphodiester bonds. We have demonstrated that this is the case in an adaptation of a previously described ligation assay (see Supplementary data; Pan and Sadowski, 1992).
Discussion
Int distribution among presynaptic complexes
The results presented in this investigation suggest that excisive synapsis occurs between two Int-containing presynaptic complexes that independently assemble on both attL and attR. Addition of Int to DNAs containing either an attL or an attR results in complexes differing from those assembled with IHF and Xis alone (compare blue and red curves in Figure 2D for attL; Figure 2F for attR). A model in which an attL complex containing 1–3 Int protomers synapses with an attR complex containing 1–3 Int protomers to form an active tetramer fits published data (Bauer et al, 1986; Nunes-Düby et al, 1989; Kim et al, 1990; Numrych et al, 1990; Kim and Landy, 1992) and the experiments reported here. However, owing to the limited precision of the effective DNA length measurements, we cannot definitively rule out the formal possibility that when formed in the presence of a partner attL site, the attR nucleoprotein complex contains only accessory proteins.
Rapid synapsis
For the intramolecular reaction studied here, in which attL and attR are held in close proximity, separated by only ∼1.3 kb of DNA, synapsis was found to be extremely rapid (1.0 min−1; Figure 3G). This contrasts with solution studies of a bimolecular reaction where synapsis requires long-distance diffusion and is thus dependent on the concentration of the DNA partners; under such conditions, it was inferred that synapsis is the slowest step in the excision reaction (Bankhead et al, 2003). In addition to diffusion, the rate at which presynaptic complexes find each other in vivo is likely determined by several factors that are not mimicked by any of the in vitro systems, such as DNA supercoiling, nucleoid structure, cation concentration, etc. However, the single-molecule experiments reported here strongly suggest that there are no intrinsic mechanistic features of the recombination pathway that make synapsis a slow step.
Commitment
Commitment to recombination occurs early in the pathway. Surprisingly, synaptic complexes formed with wild-type Int go on to recombine with ∼100% efficiency. The results with cleavage-incompetent Int mutants argue that DNA cleavage, or some closely associated subsequent step, commits the synaptic complex to recombination. Moreover, this committing step must occur before or within 1.1 min after synapsis (>0.9 min−1; Figure 5C); otherwise, we would have observed some short-lived synaptic complexes with wild-type Int.
A mechanism for the cleavage-dependent stabilization of synapsis is suggested by biochemical and structural studies showing that Int forms bridges between attL and attR complexes by binding to one DNA via its N-terminal domain and the other via its C-terminal domain. The latter domain can cleave DNA, and the resulting covalent phosphotyrosine linkage would stabilize the Int bridge (Moitoso de Vargas et al, 1989; Kim et al, 1990; Segall and Nash, 1996; Biswas et al, 2005; Radman-Livaja et al, 2005). The stabilized Int bridge(s) could, in principle, result from DNA cleavage(s) in attL and/or in attR. However, experiments with DNA substrates designed to trap cleavage events suggest that attL cleavage is likely to occur before synapsis whereas attR cleavage is likely to occur only after synapsis (Nash and Robertson, 1989).
Synaptic complexes formed with cleavage-deficient Int are expected to be kinetically less stable as they cannot form a covalent Int-DNA phosphotyrosine linkage. The data show that dissociation of these unstable complexes (0.9 min−1 from Figure 5C) generates a mixed population of ‘desynapsed' complexes (inferred from the bi-exponential lifetime distribution shown in Figure 5D). Based on the amplitude, about 35% of the complexes in this population resynapsed at a rate (1.7±0.5 min−1; corresponding to τ1 in Figure 5D) similar to the initial synaptic events (1.0±0.3 min−1; corresponding to Figure 5B), whereas other ‘wayward' complexes re-formed synaptic complexes at a much slower rate (0.2±0.1 min−1; corresponding to τ2 in Figure 5D; see Figure 6). The structures of the wayward complexes must differ from the presynaptic complex initially formed, for example, by altered distribution of Int protomers between the two att sites.
It is unlikely that the short synaptic lifetimes observed with cleavage-incompetent Int mutants are due to misfolding of or altered complex formation by the mutant proteins. The two non-allelic mutants each form normal stable complexes on single-att DNAs (this work; Nunes-Düby et al, 2002), form synaptic complexes at the same rate (e.g., Figure 5B versus Figure 3G), and assemble as stable tetramers on Holliday junctions (M Radman-Livija and A Landy, unpublished, 2003). Additionally, in other tyrosine recombination systems, the mutants used here have essentially unaltered tetrameric structures and have no known kinetic defects other than DNA cleavage (Wierzbicki et al, 1987; Jayaram et al, 1988; Parsons et al, 1988; Amin and Sadowski, 1989; Serre and Jayaram, 1992; Guo et al, 1999).
One of the striking results from these single-molecule studies is the finding that whereas only ∼50% of the att complexes formed are competent with respect to synapsis, virtually 100% of the synapsed complexes proceed through the complicated steps of DNA cleavage, strand exchange, and Holliday junction resolution to yield recombinant products. Although there are many possible reasons for the early formation of abortive complexes, for example, one or more inactive proteins, too many or too few proteins on an att site, an improperly assembled complex, or an impaired DNA substrate, it is tempting to speculate that rapid trapping of these abortive species, and/or their inability to form productive synaptic complexes, serves as a ‘functional filter' that prevents them from entering the recombination pathway where they might be prone to initiate abortive genome-damaging DNA cleavages.
Stable Holliday junction formation is rate limiting
We find that the rate-limiting step of excision occurs after synapsis, but closely precedes or is concomitant with the appearance of stable Holliday junction. Our single-molecule results are consistent with the observation that rates of stable Holliday junction formation are similar to the rates of the overall excision reaction (Bankhead et al, 2003). We conclude that once the rate-limiting barrier to forming a stable Holliday junction is overcome, resolution of the Holliday junction and the formation of product proceed rapidly.
It is likely that some conformational change stabilizes the Holliday junction and is the slow step in the reaction. Several candidates for this rate-limiting conformational change, for example, the scissoring movement of Holliday junction arms, the shift in the localized DNA bend, or the reorientation of active and inactive Int pairs, are suggested by comparison of different X-ray crystal structures of tyrosine recombinase family members complexed with four-armed DNAs (Guo et al, 1997, 1999; Gopaul et al, 1998; Van Duyne, 2001, 2002; Chen and Rice, 2003).
Irreversibility
The underlying chemical reaction catalyzed by the tyrosine recombinase family is isoenergetic and requires no high-energy cofactors (Figure 1A). For some family members, a sequential pair of forward and reverse reactions is critical to their biological function, such as 2μ plasmid copy number maintenance by Flp in Saccharomyces cerevisiae (Futcher, 1986; Cox, 1989; Jayaram et al, 2002). For excision of the phage λ chromosome, the overall reaction is driven by highly favorable protein–DNA interactions and the final product is presumed to be a highly stable nucleoprotein complex (as opposed to naked DNAs) that has a lower energy than any on-pathway intermediate in the reaction. Given the reversibility of the underlying chemistry, the irreversibility we observe at each on-pathway step of the λ excision reaction (except synapsis) is impressive. The present experiments clearly show that the overall directionality of excisive recombination is a direct consequence of an orchestrated sequence of protein–protein and protein–DNA interactions that efficiently drive the reaction forward through nearly every step.
Materials and methods
DNA substrate preparation
All DNAs used to tether beads were 1943 bp and were made using common polymerase chain reaction (PCR) techniques (see Supplementary data for details).
Protein preparation
Recombination proteins were expressed and purified as previously described for Int, IntK235A, and IntH308L (Sarkar et al, 2001), Xis (Warren et al, 2003), and IHF (Nash et al, 1987).
Bead preparation
Polystyrene beads (0.199 μm diameter; 10% solids; Seradyn Inc.) were coated with covalently coupled polyclonal anti-digoxigenin IgG from sheep (Roche) as described (Perkins et al, 2004). The coated beads were separated from free IgG on a 25 × 300 mm Sepharose CL-4B column (Sigma).
Flow chamber assembly
Glass coverslip flow chambers were prepared (Berliner et al, 1994) and the surface was derivatized with a mixture of polyethylene glycol and biotinated polyethylene glycol (Nektar), then coated with Avidin DN (Vector Labs) as described (Ha et al, 2002). Anti-dig-coated beads and DNAs were mixed in recombination buffer at a 5:1 molar ratio and incubated for 1–2 h. For surface attachment, biotinated DNAs were flowed into the chamber and incubated for 30 min before washing with 7–10 chamber volumes of recombination buffer.
Recombination assays
All recombination reactions (single molecule and test tube) were carried out in 50 mM tris(hydroxymethyl)-aminomethane at pH 8.0, 75 mM NaCl, 11.3 mM CaCl2, 1 mM EDTA, 1 mM DTT, 0.005 mg/ml fish sperm DNA (Sigma), and 0.5 mg/ml molecular biology grade BSA (Roche). Microscopically observable compaction of the DNA prevented the use of spermidine in the reaction; however, the overall rate of excision was unaffected by its substitution with CaCl2 in these (data not shown) and previously reported assays (Nash and Robertson, 1989). Reactions were stopped in the same buffer supplemented with 0.2% SDS.
Optimal protein concentrations were empirically determined in ensemble experiments using attL+attR substrate internally labeled by PCR with [32P]-deoxyadenosine. Substrate DNA was resolved from attP and attB products by electrophoresis on a 1% agarose gel supplemented with 1% Synergel (Diversified Biotech). Gels were dried and radioactivity was quantified. Average excision rates were determined using a Fuji BAS-2500 phosphorimaging scanner. Protein concentrations of 6 nM IHF, 250 nM Xis, and 60 nM Int gave maximal excision after 60 min at 22°C and were used in all single-molecule reactions.
Instrument configuration
DNA-tethered beads were observed using video-enhanced differential interference contrast light microscopy at 22°C and recorded at 30 Hz. Subsequent averaging of 64 frame blocks resulted in a time resolution of 2.13 s. Molecule-to-molecule reproducibility and long-term drift stability of the TPM experiment were greatly improved over that obtained previously (Yin et al, 1994) by incorporating an automatic focusing system into the microscope. After every set of 10 averaged frames, data acquisition was temporarily suspended and a computer-controlled stepper motor was used to refocus the image of a pre-selected surface-attached bead (Wong and Gelles, manuscript in preparation). The recordings were analyzed as described previously (Gallacher Jr and Wise, 1981; Yin et al, 1994) with the following modifications: the spatial extent of Brownian motion of each tethered bead was measured as described (Finzi and Gelles, 1995), except that image size data from multiple immobile reference beads were averaged in order to improve the signal-to-noise ratio. Brownian motion data were filtered with a three-point recursive median filter (Gallacher Jr and Wise, 1981), then converted to effective tether length using the previously reported empirical calibration method (Yin et al, 1994; Tolic-Norrelykke et al, 2004). Only those molecules that, before protein addition, exhibited a mean end-to-end distance between 1700 and 2300 bp were used for analysis. Molecules that stuck to the chamber surface (length ⩽0 bp) for more than 10 averaged frames at any time during the recording were excluded. Significance tests for changes in effective tether length were performed using a non-parametric Monte Carlo randomization method.
Single-molecule kinetics
To measure state lifetimes, effective tether length thresholds were used to discriminate between various DNA complexes. For both attL+attR and attLinvattR DNAs, molecules with tether lengths of 700–1400 bp were considered to be presynaptic, abortive, or wayward attL and attR complexes, whereas those with lengths 300–700 bp were considered to be synaptic complexes, Holliday junctions, or attB excision products. Owing to frame averaging, the effective DNA lengths of short-lived complexes (<tmin=6.3 s) could not be accurately determined and, therefore, were excluded from subsequent analysis. The remaining data were grouped in bins of width 50 s (Figures 3G, 4B, 5B, and C) or 100 s (Figure 5D). A nonlinear least-squares algorithm was used to fit the binned frequency (η) versus lifetime (t) data to a scaled exponential probability distribution function (Figures 3G, 4B, 5B, and C), 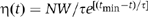 , or a scaled biexponential probability distribution function (Figure 5D)
, or a scaled biexponential probability distribution function (Figure 5D)
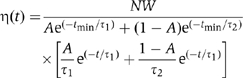
where N is the total number of observed lifetimes and A, τ, τ1, and τ2 are fit parameters (Colquhoun and Sigworth, 1995). Histograms are plotted with error bars representing the expected standard error η1/2.
Supplementary Material
Supplementary Data
Acknowledgments
We thank members of the Landy and Gelles labs for helpful discussions; Johnson Chung, Hung Wen Li, and Larry Friedman for help with intricacies of single-molecule experiments; Christine Lank for protein preparation; and Joan Boyles for manuscript preparation. We would especially like to thank Ying Tao for assistance in setting up ligation assays and Stephen Kowalczykowski for his time and comments regarding the manuscript. This work was supported by NIH grants GM43369 to JG and GM62723 and GM33928 to AL.
References
- Abremski K, Gottesman S (1982) Purification of the bacteriophage λ xis gene product required for λ excisive recombination. J Biol Chem 257: 9658–9662 [PubMed] [Google Scholar]
- Amin AA, Sadowski PD (1989) Synthesis of an enzymatically active FLP recombinase in vitro: search for a DNA-binding domain. Mol Cell Biol 9: 1987–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azaro MA, Landy A (2002) λ Int and the λ Int family. In Mobile DNA II, Craig NL, Craigie R, Gellert M, Lambowitz A (eds) pp 118–148. Washington, DC: ASM Press [Google Scholar]
- Ball CA, Johnson RC (1991) Multiple effects of Fis on integration and the control of lysogeny in phage λ. J Bacteriol 173: 4032–4038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball CA, Osuna R, Ferguson KC, Johnson RC (1992) Dramatic changes in Fis levels upon nutrient upshift in Escherichia coli. J Bacteriol 174: 8043–8056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankhead TM, Etzel BJ, Wolven F, Bordenave S, Boldt JL, Larsen TA, Segall AM (2003) Mutations at residues 282, 286, and 293 of phage λ integrase exert pathway-specific effects on synapsis and catalysis in recombination. J Bacteriol 185: 2653–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer CE, Hesse SD, Gumport RI, Gardner JF (1986) Mutational analysis of integrase arm-type binding sites of bacteriophage lambda. J Mol Biol 192: 513–527 [DOI] [PubMed] [Google Scholar]
- Berliner E, Mahtani HK, Karki S, Chu LF, Cronan JE Jr, Gelles J (1994) Microtubule movement by a biotinated kinesin bound to a streptavidin-coated surface. J Biol Chem 269: 8610–8615 [PubMed] [Google Scholar]
- Biswas T, Aihara H, Radman-Livaja M, Filman D, Landy A, Ellenberger T (2005) A structural basis for allosteric control of DNA recombination by λ integrase. Nature 435: 1059–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgin AB Jr, Huizenga BN, Nash HA (1995) A novel suicide substrate for DNA topoisomerases and site-specific recombinases. Nucleic Acids Res 23: 2973–2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushman W, Thompson JF, Vargas L, Landy A (1985) Control of directionality in lambda site-specific recombination. Science 230: 906–911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell AM (1962) Episomes. In Advances in Genetics, Caspari EW, Thoday JM (eds) pp 101–145. New York, NY: Academic Press [Google Scholar]
- Cassell G, Klemm M, Pinilla C, Segall A (2000) Dissection of bacteriophage λ site-specific recombination using synthetic peptide combinatorial libraries. J Mol Biol 299: 1193–1202 [DOI] [PubMed] [Google Scholar]
- Chen Y, Rice PA (2003) New insight into site-specific recombination from Flp recombinase-DNA structures. Annu Rev Biophys Biomol Struct 32: 135–139 [DOI] [PubMed] [Google Scholar]
- Colquhoun D, Sigworth FJ (1995) Fitting and statistical analysis of single-channel records. In Single-channel Recording, Sakmann B, Neher E (eds) pp 483–585. New York: Plenum Press [Google Scholar]
- Cox M (1989) DNA inversion in the 2μ plasmid of Saccharomyces cerevisiae. In Mobile DNA, Berg DE, Howe MM (eds) pp 661–670. Washington, DC: American Society for Microbiology [Google Scholar]
- Craig NL, Nash HA (1983) The mechanism of phage lambda site-specific recombination: collison versus sliding in att site juxtaposition. In Mechanism of DNA Replication and Recombination, Cozzarelli NR (ed) pp 617–636. New York: Alan R Liss Inc. [Google Scholar]
- Ditto MD, Roberts D, Weisberg RA (1994) Growth phase variation of integration host factor level in Escherichia coli. J Bacteriol 176: 3738–3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito D, Scocca JJ (1997) The integrase family of tyrosine recombinases: evolution of a conserved active site domain. Nucleic Acids Res 25: 3605–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi L, Gelles J (1995) Measurement of lactose repressor-mediated loop formation and breakdown in single DNA molecules. Science 267: 378–380 [DOI] [PubMed] [Google Scholar]
- Futcher AB (1986) Copy number amplification of the 2 micron circle plasmid of Saccharomyces cerevisiae. J Theor Biol 119: 197–204 [DOI] [PubMed] [Google Scholar]
- Gallacher NC Jr, Wise GL (1981) A theoretical analysis of the properties of median filters. IEEE Transcations on Acoustics, Speech and Signal Processing ASSP-29: 1136–1141 [Google Scholar]
- Gardner JF, Nash HA (1986) Role of Escherichia coli IHF protein in lambda site-specific recombination. A mutational analysis of binding sites. J Mol Biol 191: 181–189 [DOI] [PubMed] [Google Scholar]
- Gopaul DN, Guo F, Van Duyne GD (1998) Structure of the Holliday junction intermediate in Cre-loxP site-specific recombination. EMBO J 17: 4175–4187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grainge I, Jayaram M (1999) The integrase family of recombinase: organization and function of the active site. Mol Microbiol 33: 449–456 [DOI] [PubMed] [Google Scholar]
- Guo F, Gopaul DN, Van Duyne GD (1997) Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature 389: 40–46 [DOI] [PubMed] [Google Scholar]
- Guo F, Gopaul DN, Van Duyne GD (1999) Asymmetric DNA-bending in the Cre-loxP site-specific recombination synapse. Proc Natl Acad Sci USA 96: 7143–7148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha T, Rasnik I, Babcock HP, Gauss GH, Lohman TM, Chu S (2002) Initiation and re-initiation of DNA unwinding by the Escherichia coli Rep helicase. Nature 419: 638–641 [DOI] [PubMed] [Google Scholar]
- Hickman AB, Waninger S, Scocca JJ, Dyda F (1997) Molecular organization in site-specific recombination: The catalytic domain of bacteriophage HP1 integrase at 2.7 Å resolution. Cell 89: 227–237 [DOI] [PubMed] [Google Scholar]
- Jayaram M, Crain KL, Parsons RL, Harshey RM (1988) Holliday junctions in FLP recombination: resolution by step-arrest mutants of FLP protein. Proc Natl Acad Sci USA 85: 7902–7906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayaram M, Grainge I, Tribble G (2002) Site-specific recombination by the Flp protein of Saccharomyces cerevisiae. In Mobile DNA II, Craig NL, Craigie R, Gellert M, Lambowitz AM (eds) pp 192–218. Washington, DC: ASM Press [Google Scholar]
- Kim S-H, Landy A (1992) Lambda Int protein bridges between higher order complexes at two distant chromosomal loci attL and attR. Science 256: 198–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Moitoso de Vargas L, Nunes-Düby SE, Landy A (1990) Mapping of a higher order protein–DNA complex: two kinds of long-range interactions in λ attL. Cell 63: 773–781 [DOI] [PubMed] [Google Scholar]
- Kitts PA, Nash HA (1987) Homology-dependent interactions in phage λ site-specific recombination. Nature 329: 346–348 [DOI] [PubMed] [Google Scholar]
- Kitts PA, Nash HA (1988) An intermediate in the phage λ site-specific recombination reaction is revealed by phosphorothioate substitution in DNA. Nucleic Acids Res 16: 6839–6856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JA, Hatfull GF (2001) Control of directionality in integrase-mediated recombination: examination of recombination directionality factors (RDFs) including Xis and Cox proteins. Nucleic Acids Res 29: 2205–2216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis JA, Hatfull GF (2003) Control of directionality in L5 integrase-mediated site-specific recombination. J Mol Biol 326: 805–821 [DOI] [PubMed] [Google Scholar]
- MacWilliams M, Gumport RI, Gardner JF (1997) Mutational analysis of protein binding sites involved in formation of the bacteriophage λ attL complex. J Bacteriol 179: 1059–1067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moitoso de Vargas L, Kim S, Landy A (1989) DNA looping generated by the DNA-bending protein IHF and the two domains of lambda integrase. Science 244: 1457–1461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash HA (1996) Site-specific recombination: integration, excision, resolution, and inversion of defined DNA segments. In Escherichia coli and Salmonella, Neidhardt FC, Curtiss III R, Ingraham JL, Lin ECC, Low KB, Magasanik B, Reznikoff WS, Riley M, Schaechter M, Umbarger HE (eds) pp 2363–2376. Washington: ASM Press [Google Scholar]
- Nash HA, Pollock TJ (1983) Site-specific recombination of bacteriophage lambda: the change in topological linking number associated with exchange of DNA strands. J Mol Biol 170: 19–38 [DOI] [PubMed] [Google Scholar]
- Nash HA, Robertson CA (1989) Heteroduplex substrates for bacteriophage lambda site-specific recombination: cleavage and strand transfer products. EMBO J 8: 3523–3533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nash HA, Robertson CA, Flamm E, Weisberg RA, Miller HI (1987) Overproduction of Escherichia coli integration host factor, a protein with nonidentical subunits. J Bacteriol 169: 4124–4127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numrych TE, Gumport RI, Gardner JF (1990) A comparison of the effects of single-base and triple-base changes in the integrase arm-type binding sites on the site-specific recombination of bacteriophage lambda. Nucleic Acids Res 18: 3953–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes-Düby S, Tirumalai RS, Kwon HJ, Ellenberger T, Landy A (1998) Similarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res 26: 391–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes-Düby SE, Matsumoto L, Landy A (1987) Site-specific recombination intermediates trapped with suicide substrates. Cell 50: 779–788 [DOI] [PubMed] [Google Scholar]
- Nunes-Düby SE, Matsumoto L, Landy A (1989) Half-att site substrates reveal the homology independence and minimal protein requirements for productive synapsis in λ excisive recombination. Cell 59: 197–206 [DOI] [PubMed] [Google Scholar]
- Nunes-Düby SE, Radman-Livaja M, Kuimelis RG, Pearline RV, McLaughlin LW, Landy A (2002) λ integrase complementation at the level of DNA binding and complex formation. J Bacteriol 184: 1385–1394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan G, Sadowski PD (1992) Ligation activity of FLP recombinase. The strand ligation activity of a site-specific recombinase using an activated DNA substrate. J Biol Chem 267: 12397–12399 [PubMed] [Google Scholar]
- Pargellis CA, Nunes-Düby SE, Moitoso de Vargas L, Landy A (1988) Suicide recombination substrates yield covalent λ integrase–NA complexes and lead to identification of the active site tyrosine. J Biol Chem 263: 7678–7685 [PubMed] [Google Scholar]
- Parsons RL, Prasad PV, Harshey RM, Jayaram M (1988) Step-arrest mutants of FLP recombinase: implications for the catalytic mechanism of DNA recombination. Mol Cell Biol 8: 3303–3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins TT, Li H-W, Dalal RV, Gelles J, Block SM (2004) Forward and reverse motion of single RecBCD molecules on DNA. Biophys J 86: 1640–1648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollock TJ, Nash HA (1983) Knotting of DNA caused by a genetic rearrangement: evidence for a nucleosome-like structure in site-specific recombination for bacteriophage lambda. J Mol Biol 170: 1–18 [DOI] [PubMed] [Google Scholar]
- Radman-Livaja M, Biswas T, Mierke D, Landy A (2005) Architecture of recombination intermediates visualized by In-gel FRET of λ integrase-Holliday junction-arm–DNA complexes. Proc Natl Acad Sci USA 102: 3913–3920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice PA, Yang S-W, Mizuuchi K, Nash HA (1996) Crystal structure of an IHF–DNA complex: a protein-induced DNA u-turn. Cell 87: 1295–1306 [DOI] [PubMed] [Google Scholar]
- Rudy C, Taylor KL, Hinerfeld D, Scott JR, Churchward G (1997a) Excision of a conjugative transposon in vitro by the Int and Xis proteins of Tn916. Nucleic Acids Res 25: 4061–4066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy CK, Scott JR, Churchward G (1997b) DNA binding by the Xis protein of the conjugative transposon Tn916. J Bacteriol 179: 2567–2572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski PD (1993) Site-specific genetic recombination: hops, flips and flops. FASEB J 7: 760–767 [DOI] [PubMed] [Google Scholar]
- Sarkar D, Radman-Livaja M, Landy A (2001) The small DNA binding domain of λ Int is a context-sensitive modulator of recombinase functions. EMBO J 20: 1203–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segall AM, Nash HA (1996) Architectural flexibility in lambda site-specific recombination: three alternate conformations channel the attL site into three distinct pathways. Genes Cells 1: 453–463 [DOI] [PubMed] [Google Scholar]
- Serre MC, Jayaram M (1992) Half-site strand transfer by step-arrest mutants of yeast site-specific recombinase Flp. J Mol Biol 225: 643–649 [DOI] [PubMed] [Google Scholar]
- Thompson JF, Moitoso de Vargas L, Koch C, Kahmann R, Landy A (1987a) Cellular factors couple recombination with growth phase: characterization of a new component in the λ site-specific recombination pathway. Cell 50: 901–908 [DOI] [PubMed] [Google Scholar]
- Thompson JF, Moitoso de Vargas L, Skinner SE, Landy A (1987b) Protein–protein interactions in a higher-order structure direct lambda site-specific recombination. J Mol Biol 195: 481–493 [DOI] [PubMed] [Google Scholar]
- Thompson JF, Waechter-Brulla D, Gumport RI, Gardner JF, Moitoso de Vargas L, Landy A (1986) Mutations in an integration host factor-binding site: effect on lambda site-specific recombination and regulatory implications. J Bacteriol 168: 1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolic-Norrelykke SF, Engh AM, Landick R, Gelles J (2004) Diversity in the rates of transcript elongation by single RNA polymerase molecules. J Biol Chem 279: 3292–3299 [DOI] [PubMed] [Google Scholar]
- Van Duyne GD (2001) A structural view of cre-loxp site-specific recombination. Annu Rev Biophys Biomol Struct 30: 87–104 [DOI] [PubMed] [Google Scholar]
- Van Duyne GD (2002) A structural view of tyrosine recombinase site-specific recombination. In Mobile DNA II, In Craig NL, Craigie R, Gellert M, Lambowitz AM (eds) pp 93–117. Washington, DC: ASM Press [Google Scholar]
- Voziyanov Y, Pathania S, Jayaram M (1999) A general model for site-specific recombination by the integrase family recombinases. Nucleic Acids Res 27: 930–941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren D, Sam M, Manley K, Sarkar D, Lee SY, Abbani M, Clubb RT, Landy A (2003) Identification of the λ integrase surface that interacts with the Xis accessory protein reveals a residue that is also critical for homomeric dimer formation. Proc Natl Acad Sci USA 100: 8176–8181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierzbicki A, Kendall M, Abremski K, Hoess R (1987) A mutational analysis of the bacteriophage P1 recombinase Cre. J Mol Biol 195: 785–794 [DOI] [PubMed] [Google Scholar]
- Yin H, Landick R, Gelles J (1994) Tethered particle motion method for studying transcript elongation by a single RNA polymerase molecule. Biophys J 67: 2468–2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Data