


Anja Mueller was born in Bremen, Germany, in 1968. She studied biology at the University of Regensburg, graduating with a Diplom in 1993. She obtained her Ph.D. degree from Washington University in St. Louis with Professor Karen L. Wooley in the field of polymer organic chemistry in 1998. The research focused on the synthesis, characterization, and applications of fluorinated hyperbranched polymers. In the same year she joined Professor David F. O’Brien’s group to pursue postdoctoral research combining her interests in biology and chemistry, concerning the controlled drug release from liposomes by light-induced polymerization. In 2001 she joined Clarkson University as an assistant professor. Her research interests are biomaterials, the utilization of biological energy for electronic devices, and molecular electronics.

David F. O’Brien was born in Litchfield, IL, in 1936. He received his A.B. degree from Wabash College in 1958 and his Ph.D. degree with Douglas E. Applequist from the University of Illinois in 1962. After spending nearly 25 years in research at the Eastman Kodak Research Laboratories in Rochester, NY, he joined the Department of Chemistry Faculty at the University of Arizona in 1987. His research interests include the polymerization of self-organized monomers for the preparation of novel supramolecular materials and the use of site-specific polymerization for the triggered release of reagents.
I. Introduction
Hydrated amphiphiles form various phases as a function of molecular structure, temperature, concentration, and pressure.1–4 There appears to be a one-to-one correspondence between the structures observed for hydrated amphiphiles and that for block copolymers.5 Amphiphiles are characterized by having a hydrophilic headgroup attached to at least one hydrophobic tail. The unfavorable interfacial enthalpic interaction between the hydrophobic tail(s) of the amphiphile with the polar water molecules induces the former to aggregate with the hydrophobic tail(s) of other amphiphiles.4 The hydrophilic headgroup therefore separates the water from the tail(s), in much the same way that the A–B junction of a diblock AB copolymer separates the two homopolymer blocks A and B.6 Self-organized arrays of non-covalently associated amphiphiles may exist as self-supported lamellar/vesicular, various bicontinuous cubic, or hexagonal/cylindrical phases. Amphiphiles are also frequently studied as supported assemblies, e.g., monolayers at the air–water interface, LB multilayers, or self-assembled monolayers. During the past two decades or so, the understanding of each of these supramolecular assemblies has advanced significantly. This progress is a consequence of fundamental and applied research in many laboratories. The advent of methods to polymerize supramolecular assemblies—first in monolayers in the 1970s, followed by bilayer vesicles in the early 1980s, and more recently in nonlamellar phases, i.e., cubic and hexagonal phases—has led to the creation of new materials, the development of new methods, and a widening perspective on the potential applications of these novel polymeric materials. These uses include the controlled delivery of reagents and drugs, the preparation of biological membrane mimics, the separation and purification of biomolecules, the modification of surfaces, the stabilization of organic zeolites, and the preparation of nanometer colloids, among others.
The concept of an area-minimizing surface has been used extensively to describe the morphologies of amphiphile/water systems.2,7 The free energy of the system is described by the topology of the surfaces. In this analysis, a spontaneous curvature term arises purely as a result of the fact that the dimensions of the microdomain are only a few orders of magnitude greater than that of the constituent molecules. This means that the shape of the interface is influenced by the interactions on a molecular level. In order for a system to achieve equilibrium, the various terms in the free energy expression, chief of which is the mean curvature, must be minimized. This theory has been extended to describe the effects of surface charge8 and branched alkyl chains9 on the formation of nonlamellar assemblies. The distribution of a mixture of lipids in nonlamellar phases has also been investigated.10
The identification of the morphology of amphiphile/water systems traditionally relies on methods such as X-ray and neutron diffraction scattering, differential scanning calorimetry (DSC), NMR spectroscopy (e.g., diffusion, 2H, 31P), and transmission and scanning electron microscopy (TEM, SEM). With the exception of the various electron microscopy methods, the characterizations are usually indirect. Though space-group identification and unit cell dimensions are readily obtained with diffraction methods, exactly how the molecules are organized in the unit cell for the amphiphile/water system has only been recently settled.11
In principle, the polymerization of supramolecular assemblies of amphiphiles could be accomplished by at least two strategies: (a) the formation of the hydrated phase from amphiphiles containing a reactive group, followed by either linear or cross-linking polymerization of all or a portion of the organic region of the phase, or (b) the prepolymerization of the amphiphile in isotropic organic media, followed by solvent removal, polymer purification, then hydration of the linear polymer to form the desired phase. This review will emphasize the first strategy. The second approach was successfully employed for the formation of polymerized monolayers at the air–water interface and their transfer to yield LB multilayers but has been infrequently described as a method for the polymerization of self-supported assemblies of amphiphiles.12,13 This review covers the reports, up to late-2000, of the polymerization of self-supported assemblies. The review emphasizes those publications that appeared since the extensive 1988 review by Ringsdorf et al.14 Less attention is given to the numerous studies of reactive amphiphiles in monolayers or multilayers, except in those cases that aid in the understanding of hydrated amphiphilic phases. The following sections review the various methods to polymerize hydrated amphiphiles in bilayer membranes and in nonlamellar phases. In addition, the characteristics of the resultant polymers and the polymeric materials are described, if they are reported by the authors.
II. Polymerization of Hydrated Bilayers
The lamellar phase consists of extended bilayers of hydrated amphiphiles with periodic smectic-like order. However, for experimental convenience most polymerization studies have focused on lipid bilayer vesicles, also known as liposomes. A lipid vesicle is a nearly spherical lipid bilayer shell that encloses an aqueous volume. Lipid vesicles with diameters from 25 nm up to several hundred nanometers can be prepared by detergent dialysis of lipid/detergent mixed micelles, ultrasonication, or extrusion of extended bilayers.15
The bilayer shell can be composed of tens of thousands of lipids with their hydrophilic headgroups exposed to water and their hydrophobic tails aggregated in a manner to reduce exposure to water. Although the lipids employed to form vesicles are generally double-chain amphiphiles, ion pairs of oppositely charged single-chain amphiphiles can also be effective.16,17 The exceptionally low water solubility of most lipids constrains them to the bilayer, where they laterally move past one another. A key characteristic of the lamellar phase is the rate of lateral diffusion.18 The lipid diffusion coefficient, D, is ca. 10−2 μm2 s−1, when the sample temperature is below the main phase transition temperature, Tm, i.e., the transition from the solid-analogous phase (Lβ) to the liquid-crystalline phase (Lα). When the temperature is greater than Tm, then D is ca. 1 μm2 s−1. The lipid bilayer in the fast diffusion regime provides an organized structure for polymerization reactions, which is sufficiently dynamic to permit monomers to diffuse to the growing polymer chain end. Electron microscopy and laser light scattering have been used to demonstrate that polymerization does not significantly alter the shape or diameter of bilayer vesicles. However, polymerization of vesicles can dramatically alter their properties.
Polymerizable groups have been incorporated into bilayer-forming amphiphiles by chemical synthesis (see previous reviews).14,19–26 Subsequent formation of bilayer assemblies yields a two-dimensional array of the polymerizable groups. The polymerizable moiety can be positioned anywhere along the lipid tails or linked (covalently or electrostatically) to the head-group. Polymerization of the lipid tails usually leads to abolition of the bilayer Tm, whereas polymerization in the headgroup does not. Covalent linkage of the lipid tails inhibits the formation of the gauche rotamers as well as the cooperativity typically observed as lipid bilayers undergo this phase transition. In some instances, polymerizable amphiphiles clearly mimic natural lipids, e.g., phosphatidylcholine (PC) and phosphatidylethanolamine (PE), whereas others are more similar to synthetic surfactants, e.g., quaternary ammonium salts (representative examples are shown in Figure 3). Synthetic routes to polymerizable PC and quaternary ammonium lipids were reported as early as 1980, due in part to the commercial availability of synthetic intermediates. On the other hand, the lack of corresponding intermediates for PE retarded the use of polymerizable PE until a general chemical synthesis was reported in 1995.27
Figure 3.
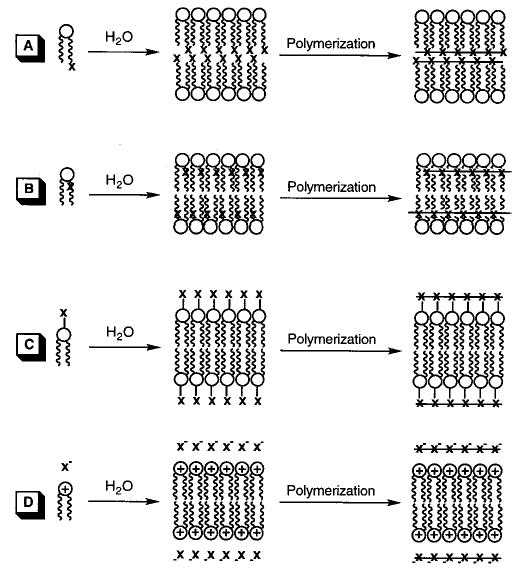
Substitution patterns for the polymerization of hydrated reactive amphiphiles that feature polymerization of the lipid tails at (A) the chain terminus and (B) near the lipid backbone or polymerization of reactive groups (C) covalently or (D) electrostatically associated with the hydrophilic headgroup.
(Reprinted with permission from ref 170. Copyright 1996 Springer-Verlag.)
A variety of polymerizable groups has been successfully employed including styryl, diacetylenyl, dienoyl, sorbyl, methacryloyl, acryloyl, and lipoyl (Figure 4). All, but the diacetylenyl group, can be polymerized in the more fluid liquid-crystalline phase (Lα). In contrast, diacetylenic amphiphiles are only polymerized efficiently in the solid-analogous phase (Lβ) or in other solidlike assemblies, e.g., tubules, and the condensed phase of Langmuir monolayers. In many instances assemblies composed of lipid diacetylenes could only be partially polymerized due to the topotactic nature of the reaction. This contrasts with the other reactive amphiphiles that may be converted to polymer in high yield (>90%). Photopolymerization is especially effective for diacetylenyl, styryl, dienoyl, and sorbyl amphiphiles, whereas thermally sensitive radical initiators are frequently used for the styryl, dienoyl, sorbyl, acryloyl, and methacryloyl compounds. Redox initiators may also be employed for these monomers.
Figure 4.
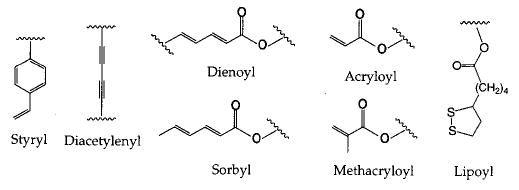
Examples of polymerizable groups which have been incorporated into polymerizable amphiphiles.
A. Acryloyl and Methacryloyl Monomers
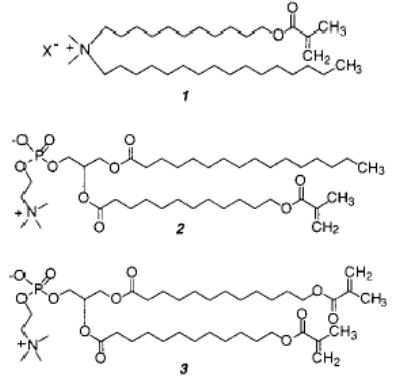
One of the first examples of polymerizable bilayer vesicles was designed by Regen and co-workers by incorporating a methacryloyl group into an ammonium amphiphile (1).28 Small unilamellar vesicles of 1 were formed by sonication and then polymerized by addition of AIBN. The resulting vesicles retained sucrose and were stable in the presence of 25% ethanol. Similar experiments were done with a mono-methacryloyl-substituted PC (2) and bismethacryloyl PC (3).29 It was shown that the polymerization of these vesicles by direct UV irradiation increased the stability of the liposomes. Polymerization of vesicles of 2 occurred more slowly. The same monomers and an analogue that incorporated the methacrylate group into the headgroup, instead of the alkyl chain, were used in a more detailed characterization of polymerized vesicle membranes.30 Bilayer vesicles of 3 did not show a main phase transition above 5 °C. Electron spin resonance measurements indicated that polymerization of the membrane caused a decrease in membrane fluidity.30 Polymerization of vesicles of 3 appeared to occur with cross-linking and had a greater effect on the membrane fluidity than polymerization of vesicles from 2. The polymerized vesicles of 3 could not be solubilized with chloroform. This early study provided the initial evidence that cross-linking polymerization of bilayer vesicles produced a different material than linear polymerization of similar bilayer vesicles.
However, it should be noted that UV polymerization of monomers that absorb at short wavelengths, such as acryloyl- and methacryloyl-substituted lipids, can result in photodegradation of the polymers. Sackmann and co-workers observed that UV photopolymerization of a methacryloyl-substituted lipid produced long polymer chains at short exposure times.31 Continued irradiation of the sample to achieve high conversion from monomer to polymer caused a progressive decrease in the size of the poly-(methacrylate lipid). In contrast, the use of thermal initiators produced long polymer chains without degradation of the polymer.
A pair of methacryloylammonium lipids 4 and 5 were prepared for an early study of the effect of linear polymerization on membrane permeability.32 The reactive group is in the hydrophilic region of monomer 4 and in the hydrophobic region of monomer 5. The polymerizations were initiated with either the water-soluble 2,2′-azobis(2-amidinopropane dihydrochloride) (AAPD) or the hydrophobic AIBN, respectively. In both cases the linear polymerization occurred with retention of the vesicle structure.
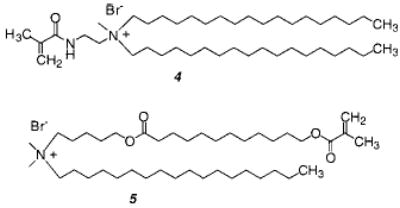
Upon polymerization, the membrane permeability of glucose decreased by a factor of 2–3. In addition, O’Brien and co-workers showed that sizable polymers, the estimated degree of polymerization was 500 (Mn = 3.5 × 105), could be formed by the radical chain polymerization of bilayer vesicles.33 Regen and coworkers found that a similar lipid gave polymers of comparable size.34 Given that the number of monomers in a vesicle is tens of thousands, it was clear that at high conversion there were tens to hundreds of polymer chains formed in each vesicle. Consequently, it was not surprising that linear polymerizations only moderately reduced the permeability of the vesicle to small solutes. Major modifications in vesicle permeability could only be accomplished by cross-linking polymerizations.32
A systematic study of the relationship between the monomer-to-initiator ratio in the vesicle and the number-average degree of polymerization (Xn) was conducted by Sells and O’Brien.35 The Xn was observed to vary from a few hundred to a few thousand monomers per polymer chain, and it was inversely dependent on the AIBN concentration at constant monomer concentration. These data were interpreted to mean that the bilayer-constrained linear polymer chains formed from 6 were terminated by primary termination at high conversion to polymer.35 A subsequent study of the rate of polymerization of bilayers of 6 also indicated that primary termination dominates at high conversion to polymer, whereas at low monomer conversions the more common bimolecular chain coupling or disproportionation are the dominant termination processes.36 These studies indicate that the growth of the polymer chains within the bilayer progressively limits the opportunity for two growing polymer chain ends to find one another. A further test of the primary termination hypothesis utilized 13C-enriched initiator to label the polymers formed during the AIBN polymerization of a mono-methacryloylPC.37 The 13C NMR spectrum of polymer chains initiated by 13CN-AIBN showed two major signals, which were assigned to head and tail addition to the methacryloyl monomer by reference to the literature and model compounds. Head addition is reaction of the AIBN fragment with the growing end of the polymer chain, i.e., primary termination.
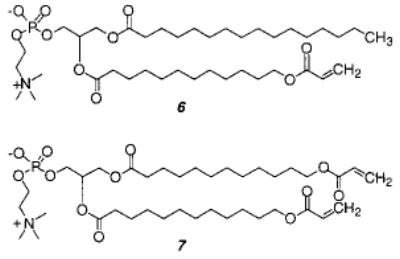
The effect of cross-linking was determined with mono- and disubstituted acryloyl PCs (6, 7).38 Both monomers were readily polymerized with AIBN at 70 °C. Because of the unequal depth of bilayer penetration of the two acryloyl groups in monomer 7, the sn-1 chain of 7 can serve as a cross-linking agent in the two-dimensional polymerization. When the bilayer membranes were composed of both 6 and 7, (30 ± 5)% of 7 was needed to achieve cross-linking of the hydrated lipid bilayer. The low cross-linking efficiency in the bilayer membranes may be a consequence of an intramolecular macrocyclization reaction of the sn-1 and sn-2 groups in the same lipid.39 Such a macrocyclization would yield a ring within a linear polymer backbone. Although such a reaction is statistically less favored in isotropic media, the constrained nature of the bilayer reduces the number of conformations available to the lipid tails, thereby enhancing the possibility of intramolecular macrocyclization.
The effect of linear and cross-linking polymerization on the lateral diffusion coefficient, D, of unreactive lipid probes was determined by fluorescence photobleaching recovery.40 Multilamellar vesicles were prepared from 6 and/or 7 plus 0.1 mol % N-4-nitrobenzo-2-oxa-1, 3-diazole-PE (NBD-PE). Prior to polymerization, the D was 3.8 ± 0.6 μm2 s−1 at 40 °C. The formation of linear poly-6 reduced the lateral diffusion of the NBD-PE to a greater extent as the length of poly-6 was increased, i.e., when Xn was 400, D was 0.60 ± 0.16 μm2 s−1 and when Xn was 1500, D was 0.23 ± 0.04 μm2 s−1. Cross-linking of the bilayers of 7 substantially decreased the diffusion of the NBD-PE probe by more than 2 orders of magnitude. Moreover, when the mole fraction of the disubstituted monomer 7 was greater than 0.3 in mixed bilayers of 6 and 7, the resulting polymerized bilayers appeared to be cross-linked, judging by the sharp decrease in D.
The effect on the rate and degree of polymerization of the change in lipid conformation and therefore lipid lateral diffusion that occurs at the Tm was examined by Lei et al.41 An Arrhenius plot of the log rate of polymerization vs T−1 showed a discontinuity near the Tm of the bilayer membrane. However, the change in reaction rate was moderate. An analysis of the activation parameters showed that the frequency factor increased when the temperature was greater than Tm (accelerating the reaction), whereas the activation energy also increased when the temperature exceeded the Tm (retarding the reaction). The modest increases in the rate of reaction with temperature were matched by moderate increases in the degree of polymerization. These studies indicate that radical chain polymerizations of acryloyl-substituted lipids, as well as sorbyl and other similar lipids, can be usefully employed in both the rapid diffusion liquidlike phase and at lower temperatures in the slow diffusion solid–like bilayer phase. Consequently, the radical chain polymerization of these monomeric lipids should be useful for the formation of polymeric films in condensed monolayers or multilayers at surfaces.
The self-organization and packing efficiency of a variety of methacryloyl monomers and polymers was determined in monolayers and LB multilayers by Ringsdorf and co-workers.12,13 One objective was to assess the effectiveness of forming the polymer by solution polymerization, then using the polymer to form lamellar structures. The three-dimensional nature of polymers makes them difficult to accommodate in two-dimensional monolayers or bilayers. This inherent organization problem was alleviated by the inclusion of flexible spacer groups to decouple the motions of the lipid alkyl chains from the motions of the polymer chain. The spacer concept has been used successfully in the synthesis of liquid-crystalline polymers.14 The monomers were based on ammonium and succinate lipids with hydrophilic spacer groups between the lipid and the methacrylate in the head-group (8). Different length hydrophilic spacers were used in either the side chain or the backbone (or both) of the resulting polymers.
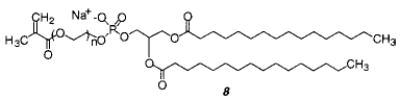
The backbone spacers were formed by copolymerization of the lipid monomer with hydroxyethyl acrylate (HEA). The incorporation of spacer groups into the side chain of the poly(lipid) made it possible to form well-ordered stable monolayers and LB multilayers. However, the monolayers were generally so well packed that only a solidlike phase was observed. The individual lipids apparently had insufficient freedom of motion to exhibit any liquidlike behavior. When the lipid monomer was copolymerized with HEA to yield polymers with both main and side chain spacers, the polymers had sufficient flexibility to form well-behaved monolayers that exhibit both liquid and solidlike phases. This strategy has been successfully used to create LB films composed of several hundred layers from reactive amphiphiles. In cases where the amphiphiles contained a chromophore, significant nonlinear optical properties were achieved by ordering the chromophore in an organized yet noncrystalline material.42
A study using glutamate-based acryloyl amphiphiles also emphasizes the effect of molecular packing on the polymerization of bilayers.43 The most efficient packing and polymerization were observed with a phenyl group in the headgroup and an ether linkage in the alkyl chain (9). There was a difference if the bilayer was polymerized in the solidlike vs the liquid-crystalline state. The alkyl chains obtained a certain degree of disorder during polymerization in the solidlike state, which slowly decreased during aging. The alkyl chains of polymers polymerized in the liquid-crystalline state did not reorder.
Lamellar phases and/or vesicles can be formed by the electrostatic association of cationic and anionic surfactant molecules known as ion-paired amphiphiles (IPA). Each surfactant moiety has a positive radius of curvature preferring to form micellar structures since the surface area of the charged headgroup is significantly larger than the cross-sectional area of the acyl chain. Kaler et al. reported the first spontaneous formation of a bilayer structure from various combinations of cetyl trimethylammonium tosylate (CTAT) and sodium dodecyl benzene sulfonate.16 When paired the electrostatic attraction between the oppositely charged surfactants reduces the effective area of the headgroup and the ion pair of the new IPA adopts a size similar to a zwitterionic PC. Monomer 10 is the single published example of a polymerizable IPA.44 Vesicles of 10 were polymerized by direct UV irradiation as well as by a water-soluble radical initiator, AAPD. Polymerized vesicles were stabilized to disruption by TX-100, NaCl, and NaI compared to the unpolymerized vesicles. Photopolymerization at 20 and 50 °C gave sizable polymers with Mn equal to 4 × 104 and 1.1 × 105, respectively. The photopolymerizations were performed in a solidlike phase, whereas the polymerization with AAPD was conducted above the Tm and gave a polymer with a Mn of 1.7 × 105.
Natural ionic lipids, e.g., the sodium salt of dialkyl phosphatidic acid, and synthetic ionic lipids, e.g., dimethyl dioctadecylammonium halide and sodium dihexadecyl phosphate (NaDHP), form bilayer vesicles with surface-associated counterions. Ion exchange of the sodium or halide counterions for polymerizable counterions either before or after vesicle formation can be used to form bilayer vesicles with an electrostatically associated reactive counterion.45–48 Vesicles composed of dimethyl dioctadecylammonium methacrylate were polymerized in the Regen laboratory by direct irradiation of the reactive counterion that was associated with the bilayer surface charges.45,47 Following the reaction, poly(methacrylic acid) was isolated by extraction of the cationic lipid and protonation of the polymer. If the monomeric counterion could be cross-linked, it was possible to observe a residual polymeric structure after the lipid was extracted. These so-called “ghost vesicles” were reported for the polymerization of surface-associated diallylamine.49 In the case of NaDHP vesicles, it was shown that charged polymers could be formed only on the outside, only on the inside, or on both sides of the DHP vesicles (Figure 6).48 Passage of NaDHP vesicles (70–100 nm diameter) through a cation exchange column containing choline methacrylate produced vesicles with this reactive counterion only on the outside of the vesicles. However, if the ion exchange preceded vesicle formation, all of the sodium ions were replaced by the choline methacrylate. Subsequent passage of NaDHP vesicles through a GPC column removed the exterior choline methacrylate, leaving vesicles with the reactive groups only on the inside of the vesicle. Photopolymerization of the reactive counterion was most effectively accomplished with UV irradiation, because water-soluble chemical initiators are usually charged resulting in ionic exchange with the electrostatically bound choline methacrylate. Nevertheless, the photopolymerization was successful. Linear polymerizations reduced the glucose permeability of the vesicles by a factor of about 3, which is similar to the effect found when vesicles of monomer 4 were polymerized.
Figure 6.
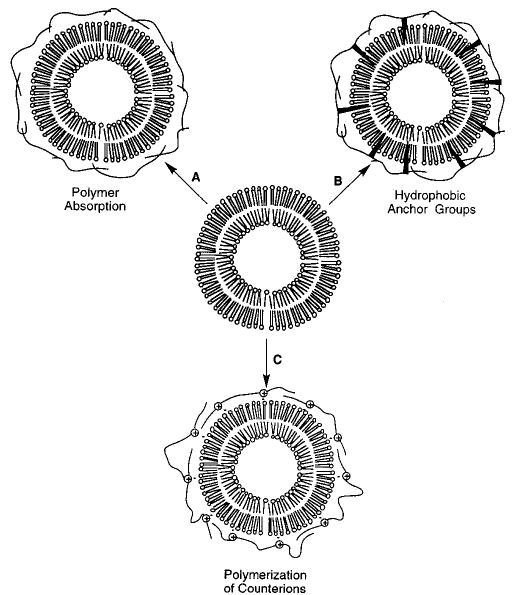
Strategies for the association of polymers with the surface of bilayer vesicles: (A) adsorption of a polymer to the lipid headgroups, (B) anchoring a polymer to the bilayer through hydrophobic groups associated with the polymer, and (C) polymerization of monomers that are electrostatically associated with the bilayer surface.
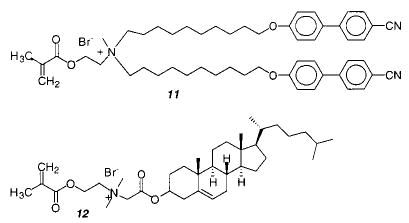
The parallel packing of amphiphilic chromophores (H-aggregation) in a bilayer can be detected by the blue shift of the UV absorption maximum. The cyanobiphenyl units of monomer 11 favor an H-aggregate structure prior to polymerization.50 However, polymerization of the methacryloyl headgroup changed the packing of the chromophores, presumably because the motions of the polymer are tightly coupled to the lipid. It would be interesting to examine the effect of a spacer group between the methacryloyl group and the lipid.
Monomer 6 has also been used for the modification of surfaces.51,52 Unilamellar liposomes were prepared and then used to form a lipid film on an alkylated glass surface. This procedure appeared to produce a monolayer with the lipid headgroup exposed to the water and the lipid tails in contact with the alkylated surface. The lipid film was thermally polymerized with AAPD to yield a modified surface film that was stable to transfer from water to air as well as to a shear force of 200 dyn/cm2 for up to an hour. This procedure could also be used to modify an alkylated hydrogel substrate.53 Monomer 6 was also used to form a polymerized PC monolayer under physiological benign conditions.54 Glass or a silicon wafer was coated with octadecyltrichlorosilane (OTS). Large unilamellar vesicles formed from the monoacryloylPC 6 were allowed to fuse onto the hydrophobic surface to form a monolayer. This monolayer was stabilized by the photoinitiated polymerization of the acrylate with visible light activation of eosin Y and triethylamine as the initiator. The resulting film was analyzed by contact angle measurements, X-ray photo-electron spectroscopic (ESCA) measurements, and polarized external reflectance IR spectroscopy. The best reaction conditions for the formation of a continuous, flat, and stable film were determined. It was shown by contact angle and desorption measurements that the film polymerized by light is more stable than the film polymerized by AAPD. Ross et al. successfully used sorbyl-substituted lipids to form uniform cross-linked bilayers on solid supports.55
A methacrylate cholesterol derivative (12) formed a stable lyotropic liquid-crystalline phase.56 Upon polymerization by free radical initiators or UV light, a phase change occurred to form stable, polymeric microvesicles. The vesicles were even more stable if acrylamide was used as a comonomer. Stable polymerized vesicles were also achieved with a variety of ammonium-based acrylate cholesterol monomers.57
The reaction kinetics of acrylates in a polymer was determined in an excimer study.58 A polyacrylate with a small amount of pyrene was spread on water under the exclusion of oxygen with a mixture of hydrophilic and hydrophobic acrylate monomer. It was determined that the rate of excimer formation is a power law expected for two-dimensional polymer melts and dense polymer solutions. The determined exponent suggests that the short-range dynamics of the monomer chains are constrained by the chains of the polymer in the mixture.
B. Dienoyl, Sorbyl, and Diene Monomers
Poly(methacrylates) are generally stiff polymers at room temperature. In contrast, poly(dienes) are rubber-like. The elastomeric properties of these polymers are attributed to low rotational barriers and weak intermolecular dispersion forces. In addition, they are normally easy to cross-link, either through the use of bis-diene comonomers in the original polymerization and/or by postpolymerization reaction of the double bond that remains after the initial polymerization. Polymerization of dienes or dienoate esters, such as monomer 13, can proceed by either a 1,4-, 3,4-, and/or 1,2-mechanism. Consequently, it is possible that different reaction conditions will produce different polymeric materials from the same bilayer membrane. Although recently more authors have taken care to establish the poly(diene) structure, to our knowledge there have not been any studies that intentionally attempted to modify the reaction in order to yield different poly(dienes).
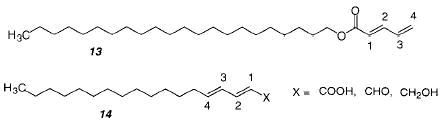
An early study of the polymerization of an amphiphilic dienoate 13 in LB multilayers found that the polymerization proceeded by a 3,4-mechanism.59 A study of similar monomers (14) in monolayers or LB multilayers found that the polymerization is dependent on the structure of the monomers.60 UV polymerization of 14 did not take place in solution, in the melt, or in a crystal but only in a monolayer. These results indicate that polymerization of these monomers requires a moderate degree of order. Laschewsky and Ringsdorf showed that polymerization of these monomers in LB multilayers occurred via 1,4-mechanism.61 A second step leads to an insoluble polymer, probably via cross-linking of the newly formed nonconjugated double bond. In another study, different configurations in the LB films of these monomers (X = COOH) led to different polymerization products, including a [2+2] cycloaddition product.62 Similar monomers were used in a study that employed UV or γ-irradiation in the crystalline state.63 In all cases, the polymerization was shown to proceed via a 1,4-mechanism. The amide-linked sorbate monomer reacted instead via a [2+2] cycloaddition mechanism when polymerized in a monolayer. A collection of polymerization conditions and results at interfaces has been published.64 A more recent study reported that the UV photopolymerization of solid-state octadeyl sorbate in the presence of air resulted in the formation of an alternating copolymer with a peroxide linkage between the monomers.65 On the other hand, the UV irradiation of crystals of (E,E)-naphthylmuconic acid in the presence of air gave polymers without peroxide linkages.66 This polymerization was found to proceed via a 1,4-mechanism. The structure of the resulting polymer was determined by 13C NMR and X-ray diffraction.
The longer conjugation length and longer wavelength of absorption (ca. 260 nm in water) of the dienoate monomers makes it straightforward to photopolymerize these monomers by direct irradiation with 254 nm light from a low-pressure mercury lamp. The absorption properties of dienoate monomers indicate a strong dependence upon acyl chain packing and media polarity. For example, at temperatures below Tm, sorbyl-substituted PCs exhibit a hypsochromic shift to shorter wavelengths (e.g., 242 nm) with a diminished extinction coefficient, indicating aggregation of the terminal sorbyl groups in the solidlike phase.67 A similar observation was reported for dienoyl-substituted PCs.68 Photopolymerization, thermal initiation, and redox initiation can be usefully employed to polymerize bilayer membranes of diene monomers. A comparison of these methods was reported by Tsuchida and co-workers for dienoyl-substituted PCs69 and by O’Brien and co-workers for sorbyl-substituted PCs.70
UV photopolymerization of the bis-dienoyl-substituted PC 16 at temperatures greater than Tm produced oligomers (Xn = 6), whereas photopolymerization at temperatures less than Tm gave somewhat longer polymers (Xn = 18).69 Polymerization of dienoyl monomer 15 at 8 °C with redox initiators gave polymers with a Xn of 28, whereas redox polymerization at 35 °C produced somewhat longer polymers (Xn = 45). Similar or slightly longer polymer chains were prepared by radical polymerization with AAPD (either photochemical or thermal decomposition) or by ionizing irradiation. While larger polymers were obtained when the photopolymerizations were performed at temperatures below the Tm, the opposite was observed for the photochemical initiation with AAPD, i.e., the Xn was greater at temperatures above the Tm. As expected the Xn varied inversely with the AAPD concentration.
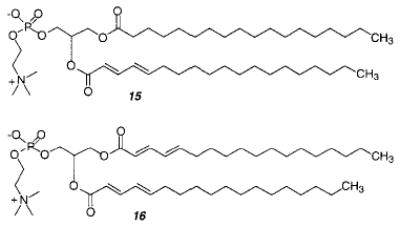
The UV photopolymerization of sorbyl-substituted PC 17 at temperatures above the Tm produced only oligomers. The rate of the photoreaction was directly proportional to the incident light intensity in a manner consistent with photoactivated addition of sorbyl monomers.70 The AIBN-initiated polymerization of monomer 17 and 18 at 60 °C produced linear and cross-linked polymers, respectively. In each case, the polymers were transesterified to remove the lipid headgroup and yield linear copolymers composed of random repeat units of methyl sorbate and methyl carboxynonanyl sorbate, which were soluble in tetrahydrofuran and analyzed by size exclusion chromatography relative to PMMA standards. The 1H NMR spectra of the transesterified polymers indicated the structures were 1,4-polymers.70 The number-average degree of polymerization (Xn) ranged from 50 to nearly 600 and was inversely proportional to the concentration of the initiator, [I]. These results, which suggest that chain termination at high conversion of SorbPC to polymer is dominated by primary termination, are similar to the behavior of acryloyl-substituted lipids in bilayers. The poly(AcrylPC)s were four times larger than the poly(SorbPC)s at a given ratio of monomer to initiator. The difference in reactivity is due to greater resonance stabilization of the sorbyl radical relative to the acryloyl radical.
Sisson et al. examined the cross-linking of bilayers as a function of the mole fraction of bis-SorbPC (18), where the reactive groups were located at the end of the lipid tails.38 The onset of cross-linking was determined by changes in lipid lateral diffusion, bilayer vesicle stability, and polymer solubility. These data indicated that a substantial mole fraction (0.30 ± 0.05) of the bis-SorbPC, as in the case of AcrylPC (see section II.A), was necessary for bilayer cross-linking. Analysis of the cross-linking and competing reactions suggested that the location of the reactive group, i.e., reaction site, in the amphiphile and therefore within the bilayer assembly influences the cross-linking efficiency. To assess this possibility the cross-linking of dienoyl-substituted phospholipids, (E,E)-DenPC 16, where the reactive diene is located near the glycerol backbone of the lipid, was compared with SorbPC. The cross-linking of (E,E)-DenPC was found to be substantially more efficient than that of SorbPC.71 It is proposed that this effect is partly a consequence of the relative probability of macrocyclization and cross-linking reactions.
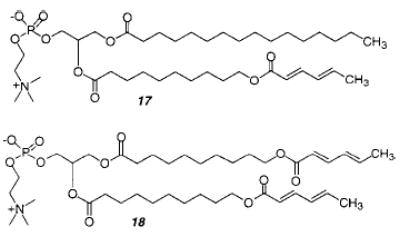
The visible light photosensitized polymerization of vesicles composed of either dienoyl or sorbyl monomers (15–18) was reported in 1997.72 These polymerizations can be sensitized to green or red light by conveniently incorporating ballasted cyanine dyes, i.e., dyes having hydrophobic chains, into the lipid bilayer. Irradiation of the vesicles with filtered light ensured that the light energy was absorbed by the dye rather than the polymerizable lipid. The effects of oxygen concentration, pH, light intensity, and temperature suggested that the sensitization reaction occurred by electron transfer from the dye to oxygen to yield superoxide anions. This may serve as the initiating species or react further to produce hydroxyl radicals, which are known to be effective radical chain initiators for lipid polymerizations. Furthermore, the degree of polymerization of the isolated poly-17 was 102, a value that is consistent with chain polymerizations. This sensitization chemistry appears to be compatible with oxygen-dependent systems such as living tissues.
The glucose permeability of bilayer vesicles composed of the dienoyl monomer 16 was determined before and after UV photopolymerization.32 Because the monomer has a reactive group in each chain, the polymerization can cross-link the vesicle. The photo-cross-linking substantially reduced the glucose permeability of the vesicles. After about 20% of the glucose was lost from the vesicles, the remaining glucose was retained for several days, even after treatment of the vesicle sample with the surfactant TX-100. These data complement another report that showed that poly-16 vesicles were impermeable to the ionized carboxyfluorescein.14 Therefore, the bilayer permeability of charged as well as neutral compounds encapsulated in suitably cross-linked vesicles is significantly lower, at least 2 orders of magnitude, than that of the unpolymerized vesicles.
Selective polymerization of only one dienoyl group in monomer 16, which is in either the sn-1 or sn-2 chain, can be accomplished by the choice of appropriate radical initiators, i.e., the hydrophobic AIBN or the water-soluble AAPD.73,74 Thermal polymerization of large unilamellar vesicles (LUV) of 16 with either AIBN or AAPD gave only 50–60% conversion of monomer to polymer, with complete polymerization occurring only if both initiators were used simultaneously.73 The polymerization of LUV of the mono-substituted dienoyl PC 15 was examined as well. In this monomer the single reactive group, which is located on the sn-2 chain, was readily polymerized using the water-soluble AAPD at 60 °C (>Tm). On the other hand, the hydrophobic initiator AIBN was less effective in initiating the polymerization. Under the same reaction conditions of temperature, monomer-to-initiator ratio, and time, the AAPD reaction gave greater than 90% conversion to polymer and AIBN produced about 10% conversion.74 The authors postulated that the diene on the sn-1 chain is located predominantly in the hydrophobic region of the bilayer, whereas the diene on the sn-2 chain is at the aqueous/hydrocarbon interface. Thus, the addition of water-soluble AAPD to the LUV initiates polymerization only at the sn-2 chain; polymerization of the sn-1 chain was accomplished using the hydrophobic initiator AIBN. These results are consistent with earlier structural and reactivity studies of glycero-phospholipids that indicate the two chains are positionally inequivalent due to the preferred conformation of the lipid in bilayer assemblies.75
The polymerization of 16 with either initiator resulted in the formation of a linear polymer. Reaction of the unpolymerized chain with the appropriate initiator or UV irradiation cross-linked the bilayer.76 The ability to selectively polymerize the sn-2 dienoyl chain in the presence of a dienoyl group in the sn-1 chain appears to require a well-ordered lipid bilayer. If small unilamellar vesicles (SUV) were used, the AAPD was more effective in initiating the polymerization of both chains. This is thought to be due to the greater lipid disorder in SUV. Polymerization of LUV of 15 with the water-soluble AAPD was shown to be dependent upon the phase transition of the lipid.68 When the reaction temperature was below the Tm, only 25–28% of monomer was lost, while at higher temperature there is a 50–60% loss of monomer. The authors postulated that at temperatures greater than the Tm, the AAPD was able to penetrate the bilayer and initiate polymerization of the sn-2 acyl chain located on the inner LUV leaflet as well as the outer LUV leaflet.
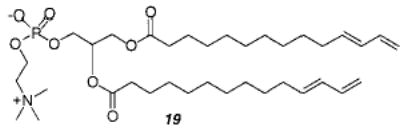
Reactive lipids have also been prepared with a diene group at the end of the lipid tails.77 The polymerization by UV or gamma irradiation of vesicles composed of monomer 19 leads to stable bilayer vesicles that were resistant to solubilization by TX-100. Electron micrographs of freeze–fractured unpolymerized vesicles showed a typical fracture plane along the midline of the bilayer, whereas the micrographs from polymerized multilamellar vesicles showed concentric circles indicating the fracture occurred across the bilayer in a manner previously reported by Roks et al. for vesicles from ω-isocyano amphiphiles.78 These data suggest the polymerization of vesicles of 19 proceeds by cross-linking between the two leaflets of the bilayer. The conformation of the reactive groups at the end of the alkyl chain was also found to be important in supported thin films of 19 on ZnSe.79 The diene group can undergo a cis/trans isomerization, which occurs through a twisted intermediate. It was suggested that this twisted transient is a diradical that initiates spontaneous bilayer polymerization. The films could also be polymerized by exposure to short wavelength UV light (diene λmax ~ 230 nm). Polymerization of these thin films disrupts the crystalline packing of the lipid tails and disorders the thin film. A comparison of the properties by infrared dichroism measurements of 19 and the corresponding phosphatidylethanolamine (PE) found that the PE exists in a lamellar liquid-crystalline phase from about 5 to 45 °C and forms an inverted hexagonal (HII) phase above this temperature when the relative humidity (RH) was greater than 90%.80 Reduction of the RH caused a reduction in the HII phase transition temperature. A further study combining data from X-ray and infrared measurements suggests that the terminal diene group gives rise to a significant component of the lateral pressure which is important for the transition from lamellar to nonlamellar phases.81 The bilayer morphology depends on the hydration of the headgroup, which does not change during polymerization, but the compressibility of the bilayer in the direction parallel to the membrane surface decreases significantly.
C. Diacetylenyl Monomers
The polymerization of diacetylenic amphiphiles in monolayers, LB-multilayers, bilayer vesicles, and tubules can be readily detected by the formation of highly colored poly(diacetylene) (PDA). This convenient characteristic made diacetylenic monomers the preferred choice for many of the early studies of monolayer polymerizations, since it was easy to detect evidence of polymer formation via the formation of red or blue PDA. It has proven more difficult to determine the extent of conversion from monomer to PDA, because of the low extinction coefficient of the monomer absorption bands. Moreover, conjugated polymers, such as PDA, are sensitive to prolonged UV light exposure.82 The topotactic nature of the polymerization limits effective polymerizations in monolayers to the more condensed region of the pressure–area isotherm,83,84 in bilayers to the slow diffusion regime found in the Lβ phase,75 or in bilayer tubules.85,86
PDAs have a polymer backbone that consists of conjugated alternating double and triple bonds, giving an absorption maximum in the visible range. Frequently there is a color change during polymerization or with a variation in sample temperature. This is usually ascribed to conformational changes in the polymer backbone. One model for the explanation of the color change predicts that the absorbance maximum is determined by the length of the overlapping π-orbitals (effective conjugation length).87 An absorption maximum at 620 nm (blue polymer) indicates a longer effective conjugation length than an absorption maximum at 540 nm (red polymer).88–90 Increasing heat or decreasing pressure introduces twists and kinks into the backbone, which disrupts conjugation.91 Theoretical calculations suggest that small rotations around the single bonds in the polymer backbone might be enough to cause this change.92,93 In those cases where the color change is reversible, it has been proposed that the change in the polymer backbone is induced by changes in the side chain conformation.94,95 The color change is irreversible if the disorder is caused by side chain entanglement, which can be locked in by cooling. Reversible color change could also be caused by the reversibility of hydrogen bonds.89
The importance of the diacetylenic group packing in various assemblies was shown by a comparison of various diacetylenic fatty acid derivatives.96 The differences in color of the resulting polymers depended on the length of the polymer as well as the packing. It was also found that large headgroups, like carbohydrates, hinder the solid-state polymerization due to the increased distance between the diacetylene units. If the diacetylene group is placed close to the amphiphile headgroup, the efficiency of polymerization may be dependent on the packing of the head-groups.96,97 In that case, packing and color change during polymerization could also result from charge interactions between neighboring headgroups.98 Unusual switching and packing behavior was achieved with the hydrazide-functionalized acid.99 This functionalized fatty acid could be UV-polymerized in dilute organic solutions, but the amphiphilic bilayers formed in water were only polymerized when the hydrazide was protonated. However, once formed, the PDA from these amphiphiles exhibited a reversal color change from blue to red as the pH was cycled from acidic to basic.
Bilayer vesicles can easily be formed from hydrated diacetylenic amphiphiles by sonication or extrusion. Sonication of diacetylenic monomer 20 produced a colorless, slightly turbid solution.100,101 Electron microscopy indicated the presence of multilamellar vesicles with diameters up to 400 nm. Polymerization of these vesicles by UV irradiation initially caused the formation of a blue product that changed to red. The sample was not thermostated, therefore, it is unclear whether the color change was due to continued irradiation or due to sample heating. Electron microscopy showed that the vesicles remained intact. Vesicles made from monomer 21 were characterized in more detail.102 The polymerization was followed by changes in the absorption spectra with time. Electron microscopy was used to demonstrate that the vesicle structure remained essentially unchanged.
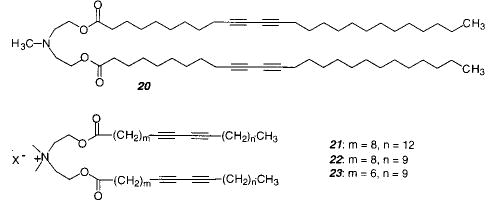
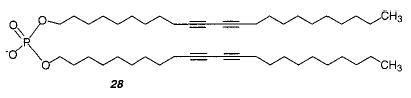
UV irradiation of unilamellar vesicles prepared from the diacetylenic PC 24 caused the formation of a red product that was insoluble in organic solvents.103 The vesicles could only be polymerized at temperatures below the Tm. The polymerization of a symmetrical dialkylammonium lipid 22 and the dialkyl phosphate 28 were much more efficient (6500 times greater rate of PDA formation) than the polymerization of 24.75 These data were the first to show that the polymerization efficiency depends on the lipid structure conformation. In addition, the polymerization of vesicles of each of the two symmetrical monomers (22 and 28) produced a blue PDA rather than the red PDA formed from monomer 24. The probable conformation of monomer 24 in bilayers makes it difficult for the two diacetylenic chains to be properly aligned to facilitate reaction with each other; therefore, monomers can only react with neighboring molecules. In contrast, the symmetrical monomers 22 and 28 have their two hydrocarbon chains properly aligned for reaction with each other.
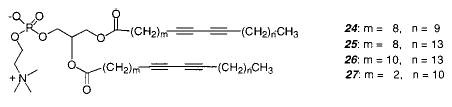
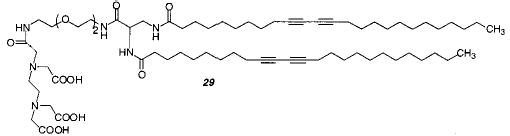
Phospholipid vesicles were also be prepared from the diacetylenic PC 26 and its mixed chain analogue.104–106 The intensity of red color produced upon UV polymerization depended on three factors: preparation procedure, irradiation time and intensity, and temperature. The highest monomer conversion was achieved with samples that had low concentrations of the lipid and were vigorously mixed or briefly sonicated. Maximum color was achieved after 3–5 min of irradiation at 4 °C, well below the Tm. The permeability of the vesicles decreased considerably after UV polymerization. The reactivity of the mono-substituted lipid was greater than the disubstituted analogue, which may be due to a change in the lipid chain packing after the first diacetylene group reacts. On the other hand, only the polymerization of the disubstituted analogue leads to an insoluble polymer, indicating the formation of a cross-linked material. A cross-linked phospholipid vesicle was also formed from monomer 24, 5% of cationic monomer 23, and 5% of monomer 29 for complexing lanthanide ions.107 By incorporating the lanthanides into stable, polymerized liposomes, lanthanide fluorescence could be used for more stable fluorometric protein detection and sensors. Unfortunately, the low molar extinction coefficient of lanthanide ions did not increase by this method.
When multilamellar vesicles composed of diacetylenic PC 26 were polymerized by UV at 0 °C, a blue suspension resulted which turned red upon heating to 50 °C.94 Upon cooling, the color reversed to blue but only if no further photopolymerization took place. Unilamellar vesicles of different sizes were also prepared, and it was determined that the efficiency of photopolymerization was sensitive to the size of the liposome.108 Small unilamellar vesicles of 24 were unreactive.
Macrocyclic diacetylene PCs of different ring sizes were prepared and shown to form vesicles which were resistant to disruption by surfactants. The lipids were prepared with an ether linkage to the glycerol backbone to increase the vesicle stability to highly acidic environments.109 Other polymerizable ether lipids are also stable to highly acidic or basic conditions that are less suitable for ester lipids.110
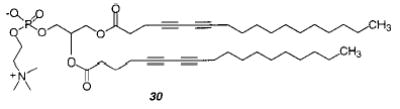
To determine the most favorable alignment for the polymerization of the two diacetylenic groups in a diacetylenic PC, the polymerization of monomer 27 and 30 were compared.111 Samples of 27 immediately turned blue during irradiation with UV light, whereas samples of 30 did not polymerize. Polymerized samples of 27 show significantly slower osmotic swelling than unpolymerized samples, indicating that polymerization reduced the bilayer permeability.
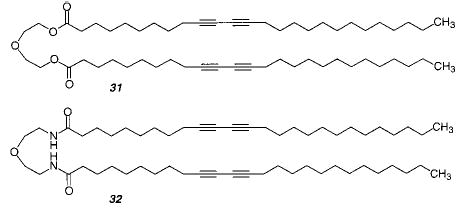
The initial studies of the polymerization of diacetylenic amphiphiles in monolayers used fatty acids. The formation of PDA was sensitive to fatty acid length, i.e., hydrophobicity, pressure, temperature, and the presence of oxygen as well as salts, e.g., CdCl2, in the subphase. To examine molecules that were more lipid-like, monolayers were also formed from 20, 31, and 32.100 Monomers 31 and 32 polymerized upon exposure to UV light in a nitrogen atmosphere (with contraction in the constant pressure isotherm). Monomer 20 exhibited decomposition of the monomers. The emergence of a red polymer correlated with a reduction in the surface area covered by the monolayer. The polymerization of monolayers of 31 or 32 was faster than previously observed for diacetylenic fatty acids.
Diacetylenic monomers were also used to form LB multilayers.112 The diacetylenic fatty acid 33 was used in the study of multilayer polymerizations. As in monolayer films, initially a blue then a red polymer were formed. Further UV radiation led to the degradation of the polymer. Multilayers formed from diacetylenic fatty acids, phospholipids, and amphiphilic esters were found to be stable to oxygen,113 because the oxygen-mediated radiation damage was limited to the top layer. On the other hand, an isosbestic point was usually not found in the UV–vis spectra of polymerized multilayers of diacetylenic acid 34,114 indicating that multiple products were formed during the polymerization. The quantum yield of the UV photopolymerization of 34 was larger than 10, indicating that a single photon initiates a polymer chain.114
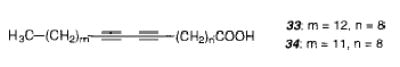
When multilayers of 34 were annealed at different temperatures before the polymerization, different colored polymers were obtained.115 Annealing the sample at temperatures up to 50 °C resulted in a blue film, annealing from 64 to 100 °C yielded a red film, whereas annealing from 160 to 210 °C resulted in a film with a broad absorption between 540 and 450 nm. Only when the sample was annealed below 50 °C was the color change reversible.
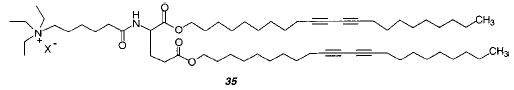
Solvent casting of lipid vesicles can yield ordered multilayers.116,117 These methods were developed by Kunitake and co-workers to form free-standing multilayer films. To assess whether cast multilayer films were well enough ordered to permit efficient polymerization of diacetylenic amphiphiles, multilayer films were prepared from diacetylenic monomers.118,119 A cast film, composed of the diacetylenic glutamate lipid 35, was irradiated with UV light. A deep blue, 10 μm thick, free-standing, flexible polymeric film resulted. The extent of polymerization of the lipids was estimated to be 75–80% after 3 min exposure. The PDA films exhibited a reversible thermochromic effect. Efficient PDA formation was favored by highly polar or hydrogen-bonding monomers, which appeared to favor well-ordered lattices of the monomers in the multilamellar assembly.
During the early study of hydrated diacetylenic PC 26, tube-like structures, as well as vesicles, were found in the freeze–fracture electron micrographs.120 These structures, termed tubules, were found in optical micrographs as well, thereby demonstrating the structures were not an artifact of sample preparation.85 In some cases surface features were observed that suggested that the tubules consisted of a rolled-up bilayer sheet, akin to the previously reported cochleate cylinders of anionic lipids in the presence of divalent cations.121 The discovery of tubules lead to detailed studies of their structure, formation, and applications.25,122 The yield, diameter, and length of the tubules were found to depend on the preparation conditions. When liposomes made from 24 were prepared above their Tm and then cooled, the tubules obtained were observed to vary in diameter from 0.3 to 1 μm with lengths up to hundreds of micrometers.85,123 The thickness of the tubule walls can vary from two to tens of lipid bilayers.123 When concentrated solutions of lipid in ethanol were injected into water, helical tubules were obtained.86 A degree of control over the diameter and length of the helical structures was achieved by choosing the monomer concentration, temperature, and solvent for the precipitation. Tubules made from diacetylenic PCs, such as 24, can be photopolymerized to various extents but not always to high conversion.124
To determine how the tubules from lipid 24 formed, rapid quench freeze–fracture electron microscopy was employed.125 Transitional structures of liposomes partially wrapped around nascent tubules were found suggesting that lipid bilayers from the liposomes continuously transfer to the tubules. Circular dichroism was used to determine if the monomers in the tubules packed in a chiral manner.126 Tubules composed of the same lipid, yet made by different techniques, were optically active. If the tubules were made from optically pure enantiomers, they had the opposite handedness. Therefore, it was concluded that chiral packing occurred in a manner similar to wind up a helical ribbon of lipids. The relationship between tubule helix handedness and phospholipid chirality could not be observed during the first seconds of tubule formation.127 An enantiomer of 24 yielded an equal amount of left- and right-handed helices at the beginning of tubule formation but progressively grew into multilamellar tubules with a single handedness.
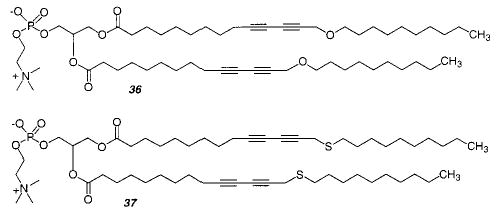
A comparison of the structures formed from monomer 24, 36, and 37 indicated that interactions between the diacetylenes in the acyl chains are important for tubule formation.128 An acyl chain with a diacetylene is rigid and has a tilt toward the bilayer axis, which can contribute to the observed chirality of the tubule. The tilt can be changed by changing the electronic structure of the diacetylene. When one oxygen was inserted β to the diacetylene (36), the resulting tubules were more flexible than tubules formed from the parent monomer 24 and had a wide variety of diameters. When the oxygen was replaced with sulfur in 37, thin, long, and flexible ribbons were formed instead.
The morphology of hydrated diacetylenic aldonamides was determined by electron microscopy by taking advantage of the electron density of the diacetylene group.129–131 The microstructures formed from these single-chain aldonamides depended on the structure of the aldonamide headgroup. Headgroups based on the following aldoses favored the formation of tubules and helices: D-galactose, D -glycero-L-mannose, L-lyxose, L-mannose.131 In contrast, sheets were formed when the headgroup was L -threose or D-glycero- D -glucose. If the alkyl chain was changed from the 12-carbon N-dodeca-5,7-diynyl to the saturated dodecyl, then tubule morphologies were still found for many aldonamides. The authors proposed that the morphology is strongly affected by the hydrogen-bonding patterns of the hydrophilic head-groups.131 Clearly, the formation of tubules was not completely dependent on the presence of diacetylene groups in the hydrophobic tail of these compounds.131–133
Anionic glucophospholipids were also observed to form tubules.134 Tubule formation was attempted with lipids with different sugars as the headgroup. The glycolipids with glucose attached at the O-6 position yielded tubules. When the position of the hydroxyl groups or the negative charge was changed, structures such as micellar rods, scrolled bilayers, and twisted ribbons were formed instead. The tubules were stable at higher temperatures when the fluorinated analogues were used. The fluorinated alkyl chains also increased the curvature and therefore decreased the diameter of the resulting tubules.
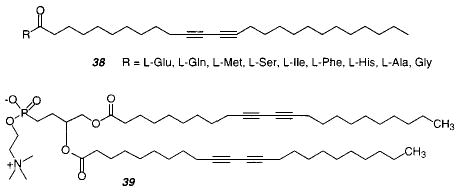
Other microstructures were formed by the polymerization of amino acid-substituted diacetylene fatty acids 38.135 Fibers, ribbons, and helical ribbons were obtained as microstructures. The structure was dependent on which amino acid was used as the headgroup. Helical and curved structures were only obtained with chiral headgroups. Blue PDA was formed upon exposure to UV light. A blue to red chromatic transition of the polymer was found when the headgroup was ionized by changing the pH. The authors propose that the generation of charge at the surface reduces the hydrogen-bonding interaction, which affects the extent of electron delocalization along the conjugated PDA backbone.
The phosphonate analogue 39 of PC 24 yields fragile and deformable tubules.136 Helical ribbonlike structures were also found. The tubules can be polymerized by exposure to ionizing radiation, yielding a deep purple color. The conversion of tubules into helical ribbons was found with an approximately 1:1 mixture of 39 with the nonpolymerizable 1,2-bis-(dinonanoyl)phosphatidylcholine (DNPC).137 The originally formed tubules were 50–60 nm in diameter and up to 100 μm long. When this phase remained at ambient temperature for more than a few hours, a gradual transformation to a gel phase with helical ribbons occurred. The rate of transformation was temperature and concentration dependent and did not occur below 4 °C.
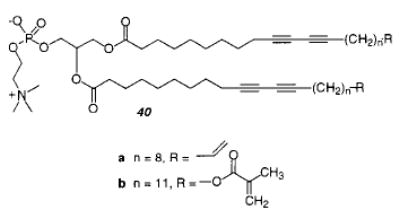
It was possible to stabilize tubules by cross-linking them by including another functional group (see section II.E) at the end of the acyl chains of the lipids (monomer 40).138 Depending on the temperature during UV irradiation, the tubules retained their morphology during polymerization, even though their shape became a little more irregular. The resulting tubules were stabilized to ultrasonication, lyophilization, and redispersion.
The chirality of lipid tubules was investigated in some detail. A theory for the formation of cylindrical tubules of chiral lipid bilayers predicts the radius and average tilt direction from a chirality parameter q and phase transition temperature.139 This theory was tested in a circular dichroism study.126 The effect of solvent on the chirality of the tubules was investigated with tubules from monomer 24 and was found to be consistent with the theory.140 Circular dichroism and electron microscopy showed that single bilayer tubules were prepared from a methanol/water mixture, whereas multilayer tubules were formed from other alcohols. When the concentration of lipid was increased, tubules formed in methanol/water tended to form multilayer structures.141 When ethanol or other alcohol was added to the solvent mixture, the tubules were narrower with thicker walls and less chirality. When the diacetylene group in the lipid is placed further away from the headgroup, with an even number of the methylene groups between the ester and the diacetylene, the order in the lipid packing was increased.
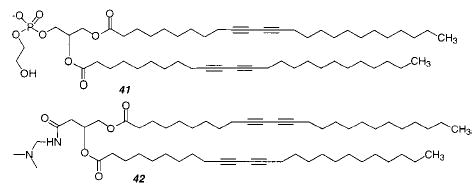
Tubules have also been formed from a lipid with biotin attached to the headgroup. The biotin was sufficiently effective in binding the protein streptavidin that 2-D crystals were formed.142 The crystal structure could be readily solved because the cylindrical nature of the tubule simultaneously presented the protein in several orientations relative to the probe beam. Lipid tubules have also been used to template the deposition of metals on the exterior surface of the tubule.143,144 Zerovalent palladium deposited on tubules prepared from 41 was used as a catalyst for electroless plating of nickel onto the tubules.144 A similar lipid (42) was used for the electroless deposition of either gold, nickel, or copper.145 Metallized tubules could be oriented in a magnetic field.145,146 Lipid–iron oxide composites of different structures and compositions were formed from a variety of galactocerebroside derivatives.147 Tubules, disks, and fibers were formed from the lipids and used as templates for iron plating under varying reaction conditions. Tubules made from 24 were treated with the anionic polymer poly(styrene-sulfonate), followed by the cationic polymer poly-(ethyleneimine) and the adsorption of anionic silica spheres.148 This resulted in the formation of caps for the tubules. When a small amount of 41 was mixed into 24, a helical band of silica spheres showed the position of the negatively charged lipid in the tubule. This is the first time the composition and structure of the lipids in a mixture could be imaged directly.
D. Other Monomers
Regen and co-workers demonstrated that vesicles could be polymerized by oxidation of thiol-containing lipids, such as the bis-thiol PC (43), to yield disulfide bonds between lipids. This monomer yields linear polymers with an estimated Xn of 25. Vesicles composed of 43 or other thiol PCs could be oxidatively polymerized and then reductively depolymerized multiple times.149,150 These studies demonstrated an effective strategy for the reversible polymerization–depolymerization of lipid vesicles. It appears that this method could be utilized in other lipid assemblies. The polymerized vesicles exhibited a greater colloidal stability and were more resistant to disruption by added surfactants than the unpolymerized vesicles. In addition, the lateral diffusion of an nonpolymerizable fluorescent lipid probe was retarded about an order of magnitude after the polymerization of vesicles of 43.
Subsequently, the same research group introduced lipoyl-substituted PCs (44, 45). The bis-lipoyl PC 45 can be polymerized by the addition of catalytic amounts of dithiothreitol (DTT) or other reducing agents to yield cross-linked polymeric vesicles which were stable to surfactants.151 The DTT appears to initiate the polymerization by production of an initial population of lipid thiol groups, which can react with the disulfide group in the lipoyl PC via thiol–disulfide exchange. After freeze-drying the polymerized vesicles to remove the water, the recovered poly-45 was insoluble in chloroform or chloroform/methanol. The permeability of vesicles before and after polymerization was determined for varying mixtures of mono-lipoyl PC 44 and bis-lipoyl PC 45.152 The cross-linking polymerization of bis-lipoylPC vesicles reduced their permeability to sucrose by a factor of 50. As the mole fraction of 44 was increased the cross-linking density in the bilayer was expected to decrease with a corresponding decrease of the effect of polymerization on the bilayer permeability. In fact, at an initial ratio of 44 to 45 of 4 to 1, vesicle polymerization did not substantially alter the bilayer permeability. This suggests that insufficient bis-lipoyl PC was present to effectively cross-link the bilayer vesicle.
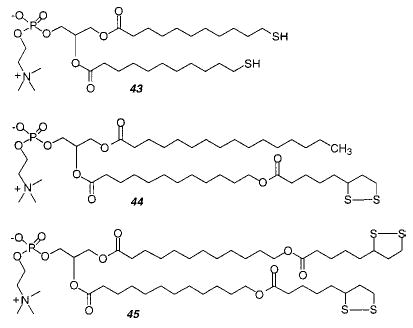
Liposomes composed of bis-lipoyl PC 45 and a few mole percent of a mono- or bis-thiolPC can be polymerized by simply increasing the pH of the aqueous suspension to pH 8.4.153 The pH change increases the nucleophilicity of the thiol group, which initiates the ring opening polymerization of bis-lipoyl PC 45. These mild polymerization conditions are obviously very attractive for the formation of lipid assemblies that contain sensitive components. Moreover, the authors showed that the polymerization could be slowed and essentially stopped by decreasing the sample pH. This feature permits the formation of liposomes with a continuum of polymeric and monomeric lipid content.
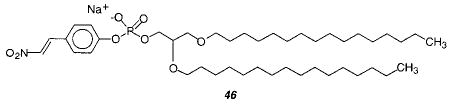
Engberts and co-workers designed a phospholipid 46 with a β-nitrostyryl group attached to the phosphate.154 Upon exposure to UV light the styryl lipids react to form oligomers. Interestingly, after vesicle formation the exterior β-nitrostyryl group can be hydrolyzed at high pH as long as the vesicles are at a temperature below the Tm. Under these conditions the rate of lipid flip-flop was too slow to compete with the hydrolysis reaction. Therefore, the vesicles of 46 could be prepared, hydrolyzed at the exterior surface, and then oligomerized preferentially at the inner leaflet of the vesicle. These novel polymerized vesicles provide a new tool to examine the characteristics of vesicle reactions, such as vesicle fusion.155
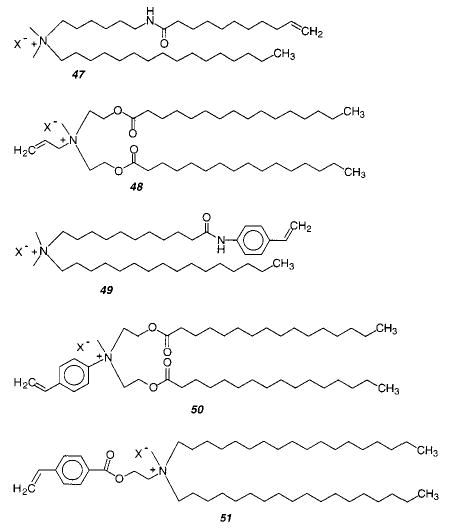
Fendler, Tundo, and co-workers described the synthesis and polymerization of vinyl- and styryl-substituted lipids. Amphiphiles were prepared with vinyl (47, 48) or styryl (49, 50) groups in either the hydrophobic tail or the more hydrophilic head-group.156,157 These amphiphiles were generally based on a quaternary ammonium structure. Vesicles were prepared by ultrasonication, and either thermal initiators or direct irradiation with UV light was used to polymerize the vesicles. The authors demonstrated that the polymerized vesicles exhibited enhanced colloidal stability. A comprehensive study of the kinetics of the photopolymerization of vesicles composed of the styryl monomers showed that the polymerization process was faster in the organized media of the bilayer than when the monomers were in isotropic solution.158 Moreover, the rate of photopolymerization was independent of vesicle concentration, thereby conclusively demonstrating that the polymer chain growth was limited to individual vesicles. Analysis of the polymerization data indicated that the lifetime of the styryl radical during the vesicle polymerization was 17 ms, the rate constant for propagation was 103 s−1, and the quantum efficiency of free radical formation was 0.1.158 The kinetic data also indicated that the average kinetic chain length of the polymers was 20. A subsequent study of the hydrolyzable styryl monomer 51 found that the Xn varied from 10 to 40 depending on the vesicle composition.159
Styryl lipid monomer 52 can be polymerized with an amphiphilic photoinitiator 53 with exposure to UV light. The authors claim the formation of very high molecular weight linear polymers.160 The observed polymer size was attenuated by the introduction of chain transfer agents. Given the reported size of the polymers and the number of monomers at the exterior surface of the vesicle, as well as a similar number on the interior surface of the vesicle, it would appear that almost all of the monomers at these sites in each vesicle react to form the observed polymer. This remarkable observation suggests that electrostatically associated monomers more readily diffuse to the growing polymer chain than covalently linked reactive groups.
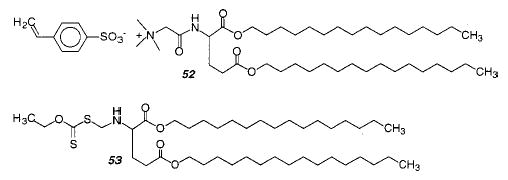
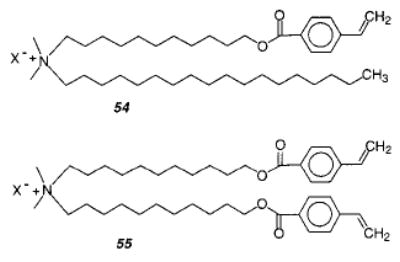
The mono- and bifunctional styryl monomers 54 and 55 were used to study the formation of vesicle–polymer colloids.161 First, vesicles of 54 were formed and polymerized with the photoinitiator 2,2-di-methoxy-2-phenylacetophenone (DMPA) using a 351 nm laser, which resulted in complete monomer conversion. The liposomes were not stable to surfactant lysis. Liposomes with only 55 could not be formed, but the polymerization of liposomes with a composition of 54:55 = 2:1 yielded liposomes that were stable toward surfactant lysis. The polymerization of styrene or divinylbenzene (DVB) in a bilayer membrane of 54 was investigated. This covalently linked the polymer within the vesicle matrix and prevented the typical phase separation between polymer and vesicle matrix. However, when the cross-linking polymerization of styrene and DVB in 54 was attempted, the resulting vesicles were strongly deformed and wrinkled. When the vesicles were cross-linked with DVB after they were prepolymerized with styrene, the desired thickened, stable vesicle–polymer colloid was formed.
E. Heterobifunctional Monomers
Heterobifunctional monomers have two different reactive moieties with distinct reactivities. This difference may be reflected in the rate and degree of polymerization as well as the type of initiators that can be usefully employed. The distinct location of each reactive group within the assembly inhibits copolymerization between the groups even if they have similar reactivities. A heterobifunctional single chain amphiphile that contained a diacetylene and an acetylene group was reported by Nezu and Lando.162 LB techniques yielded multilayers of the heterobifunctional fatty acid. The fatty acid could be polymerized in the condensed phase by γ-ray irradiation to yield polydiacetylene; however, the isolated acetylene did not polymerize under these conditions. Singh et al. reported the synthesis of a heterobifunctional phospholipid (40) with a diacetylene and either a vinyl or methacryloyl in each lipid tail.138 The polymerization of these heterobifunctional phospholipids was performed in the Lβphase by first UV then γ-ray irradiation in a sequential fashion. The nature, degree of polymerization, and extent of polymerization of each individual group was not reported.
It is interesting to consider the polymerization characteristics of an amphiphilic monomer with two reactive groups in a single chain. The behavior of such monomers might shed light on the competitive pathways for polymer chain growth for reactive groups located in different regions of organized assemblies. This intriguing prospect of selective polymerization of reactive groups was explored with the novel heterobifunctional lipid 1-palmitoyl-2-(2,4,-12,14-tetraenehexadecanoyl)PC (56), which contains a dienoyl group conjugated with the ester carbonyl of the fatty acid chain and a 1,3-diene located near the acyl chain terminus of the same chain.163,164
Because the two diene groups are located in regions of different polarity, it was possible to perform either simultaneous, selective, or sequential polymerizations. The selective polymerization of either group was used in tandem for sequential polymerization. Two possible modes of reaction were distinguished for vesicles of 56 by surfactant treatment of the polymerized vesicles.164 If the reaction path of the second group to react is uncorrelated with the reaction path of the first group, then cross-linking should occur. However, if the second reaction occurs preferentially with an adjoining group in a repeat unit of the polymer formed in the first reaction, then a ladder-like polymer is possible. Sequential polymerization of bilayer vesicles of 56 was performed by either (1) selective photopolymerization of the dienoyl, followed by redox polymerization of the diene, or (2) selective AIBN polymerization of the diene group, followed by redox polymerization of the dienoyl. Persulfate production of hydroxyl radicals was used for simultaneous polymerization. Each of the three modes of polymerization gave polymerized vesicles that were disrupted by Triton X-100, thereby indicating a lack of cross-linking. These results show that both groups preferentially react with the same neighbor lipid. An alternative interpretation, that their reactions occur in nonoverlapping domains of the bilayer, was excluded by the observed high conversion to polymer.
The ladder-like polymers formed by polymerization of bilayers of lipid 56 are distinctive because of the relatively long aliphatic link between the two polymer chains. These results suggest that properly designed amphiphilic monomers could be used to prepare novel polymer architectures, such as copolymers where the two components are essentially parallel blocks, e.g., lipid 56 yields two poly(1,4-diene)s. In a similar manner it should be possible to form parallel blocks of different polymers. The preferred ladder-like mode of polymerization of monomer 56 is attributed at least in part to the relatively short spacer link between two reactive moieties, i.e., six methylenes. At some separation distance, the congruence of the preferred reactivity of the two groups will diminish to the point that the polymerization of both groups will be uncorrelated, resulting in the formation of cross-linked polymers instead of ladder-like polymers.
Liu et al. synthesized two sets of heterobifunctional PCs with variable length spacer groups (7, 9, or 11 atoms in length) between a dienoyl group and either a sorbyl or acryloyl group (57, 58).165 Liposomes were prepared from each lipid and polymerized via radicals generated from a redox pair. The resulting polymerized liposomes were stable to the addition at least 12 equiv of the surfactant, Triton X 100, which is more than sufficient to solubilize an un-cross-linked liposome. On the other hand, photopolymerization of these lipids did not yield un-cross-linked liposomes. These data indicate that lipids of this general design can be employed to cross-link lipid assemblies of various types, including lamellar and nonlamellar phases as well as bilayers on solid supports. This is a consequence of the uncorrelated reaction pathways of the two reactive groups per lipid. The fact that a spacer length of only seven atoms between the reactive groups in these lipids is sufficient to favor decorrelation of the growth of the two polymer chains, whereas a spacer of six atoms in 1-palmitoyl-2-(2,4,-12,14-tetraenehexadecanoyl) PC (56) results in correlated polymer chain growth, suggests that the flexibility of the ester linkage aids in decoupling the polymer chain growth of the poly(dienoyl) and either the poly(sorbyl) or poly(acryloyl). Further study is necessary to provide clearer insight into the nature of polymer chain growth at two ends of heterobifunctional monomers.
F. Polymerization and Domain Formation
Epifluorescence microscopy is frequently employed to characterize lipid domains in phospholipid monolayers. The smallest domains observed by fluorescence are at least a few micrometers in diameter. The introduction of AFM and complementary microscopies now permits the observation of submicrometer domains in suitable multilayer samples. The sizes and shapes are dependent on the lipid composition, temperature, pressure, as well as electrostatic interactions. The domain shapes exhibit smooth boundary lines or fractal structures. The early reviews by Möhwald166 of phospholipid monolayer domains at the air–water interface and by McConnell167 of structures and transitions in these monolayers are valuable entry points for this literature.
The best understood bilayer lipid systems at equilibrium are those composed of two phospholipids. Both phosphatidylcholine (PC)–PC systems168 and PC–phosphatidylethanolamine (PE) systems169 have been reviewed. Binary lipid systems where the lipid chain lengths vary by less than four carbons in length exhibit nearly ideal mixing. Increased differences in chain lengths lead to immiscibility of the two lipids in the solid phase. When the lipids differ significantly in their Tm, the phase diagrams are complicated due to the immiscibility of the lipids in the solid phase region. The lipid chain substitution pattern, i.e., unsaturation, methyl branches, ester or ether groups, and rigid entities such as diacetylenes, will also influence the lipid phase behavior. However, only the effects of unsaturation have been systematically studied to date. Binary systems of neutral and charged lipids tend to exhibit ideal behavior due to the electrostatic repulsion between the charged lipids.
The most complete insights into the behavior of mixtures of nonpolymerizable lipids have come from the phase diagrams of these systems. Phase diagrams of lipid mixtures have provided important reference points for the polymerizable lipid systems, even though few phase diagrams have been reported for polymerizable lipid mixtures. Despite this deficiency the polymerization studies have revealed several important phenomena. Armitage et al. reviewed the literature describing domain formation of mixtures of polymerizable and nonpolymerizable lipids.170 Two circumstances are well established: the phase separation of a reactive monomeric lipid and a nonreactive lipid before or after lipid polymerization.
1. Polymerization of Preexisiting Domains
The formation of lipid domains in binary mixtures of a polymerizable lipid and a nonpolymerizable lipid is well established for diacetylenic lipids. The rigid diacetylenic unit facilitates the formation of enriched domains in the condensed phase of monolayers or the solid-analogous phase of bilayers. Since diacetylenes polymerize most readily in solidlike states, most studies have focused on conditions that favor domain formation. Phase separation was not observed when ionic diacetylenic lipids were mixed with zwitterionic PC.171,172
Ringsdorf and co-workers were the first to describe the polymerization of phase-separated monolayers composed of a diacetylenic PC and a nonpolymerizable lipid, distearoyl PE (DSPE).173 Phase separation was also investigated in bilayer systems.171,172 Evidence for phase separation was obtained in calorimetric studies and substantiated by the photopolymerization characteristics of the lipid mixtures. A zwitterionic diacetylenic PC, e.g., 24, phase separated from nonpolymerizable PC co-lipids at temperatures below their Tm. However, at temperatures greater than the Tm, the reactive diacetylenic PC and non-polymerizable PC were more miscible. The cationic diacetylenic lipid 21 appeared to be miscible with nonpolymerizable PCs. Collaborative studies between Sackmann and Ringsdorf employed electron microscopy to visualize phase separation of polymerizable and nonpolymerizable lipids in giant vesicles.174 Mixed vesicles composed of a diacetylenic PC and DPPC were used. At 20 °C and below 50 mol % of the diacetylenic PC, the micrographs revealed domain formation. When the diacetylenic PC was present in greater than 50 mol %, phase separation was not observed in the micrographs; however, the presence of domains too small to detect by the technique cannot be excluded.
The behavior of mixed monolayers or bilayers of dienoyl-substituted lipids and other amphiphiles has also been extensively investigated. Because the dienoyl lipids do not contain a rigid group in the middle of the hydrophobic chains, they tend to be miscible with other lipids over a wide range of temperatures and compositions. To decrease the lipid miscibility of certain dienoyl amphiphiles, Ringsdorf and coworkers utilized the well-known insolubility of hydrocarbons and fluorocarbons.175 Thus, two amphiphiles were prepared, one with hydrocarbon chains and the other with fluorocarbon chains, to reduce their ability to mix with one another in the bilayer. It was necessary to demonstrate that the lipids form a mixed lipid bilayer rather than independent structures. Elbert et al. used freeze–fracture electron microscopy to demonstrate that a molar mixture of 95% DMPC and 5% of a fluorinated amphiphile formed phase-separated mixed bilayers.175 Electron microscopy and vesicle permeability studies were used to clearly demonstrate the existence of hydrocarbon- and fluorocarbon-rich domains within the bilayers. In one study the polymerizable lipid was a hydrocarbon-based zwitterionic dienoyl lipid 59, which was combined with a nonpolymerizable fluorocarbon lipid 60.14 The dienoyl lipid domains were then photopolymerized in a manner that should result in cross-linking of the hydrocarbon lipids. Following the polymerization the double-chain disulfide derived from oxidized cysteine was selectively reduced by the addition of dithiothreitol (DTT). This reduction of the disulfide bond produced a single chain thiol, which dissolves in water leaving permeability paths in the polymerized bilayer wall. A similar set of experiments was preformed with a partially fluorinated polymerizable muconyl lipid 61 and a hydrocarbon-based cystine 62.14 After the fluorinated domains were photopolymerized, the vesicles were combined with DTT to reduce the disulfide linkage in 62 and initiate the release of encapsulated eosin.
The reductive cleavage of cystine amphiphiles illustrates the potential utility of phase-separated bilayers for the stimulated release of encapsulated reagents. Ringsdorf coined the phrase “uncorking the liposome (vesicle)” to graphically describe the process.176 A partially polymerized vesicle with labile domains is uncorked by a chemical or physical process to yield multiple holes in the otherwise stabilized polymeric bilayer. Electron micrographs of such “uncorked liposomes” were obtained by scanning electron microscopy and are reported in the 1988 review by Ringsdorf et al.14 The great interest in the ability to selectively release encapsulated reagents from vesicles has led to the design of other methods to trigger the opening of labile domains in an otherwise stabilized vesicle. These include hydrolytic (enzyme mediated), photochemical, and redox processes. Most of the reported examples of these methods involve polymerization-induced domain formation.
2. Polymerization-Induced Domain Formation
Thze polymerization of reactive lipids in two- or multicomponent membranes can cause the separation of the polymerized lipid and the co-lipid(s) into enriched domains. Several factors may be important for the phase separation process including the nature and size of the polymer chains, the relative location of the polymer chains within the bilayer, and the composition of both the lipid hydrophilic headgroup (charged or neutral) and the lipid hydrophobic tails. Most of these variables have not been systematically characterized. Therefore, the studies to date are grouped into those that examined (1) bilayers composed of two neutral lipids (neutral–neutral) and (2) bilayers composed of a charged lipid and a neutral lipid (charged–neutral).
Bilayers of Neutral Lipids
Tsuchida and coworkers reported the polymerization behavior of lipid membranes composed of the bis-DenPC 16 and dipalmitoylPC (DPPC).177,178 The similarity of chain length of the fatty acid chains, 18 carbons and 16 carbons, respectively, favors the formation of a mixed lipid phase. Vesicles of 16/DPPC (1:1) were prepared by ultrasonication, and both dienoyl groups were polymerized by exposure to UV light. Evidence for polymerization enhancement of phase separation was obtained by freeze–fracture electron microscopy of the polymerized vesicles, which revealed domains of the Pb′(ripple) phase of DPPC and patches of smooth surfaces. The latter were assigned to domains of poly-16. Similar micrographic images were obtained whether the polymerization was performed on a partially phase-separated sample at room temperature or a homogeneous lipid sample at 50 °C, indicating that a cross-linking polymerization of a neutral lipid can cause its phase separation from another neutral lipid.
The formation of domains in two-component vesicles was effectively demonstrated by electron microscopy of partially solubilized vesicles. Ohno et al. coined the term “skeletonized” vesicles, i.e., porous vesicles, for the resulting supramolecular structure.179 Skeletonized vesicles were prepared from mixtures of 16 and DPPC, where the mole fraction of DPPC varied from 5 to 25 mol %. Takeoka et al. analyzed the release of entrapped water-soluble molecules in order to assess the size of the pores formed in the vesicle bilayer.180 They determined the ease of release of dextrans of various molecular weights and radius of gyration from the skeletonized vesicles. Vesicles of a particular composition were polymerized, and the nonpolymerized domains were dissolved with sodium dodecyl sulfate. If the entrapped dextran was not released, then the largest pore sizes were too small to permit facile release of the marker. Larger dextrans were released from vesicles having greater mole fractions of DPPC. The authors estimated the diameter of the largest pores to be 3 or 4 nm, when the mole fraction of DPPC was 0.10 or 0.20, respectively. Polymerization in the Lα phase appeared to yield smaller lipid domains than when the polymerization was performed at lower temperatures where the two lipids were partially phase separated prior to polymerization.
Tyminski et al. found that a 1:1 molar mixture of a neutral phospholipid, e.g., DOPC, and the neutral bis-DenPC (16) formed stable homogeneous bilayer vesicles prior to polymerization.181,182 Following photopolymerization the lipid vesicles were titrated with the trans-membrane protein bovine rhodopsin in octyl glucoside micelles in a manner that maintained the octyl glucoside concentration below the surfactant critical micelle concentration. The resultant bilayer vesicles were purified on a sucrose gradient by ultracentrifugation to yield a major lipid/rhodopsin band with a lipid-to-rhodopsin ratio of 100. Almost all of the rhodopsin was successfully inserted into the vesicles of poly-16 and DOPC. A control experiment showed that rhodopsin could not be inserted into vesicles composed of only poly-16. Rhodopsin incorporation into the poly-16/DOPC bilayer vesicles appeared to occur preferentially at enriched domains of the DOPC. A large fraction of the rhodopsin inserted into these vesicles was both photochemically and enzymatically functional. Moreover, the poly-PC lipid surface was shown to be biocompatible even after polymerization of 16.
After the initial studies demonstrated that mixed lipid bilayers could be separated into domains of poly-PC and unreactive PC domains, O’Brien and coworkers reasoned that the polymerization of vesicles composed in part of polymorphic unreactive lipids could immediately destabilize the bilayer vesicle.183,184 This premise was based on the accumulated evidence that lipids such as dioleoylPE (DOPE) can form a variety of hydrated lipid morphologies. Processes that form enriched domains of PE in bilayers result in destabilization of the bilayer (lamellar) structure with the formation of precursors to nonlamellar phases. The effect of photopolymerization on bilayer stability was experimentally examined with vesicles composed of the photosensitive bis-SorbPC 18 in combination with either DOPE or DOPC.
Vesicles composed of 18/DOPC containing the water-soluble fluorescent marker calcein were stable throughout the photopolymerization of 18, even though nearly 85% of 18 was polymerized. In contrast, the photolysis of vesicles of 18/DOPE caused the release of the calcein. Only oligolamellar or multilamellar 18/DOPE vesicles were efficiently photodestabilized at the vesicle concentration employed. Bilayer contact of PE-rich domains of separate vesicles is a prerequisite for bilayer destabilization.185,186 Bilayer contact within a vesicle (intravesicular) is possible between lamellae, if the vesicle consists of more than one bilayer. Bilayers of randomly distributed PE/PC lipids are repelled from neighboring bilayers by strong hydration forces.187 Processes which promote phase separation and cause the enrichment of domains of the poorly hydrated PE decrease the repulsive forces in these regions and facilitate bilayer contact. Contact between apposing bilayers enriched in PE in oligolamellar vesicles can lead to the formation of interlamellar attachment(s) (ILA). It has been proposed that an ILA is a hourglass-shaped bilayer attachment between two of the original lamellae that effectively fuses these bilayers.188 Under appropriate circumstances ILAs can proceed to form long-lived arrays which take on the characteristics of an inverted cubic phase. 31P NMR was successfully used to demonstrate the progressive formation of an isotropic NMR signal and the decrease of the lamellar signal.189 This signal was associated with the formation of lipid structures with isotropic symmetry, such as ILAs and inverted cubic phases. X-ray diffraction data confirmed that the NMR signal was due to the formation of an inverted cubic phase.189 The development of an ILA(s) that connects the bilayers of an oligolamellar vesicle provides an aqueous pathway for the leakage of water-soluble encapsulated reagents. ILAs have been proposed as critical intermediates in the fusion of bilayer vesicles with one another as well as precursors to the formation of nonlamellar phases, e.g., inverted cubic structures. The bilayer reorganization suggests that the photopolymerization of 18/DOPE large unilamellar vesicles (LUV) could usefully initiate vesicle-vesicle fusion.
The fusion of vesicles requires bilayer contact, followed by lipid reorganization in a manner that allows the mixing of the vesicle encapsulated aqueous contents in addition to a mixing of the lipids from each of the vesicles. Both the rate and extent of vesicle fusion are a function of several factors including temperature, lipid composition, and pH. Bennett and O’Brien demonstrated that the photopolymerization of 18 in 18/DOPE LUV significantly reduced the critical fusion temperature, i.e., the temperature threshold for the onset of rapid fusion.190 They utilized fluorescent probes to study the interaction of photolyzed LUV of 18/DOPE with either photo-lyzed or dark (unpolymerized) 18/DOPE LUV. Both lipid mixing and aqueous contents mixing between the donor and target LUV was enhanced by the photoactivated polymerization of 18. Fusion of vesicles has been described by a mass action kinetic model in which the first step is aggregation of two stable vesicles to form a dimer and the second step is the actual fusion process which produces the fusion product.191 The kinetics of the overall fusion process depends on the rates of both aggregation and the fusion event. Polymerization of 18/DOPE bilayer vesicles is proposed to induce fusion by facilitating both aggregation and subsequent fusion. Polymerization induces lipid phase separation with consequent decreased hydration of PE-rich domains and decreased intrinsic radius of curvature (R0)7 of the lipids composing the PE-rich domains. Since the domains of monomeric lipids contain only small fractions of PC, the hydration of these surfaces is diminished. The decreased R0 of the PE domains is a consequence of both the increased ratio of DOPE to monomeric-PC and the decreased membrane hydration. Attractive hydrophobic forces promote adhesion of the membranes at these sites. After adhesion, formation of fusion intermediates is facilitated by the decreased R0 of the lipids composing the apposed membrane leaflets.188
Bilayers of Neutral and Ionic Lipids
Cross-linking polymerizations of homogeneous mixtures of polymerizable and nonpolymerizable lipids can produce lipid domains when both lipids are neutral or even if one of the lipids is charged. Gaub et al. showed that the polymerization of a 1:1 molar mixture of 16 and DMPC in the fluid phase caused the formation of a DMPC-rich phase and polymeric lipid domains.192 These experiments showed that the polymerization of charged lipids could occur despite the electrostatic repulsion of the monomers of 16.
Armitage et al. demonstrated that polymerization of neutral lipids could serve to concentrate nonreactive charged colipids into domains.193 The high lateral mobility of lipid monomers permits their diffusion to the growing polymer chain ends even if the bilayers are composed in large part of nonreactive lipids. The polymerization-induced formation of enriched domains of ionic lipids was demonstrated by interactions of molecules at the bilayer surface that are electrostatically associated with the nonpolymerizable ionic lipids. Vesicles composed of a neutral polymerizable PC, bis-AcrylPC 7, and the nonreactive anionic 1,2-dioleoyl phosphatidic acid (DOPA), which can bind cationic dyes to the bilayer surface, were polymerized to different extents. The efficiency of electronic energy transfer between a pair of donor and acceptor dyes was used to probe the effect of polymerization on the average distance of separation between the dyes. Calculation of the energy transfer efficiency revealed that the quantum efficiency was 0.09 for the unpolymerized vesicles whereas it increased to 0.71 for the completely polymerized vesicles. The increased efficiency of energy transfer at the vesicle surface was a direct reflection of the increased local concentration, i.e., domain formation, of the bilayer anionic binding sites.
III. Polymerization of Nonlamellar Phases
The initial goal of the research on lipid nonlamellar phases, including the various bicontinuous cubic (QII) phases and the inverted hexagonal (HII) phase, was to demonstrate a general polymerization strategy for the stabilization of these three-dimensional lyotropic liquid crystals. Their potential utility depends in part on the stabilization of these phases in order to expand their rather limited useful temperature and concentration ranges. Generally the HII phase is found at higher temperatures and lower water content than the Lα phase and the QII phases occur at intermediate temperatures and concentrations. Srisiri et al. compared the polymerization of a reactive lipid 63 in the Lα phase and the HII phase.194 The number-average degree of polymerization of linear polymers obtained from the same lipid in these two phases depended strongly on the initiation chemistry but was insensitive to the lipid phase. Thus, the degree of polymerization for the chain reaction initiated by the redox couple, sodium persulfate and sodium bisulfite, was 250 ± 30 for the lamellar phase and 293 ± 40 for the HII phase. The rates of polymerization were similar as well. The rate constants for propagation and termination are likely to depend on the rate of diffusion of monomeric lipid to the growing radical chain end. Since the lipids in both the Lα and the HII phases diffuse rapidly, i.e., diffusion coefficients of 1 μm2 s−1 or more,195 it appears reasonable that the polymerization processes in the two phases would yield similar results. Consequently, many of the lessons learned from the extensive polymerization studies of the Lα phase can be applied to the higher temperature nonlamellar phases. Thus, for example, diacetylenic lipids that only polymerize in the slow diffusion regime do not appear to be suitable for polymerization of nonlamellar phases.
The design of suitable polymerizable lipids was crucial to the success of the research. It is desirable to be able to perform the polymerizations at a variety of temperatures as dictated by the phase behavior of the lipid, thereby limiting the use of thermal initiation chemistries and favoring photo and redox initiation. Consequently, reactive groups, such as diene, styrene, and acryloyl, have received the most attention to date. Unlike lamellar phase polymerizations, the location of the reactive group in the lipid and the assembly requires careful consideration. The placement of the group on either or both the lipid tail(s) near the lipid backbone, i.e., the glycerol unit in the case of phospholipids, appeared to be the most promising because covalent linkage of lipids near the backbone should have less effect on the important forces that act at the headgroup and tails of the lipids. Furthermore, the use of a diene group conjugated with the acyl chain carbonyl does not interfere with the biocompatibility of the lipid–water interface. These factors were used in the initial design of suitable lipids for the successful polymerization of nonlamellar phases.196 The selection of lipid structures also included consideration of the spontaneous radius of curvature, R0, of the designed lipid by reference to nonpolymerizable lipids reported in the biophysical literature.2,7 These data were used to predict the mesomorphic behavior of lipids or lipid mixtures. Lipids with small absolute R0 values require less thermal energy to undergo Lα/HII transitions, whereas those with large absolute R0 values form stable Lα phases even at high temperatures. Hydrated lipids with intermediate absolute R0 values are likely to form QII phases at moderate temperatures. Unsaturated or chain-substituted PE lipids are well-known for their small R0 values, whereas compounds with intermediate values include certain PC/PE mixtures, N-methylatedPEs, monoacylglycerols (MAG), among others.1
A. Bicontinuous Cubic Phases
The stabilization of cubic phases, which are bicontinuous with respect to the polar (aqueous) and nonpolar (lipid) regions, is especially interesting because these phases are organic analogues of zeolites. Mathematical descriptions of the minimal surfaces of the three bicontinuous cubic morphologies found in amphiphile/water systems were reported by Benedicto and O’Brien.5 Inspection of the minimal surfaces (Figure 17) and the complementary three-dimensional labyrinth nets (Figure 18) reveals the interpenetrating nature of the two distinct water channels. The three phases represented in the figures are termed double diamond (D), gyroid (G), and primitive (P), which correspond to the Pn3m, Ia3d, and Im3m space groups, respectively. Successful polymerization of the lipid domains in these mesophases should yield materials with interpenetrating water channels whose surfaces might be functionalized with chemical or biological labels or reagents in a suitable manner for diagnostics, separations, and conversions. The size of the aqueous channels, which is a function of the lipid composition and the phase, can range from ca. 4 to 20 nm in diameter, large enough to accommodate certain water-soluble macromolecules. It is known that Schoen’s normalized surface-to-volume ratio increases from P to D to G.197 That means that the Ia3d needs the least amount of water, that the Pn3m requires more water, and the Im3m requires the most water.
Figure 17.
Mathematically generated minimal surfaces that represent the three bicontinuous cubic morphologies associated with hydrated lipids. These surfaces correspond to the lipid portion of the respective phases (space groups from left to right, Pn m, Ia d, and Im m) (ref 5).
Figure 18.

Representation of the three-dimensional networks that are complementary to the surfaces shown in Figure 17. These labyrinths correspond to the water portion of the respective phases obtained from hydrated lipids.
Lee et al. described the polymerization of a pre-formed QII phase as well as HII phase in 1995.196 Theyused a pair of lipids, a mono-DenPE (63) and a bis-DenPC (64) in a 3:1 molar ratio, to achieve an intermediate average R0 value for the mixture. It was already known that certain mixtures of the PE and PC form the D phase.1,198 Both X-ray diffraction and 31P NMR spectroscopy were used to partially characterize the phase behavior of the hydrated lipid mixture. The sample remained lamellar until 55 °C and transformed into a cubic phase upon thermal cycling between 50 and 60 °C. When the sample was cooled below 55 °C, the lamellar phase was reformed. The observed basis vector length of 12.2 nm for a sample (300 mg lipid/mL) incubated at 60 °C for 12 h was consistent with the Pn3m space group. The 31P NMR spectrum of a similar sample at temperatures below 55 °C exhibited a line shape consistent with the Lα phase, but after incubation at 60–65 °C the sample spectrum was simply a narrow (2 ppm fwhm) isotropic peak as expected for a QII phase. Polymerization studies of this QII phase were performed at low lipid concentration (25 mg lipid/mL), i.e., excess water. Several samples were incubated at 60 °C for 6 h or more with potassium persulfate and sodium bisulfite. The loss of monomer was determined by absorption spectroscopy. Following the polymerization the water was removed by freeze-drying, then the polymerized sample solubility was tested in organic solvents. When the conversion to polymer in the sample was greater than 80%, the freeze-dried polymer was insoluble in several organic solvents, including hexafluoroisopropanol (HFIP). This evidence for sample cross-linking is consistent with the observations that 12.5 mol % of a bis-DenPC, in a mixture of mono-DenPC and bis-DenPC, is sufficient to cross-link the lamellar phase.71 The hydrated cross-linked samples remained in the QII phase even after they were cooled below the original phase transition temperature at 55 °C. Polymerized samples were stable during storage near 0 °C for prolonged periods. This confirms the original hypothesis that cross-linking of nonlamellar phases would broaden the temperature range of the mesophase. To gain further information about the new materials, both cryo and negative staining TEM were used to examine the morphology of the QII phase before and after polymerization.196 Cryo-TEM showed that the hydrated lipid mixture at room temperature consisted of multilamellar and unilamellar liposomes, whereas the morphology completely changed to a cubic lattice after incubation at 60 °C. The polymerized QII phase was stained with osmium tetroxide, embedded, and microtomed to yield thin 90 nm sections. The microscopy images showed polydomain structures with repetitive regions ascribed to aqueous channels with a diameter of 6 ± 1 nm. The rapid diffusion of the stain through the cubic phase sample was readily observed, indicating the water channels of the polymerized QII phase were readily accessible to compounds added at the periphery of the sample.
QII phases are also formed from monoacylglycerols (MAG) alone or in combination with diacylglycerols (DAG).1,199–201 A method to synthesize both a polymerizable dienoyl MAG and a DAG was developed to prepare for studies of QII phase polymerization.202 More recently, Srisiri et al. showed that when polymerizable MAG 65 was combined with the corresponding DAG 66 in a 9:1 molar ratio, an optically clear QII phase was formed at room temperature.203 Investigation of the phase behavior using cross-polarized light, 2H NMR spectroscopy, and X-ray diffraction found a well-defined cubic phase existed from no more than 5–45 °C. The X-ray diffraction pattern corresponded to a QII phase with Ia3d symmetry and a unit cell size of 13 nm at 25 °C. Polymerization of the reactive amphiphiles increased the stability of the assembly with retention of the QII phase. In this case the lipids were hydrated at 5 °C with deoxygenated aqueous hydrogen peroxide to form the QII phase, which was then incubated at 45 °C for 2 days to polymerize the hydrated lipids giving an optically clear product. The loss of monomers was greater than 90%. A sample of polymerized QII phase was freeze-dried and then dissolved in common organic solvents, demonstrating that the polymer was not cross-linked. The molar ratio of the bis-substituted dienoyl DAG 66 to the MAG 65 was insufficient to cross-link the mesophase. The number-average degree of polymerization was 200 ± 20. The isotropic nature of the linearly polymerized QII phase was observed up to at least 70 °C, whereas the unpolymerized QII phase was only stable to 45–50 °C.
This QII phase has been shown by NMR diffusion measurements to consist of bicontinuous lipid and water regions.204 Pulsed field gradient NMR spectroscopy was used to determine the water diffusion coefficient prior to polymerization of the dienoyl MAG 65 and dienoyl DAG 66 mixture. It was 1.0 ± 0.4 × 10−10 m2/s, an order of magnitude smaller than the diffusion of pure water (2 × 10−9 m2/s).203 This reduction in water diffusion was attributed to the non-Newtonian flow of water molecules in the interpenetrating network of water channels in the QII phase because of the interaction of the water molecules with the hydrophilic lipid headgroups. The diffusion of water inside the QII phase formed from monoolein is similarly retarded.199 The diffusion coefficient of water molecules after polymerization of the QII phase was 1.2 ± 0.2 × 10−10 m2/s, which is essentially the same as before the polymerization. These data indicate that polymerization of the lipid domains of QII phases composed of dienoyl-substituted lipids does not significantly perturb the water channels. This suggests that the water channels in appropriately polymerized QII phases could be elaborated to provide specific binding sites for water-soluble macromolecules. The reports of Lee at al.196 and Srisiri et al.203 introduced a successful general strategy for the stabilization of bicontinuous cubic phases. The ability to form stable organic zeolites with aqueous pore sizes that are an order of magnitude larger than those observed with inorganic zeolites should also provide opportunities for the creation of new composite materials.
B. Inverted Hexagonal Phase
The most commonly studied nonlamellar lipid phase is the HII phase, which is composed of aqueous columns encased in a monolayer of lipid and arranged in a hexagonal pattern (Figure 20).3 The polar head-groups are well-ordered at the water interface, whereas the lipid tails are highly disordered to fill the volume between the columns of water. Two complimentary strategies have been employed to polymerize the lipid domains of the HII phase. In one, the reactive groups were incorporated into the tails of the amphiphiles,205,206 whereas in the other the reactive groups were placed adjacent to the backbone of the amphiphile,196,207 as discussed earlier for the QII phase. The approach introduced by Gin and co-workers results in partial or complete cross-linking of the amphiphiles in the HII phase and yields an ordered polymer matrix.208 The alternative strategy (described below), reported by O’Brien and co-workers, polymerizes the individual unit cells of the HII phase in a manner that permits their subsequent separation.207
Figure 20.
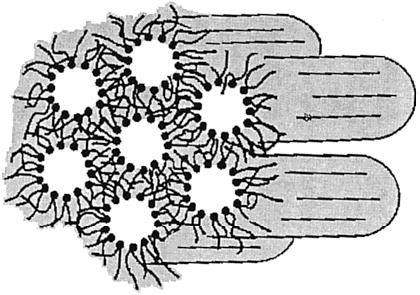
Schematic representation of the HII phase. Note the disordered tails of the lipids and the headgroups surrounding a water channel. Two strategies have been used to polymerize the HII phase. The polymerization of monomer 67 cross-links the phase and yields a mesoporous solid (ref 205). The polymerization of monomer 70 cross- links the individual unit cells of the HII phase (ref 207).
A polymerizable monomer 67 was prepared by substitution of the three hydroxyl groups on gallic acid with hydrophobic chains terminated with acryl-oyl groups. Hydration of the ionized acid produced a lyotropic mesophase, which was identified as a hexagonal phase by low-angle X-ray diffraction.205,209 The size of the unit cell was sensitive to the nature of the metal ion. When the metal ion was Na(I), Ni-(II), Co(II), or Cd(II) the d100 was 35.0 ± 0.7 Å, whereas it was 30.5 ± 0.4 Å for metal ions Eu(III) and Ce(III).209 The unit cell size of the HII phase formed from a monomer with divalent cations was independent of the metal ion size. However, the stronger lanthanide–carboxylate interactions appear to decrease the size of the monomer headgroup in a manner that permits closer packing of the hydrated amphiphiles. The observed reduction in unit cell sizes correlated with the IR characteristics of the materials. HII phases composed of monomer 67 with these counterions, water, and the photoinitiator, 2-hydroxy-2-methylpropiophenone, in a ratio of 85/10/5 (w/w/w) were cross-linked by overnight irradiation of the sample under nitrogen. The reaction was followed by changes in the acrylate IR bands, although the percent conversion to polymer was not reported. The polymers containing Ni(II), Co(II), and Ce(III) were paramagnetic, and the susceptibilities were determined. The Eu(III) polymer luminesced at 593 and 615 nm when excited at 370 nm. The observation of Eu(III) emission indicates the Eu(III) ions, which are presumed to be in the HII phase channels, have few if any water molecules in their primary coordination sphere.209
A nanocomposite of a polymerized HII phase with poly(p-phenylenevinylene) (PPV) in the channels was prepared by cross-linking polymerization of a mixture of monomer 67, the photoinitiator 2-hydroxy-2-methylpropiophenone, and a water solution of a PPV precursor in a 8/1/1 ratio (w/w/w).205 The polymeric film was 100 μm thick. Low-angle X-ray diffraction data of the thin film was consistent with a hexagonal phase both before and after the photopolymerization. The unit cell dimensions of the polymeric material were slightly smaller than those of the unpolymerized sample. The PPV precursor, poly(p-xylylenedimethylsulfonium chloride), was dissolved in the aqueous domains of the HII phase. Partial conversion of the precursor to PPV occurred during the photopolymerization, whereas a more complete conversion was achieved by heating the nanocomposite film at 220 °C for 4 h. The sample fluorescence increased with the heating time in a manner consistent with the generation of PPV. The PPV fluorescence of the nanocomposite, which contained no more than 0.04 wt % PPV, was greater per unit volume than that of pure PPV. These data indicate that the PPV chains in the HII phase channels were considerably more fluorescent than bulk PPV. Because the emission from conjugated polymers is known to be enhanced when the polymer interchain separation distance is increased by solubilization, copolymerization, or composite formation,210 it is likely that the enhanced fluorescence of the PPV in the ordered nanocomposites is due to the isolation of the polymer chains in the nanocomposite channels.
Gin and co-workers demonstrated that polymer–inorganic nanocomposites can be formed by condensation of tetraethyl orthosilicate (TEOS) in the aqueous channels of the polymerized HII phase.206 The hexagonal phase was formed from a sodium p-styryloctadecanoate 68 monomer, the cross-linker divinylbenzene (5–10 wt %), a photoinitiator (2 wt %), and 2–20 wt % water. A similar hexagonal phase could be formed in the presence of TEOS and its photoinitiator, diphenyliodonium chloride. A thin film of the sample was polymerized by exposure to 365 nm light. Evidence of monomer loss was obtained from the decrease in the styryl absorbance at 254 nm. Solid-state 29Si MAS NMR provided evidence for TEOS condensation in the cross-linked polymer film, thereby demonstrating that the cross-linking of the amphiphilic styrene and the sol–gel precursor can proceed at the same time in the respective hydrophobic and hydrophilic domains of the HII phase. Another inorganic–organic composite was prepared by reaction of the HII phase of the Cd(II) salt of monomer 68 with hydrogen sulfide gas to produce CdS particles in the nanochannels of the HII phase.211 The same amphiphilic monomer was used to create nanoporous polymeric films for heterogeneous catalysis.212 Because the aqueous pores of the resultant HII phase are lined with closely packed carboxylate groups, it is likely that the basicity of the carboxylate is increased. This possibility was confirmed through an examination of the base-catalyzed Knovenagel condensation of ethyl cyanoacetate with benzalde-hyde. The reaction proceeded more rapidly in the inverted hexagonal phase than in an otherwise equivalent lamellar phase. Moreover, mesoporous materials such as NaY and MCM-41 did not show any significant catalytic activity under the same conditions.
A new type of monomer, based on gallic acid, was recently reported to form the HII phase at room temperature. Hoag and Gin developed a synthetic route toα,ω -bromodienes, which were used to alkylate the three hydroxy groups on methyl gallate.213 After hydrolysis of the ester group, the resulting trisubstituted gallic acid salt 69 was used in photopolymerization experiments to cross-link the HII phase. Lipids with terminal diene groups have previously been used to polymerize lamellar phases.77,79
The biophysical literature indicates that PE lipids with unsaturated or branched acyl chains are good candidates for formation of the HII phase.214 This information was used to design dienoyl-substituted PE for the preparation of HII phases. Among the various polymerizable PE lipids prepared to date, the mono-DenPE 63 and bis-DenPE 70 are noteworthy. The transition temperature (TH) for the lamellar HII phase for hydrated mono-DenPE 63 was 55 °C. Consequently, it was especially useful for the comparative study of polymerizations in the lamellar and HII phase described earlier.194 In contrast, the TH of hydrated bis-DenPE 70 was below 0 °C.207 The HII phase of this hydrated PE persisted to a least 80 °C. Therefore, this bis-substituted PE was a good candidate for cross-linking polymerization around and along the water columns of the HII phase at a variety of temperatures. A particularly interesting characteristic of the HII phase is the decrease in the unit cell dimension of the hexagonal lattice as the sample temperature is increased. Reduction in the radius of the water channel accounts for most of the change in this lattice vector.215 It appeared that a successful polymerization strategy of the lipid domains could lock-in the column diameter rendering the unit cell of the HII phase insensitive to subsequent changes in temperature. The HII phase of bis-DenPE 70 in water (1/1 w/w) showed a typical reduction in unit cell size over the temperature range examined (near 0–80 °C). Polymerization to high conversion was accomplished with redox initiators, e.g., potassium persulfate/sodium bisulfite or hydrogen peroxide. Photopolymerization was not used because the sample optical density was too great for uniform exposure and polymerization of the sample. Characterization of the polymerized material by X-ray diffraction and 31P NMR indicated that it remained in a hexagonal phase with a unit cell diameter of 7 nm. As predicted, temperature cycling (over at least 40° range) of the polymerized HII phase did not alter the dimensions of the unit cell.207 Similar observations were reported for a HII phase sample composed of a mixture of polymerizable PE and PC.196 These data indicate that the dimensions of the unit cell could be varied by choice of temperature and then locked-in to prepare cross-linked tubes of selected diameters. The polymerization of the HII phase composed of 70 proceeded by cross-linking of the individual tubes, because the polymerized sample could be dispersed in volatile organic solvents then spread as a monolayer of the polymerized unit cells, i.e., tubes of poly(lipid) each surrounding a water core.207 Light scattering of a dispersed sample indicated that some of the objects were ca. 200 nm in length, i.e., an aspect ratio of 28 for a individual tubes with a unit cell diameter of 7 nm.
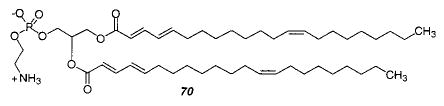
The polymerization of suitably designed lipids in both the HII and QII phases has proven to be an effective means to enhance the stability of these lyotropic liquid crystals. The research since the initial publication in 1995 provides an initial understanding of the polymerization process as well as initial insights into the possible utility of these new materials. Reasonable rates and degrees of polymerization can be achieved under suitable conditions. The stabilized QII phases offer large water channels that appear to be able to accommodate water-soluble macromolecules. There is also considerable interest in the prospect of using QII phases as biocompatible encapsulating and controlled release media. The smaller water columns of the HII phase have already been shown to be suitable for catalytic sites as well as the creation of elongated nanocomposites composed of minerals or polymers. Continued progress in these research areas will require interdisciplinary contributions of biological, polymer, and surface sciences.
IV. Outlook
It should be clear from this review that the polymerization of lamellar and nonlamellar phases has attracted the interest of many research groups over the past two decades. Their combined efforts have introduced a wide range of chemistries to be able to prepare the monomeric amphiphiles and to successfully use these new compounds to polymerize pre-formed ensembles of molecules. In addition, the increased understanding of the polymerization process in the confined space of monolayers, bilayers, etc., now makes it possible to control these reactions to prepare new materials in a reproducible manner. The materials properties are, of course, sensitive to the extent of cross-linking. Fortunately, recent studies have revealed some of the requirements for cross-linking in lamellar and nonlamellar phases. Obviously more remains to be learned. However, it is already possible to prepare molecular objects such as spheres and rods by the cross-linking of bilayer vesicles and the unit cell of the inverted hexagonal phase, respectively. Moreover, the cross-linking of thin bilayer films of reactive phosphatidylcholines yields uniform, biocompatible, robust films that offer interesting possibilities for surface modification.55
We conclude this review by briefly discussing a couple of the potential uses of polymerized lamellar or nonlamellar phases. These include materials for diagnostics and sensors, the controlled delivery of drugs or other reagents, the modification of surfaces, and the preparation of nanocomposites, among others. The critical contribution of the polymer varies widely with the application. In some cases a polymerized diacetylenic bilayer serves as a colorimetric sensor, whereas in other instances the initiation of the polymerization process can be used to trigger the release of encapsulated agents from vesicles.
A. Polydiacetylene Colorimetric Sensors
The deep blue or red color associated with polydiacetylenes has long attracted interest because of the potential for using the shift from blue to red to detect specific changes in the vicinity of the polymer. Early examples correlated increased temperature with this colorimetric change.112,118,216
Glycolipids in cell membranes mediate cell recognition, antigenicity histocompatibility, and protein affinity. Biological recognition could be mimicked in polymerized vesicles by incorporating a diacetylenic lipid bearing a carbohydrate, e.g., glucose, headgroup into vesicles composed of other diacetylenic lipids.217 After polymerization, the red PDA vesicles were agglutinated by the addition of a solution of concanavalin A (Con A) to the suspension of vesicles. The red precipitate was then redispersed by addition of an aliquot of a solution of α-methyl mannopyranoside, which has a higher affinity for Con A than glucopyranoside.
Direct colorimetric detection of the recognition of a ligand by a membrane-associated receptor was described by Charych et al.218 The color of a supported monolayer of PDA containing a small percent of lipid-linked silaic acid was found to change, in a dose-dependent manner, from blue to red upon binding the viral lectin, hemoglutinin.219 The color change is proposed to occur due to a reduction on the effective conjugation length of the PDA, once the macromolecule binds to the polymeric film surface. Other macromolecular assemblies, such as toxins, and macromolecules, such as enzymes, can be detected colorimetrically by appropriately designed PDA vesicles.220–223 A review that describes the choice of diacetylenic lipids for the formation of these chromic PDA vesicles has appeared.96 These assays have been successful with macromolecular analytes at high concentration. A particularly interesting extension of the PDA colorimetric strategy to the detection of small compounds relies on the coupling of an enzyme to the PDA assembly. The enzyme hexokinase was synthetically coupled to a PDA film.224 Hexokinase undergoes a conformational change upon glucose binding at the active site. The resultant change in enzyme structure perturbs the PDA film leading to a measurable color change. The specific nature of the interaction was shown by the absence of a color change when other sugars were used. Recently, it was reported that millimolar concentrations of cations could be detected by the PDA color change associated with vesicles composed of a PDA, a co-lipid, and a selective ionophore.225 The second issue, the inherent sensitivity of colorimetric methods are limited to relatively high concentrations of analyte, appears to be more challenging. However, an interesting approach to expanding the concentration range of PDA-based sensors could take advantage of the difference in fluorescence of the blue and red forms of PDA.226
B. Polymerized Liposomes for Drug Delivery
Vesicles composed of phospholipids, i.e., liposomes, have been extensively investigated as vehicles for the protection and delivery of therapeutic agents in the blood stream. The pharmacokinetics of drugs is significantly enhanced by encapsulation and delivery from liposomes. Both polymerized liposomes and polymerizable liposomes have been examined for drug delivery. In the former instance, polymerized liposomes offer significantly improved in vivo and in vitro stability. Appropriate design of polymerized liposomes can modulate the permeability of drugs. Linear polymerization of liposomes causes a moderate decrease in permeability, whereas substantial reductions in permeability can be achieved by cross-linking of liposomes.32 The observed effect of bilayer cross-linking on permeability depends on the cross-link density and the size of the encapsulated permeant.
The potential biomedical utility of any polymerized liposome preparation will depend on its toxicity. Regen and Juliano collaborated in a series of investigations to determine the toxicity of liposomes prepared from bis-methacryloylPC (3).29 Polymerized liposomes of 3 in plasma caused very little platelet aggregation.227 The uptake of both polymerized and unpolymerized liposomes of 3 were examined in mouse peritoneal macrophages in tissue culture.228 A radioactive lipophilic marker incorporated into the liposomes was metabolized at a similar rate regardless of whether the liposomes were polymerized or not. The polymerized liposomes were not toxic to the macrophages, whereas unpolymerized liposomes of 3 were toxic to the cells. When polymerized liposomes of similar composition were administered intravenously to mice, the authors observed no signs of acute toxicity after 3 weeks.229 These studies indicate that polymerized liposomes from methacryloyl lipids are well tolerated by cells and mice whereas the monomeric 3 is not. The reasons for toxicity of the monomeric lipid are not clear. More recently, a tissue culture study of the HeLa cell uptake of liposomes composed of DOPE and the photopolymerizable lipid, bis-SorbPC (18), did not find evidence of toxicity by the unpolymerized liposomes.230 Moreover, cross-linked liposomes prepared from 17 did not diminish the viability of mucosal cells in culture.231 The studies to date demonstrate that each polymerizable lipid or polymerized liposome composition will need to be examined for toxicity and that at least some of these reactive lipids can be considered for biomedical purposes.
The triggered release of water-soluble encapsulated agents from liposomes has been successfully demonstrated by the application of radiation or other forms of energy to polymerize appropriately designed liposomes. The concept was advanced by Ringsdorf as the “uncorking of liposomes”,176 as discussed in section II.F.1. O’Brien and co-workers have shown that the photopolymerization of a subset of lipids that compose conventional183,184,190 as well as sterically stabilized liposomes232,233 can trigger the release of water-soluble fluorescent markers (see section II.F.2 for discussion of the composition and polymerization of the liposomes). Sterically stabilized liposomes are liposomes with surface-attached poly(ethylene glycol) groups. The PEG groups of these liposomes diminish their uptake from the bloodstream by the reticuloendothelial system. Quite recent studies have shown that encapsulated therapeutic agents, e.g., doxoru-bicin, can be released from sterically stabilized liposomes by clinically relevant doses of radiant energy. These observations are especially encouraging because of the capability of sterically stabilized liposomes to accumulate at tumor sites.234–237 The increased permeability of the vasculature at these sites allows sterically stabilized liposomes to escape the capillaries to reach the tumor interstitial space. However, once the sterically stabilized liposomes are at the tumor site, the PEG groups can interfere with rapid release of the encapsulated reagents. Consequently, it continues to be important to find methods for the triggered release of reagents from PEG–liposomes.
Another approach to drug delivery is the attachment of the drug to the bilayer wall of a polymerized liposome. Stable liposomes were formed by polymerizing a tocopherol-containing lipid analogue via a methacrylate function. The liposomes were formed by ultrasonication and polymerized by radical polymerization. The vesicles were stable for months. The tocopherol liposomes effectively functioned as an antioxidant.238
C. Nanocomposites
The ordered structure of monolayers, bilayer vesicles, tubules, and the inverted hexagonal phase has stimulated considerable interest in the formation of mineralized or metallized composites. Much of this research was reviewed by Fendler.239 Here, we highlight those studies that have utilized polymerizable amphiphiles. Over a decade ago, it was already known that metal particles could be formed at the surface of bilayer vesicles by electroless deposition240 and that the interior of vesicles could be employed to precipitate minerals.241–243 Fendler and co-workers used polymerized vesicles with entrapped cadmiun sulfide to photochemically generate hydrogen.242 Markowitz et al. used membrane-bound palladium in the interior of polydiacetylenic vesicles to catalyze the formation of gold or cobalt nanoparticles.244 The methods were similar to those used earlier for the electroless deposition of metals on tubules.144 The use polymerized bilayer vesicles for metal particle formation was proposed to be superior to conventional vesicles for two reasons. First, the polymerized vesicles provided a relatively stable environment for the formation and protection of the nanoparticles in a manner that inhibits agglomeration. Second, the PDA formation in the bilayer caused a sufficient increase in the membrane permeability of cations and anions to facilitate efficient growth of the particles.
Earlier it was noted that tubules formed from diacteylenic lipids or other amphiphiles could serve as templates for the growth of metals along their external and internal surface.143,245 The water channel of the inverted hexagonal phase offers somewhat similar sites for the formation of elongated metal or mineral deposits. The initial realization of these possibilities206,211 presages ample opportunities for the formation of rodlike materials for various applications.
V. Abbreviations
AAPD 2,2′-azobis(2-amidinopropane dihydrochloride)
AcrylPC acroyl-substituted PC
AFM atomic force microscopy
AIBN 2,2′-azobis(isobutyronitrile)
CON A Concanavalin A
CTAT cetyl trimethylammonium tosylate
D double diamond
DAG diacylglycerol
DenPC dienoyl-substituted PC
DMPC dimyristoyl PC
DNPC 1,2-bis(dinonanoyl)PC
DOPA dioleoyl phosphatidic acid
DOPC dioleoyl PC
DOPE dioleoyl PE
DPPC dipalmitoyl PC
DSPE distearoyl PE
DTT dithiothreitol
DVB divinylbenzene
ESCA X-ray photoelectron spectroscopy
G gyroid
GPC gel permeation chromatography
HII inverted hexagonal
HEA hydroxyethyl acrylate
HFIP hexafluoroisopropanol
ILA interlamellar attachments
IPA ion-paired amphiphile
LB Langmuir–Blodgett
LUV large unilamellar vesicle
MAG monoacylglycerol
MAS magic angle spinning
MCM41 commercially available silicon zeolite
NaDHP sodium dihexadecyl phosphate
NBD-PE N-4-nitrobenzo-2-oxa-1,3-diazole-PE
OTS octadecyltrichlorosilane
P primitive
PC phosphatidylcholine
PDA poly(diacetylene)
PE phosphatidylethanolamine
PMMA poly(methyl methacrylate)
PPV poly(p-phenylenevinylene)
QII bicontinuous cubic
SEM scanning electron microscopy
SorbPC sorbyl-substituted PC
SUV small unilamellar vesicle
TAPP tetra(trimethylammonium phenyl)porphyrin
TEM transmission electron microscopy
TEOS tetraethyl orthosilicate
TX-100 Triton X-100
Figure 1.
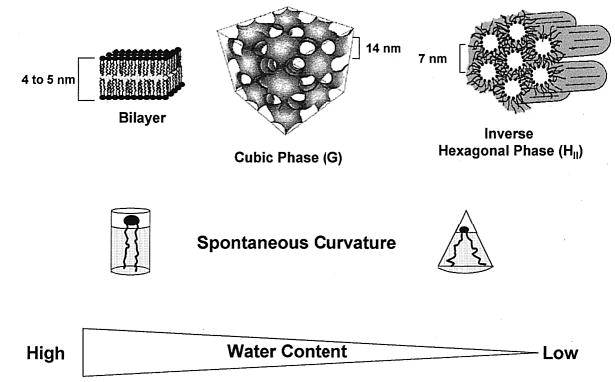
Representations of lamellar and nonlamellar phases of hydrated amphiphiles. (Left to right) The phase changes from lamellar to a bicontinuous cubic (QII) phase and to the inverted hexagonal (HII) phase, as the spontaneous curvature of the hydrated amphiphile increases, and the water content of the sample decreases. The approximate repeat distances are shown for each phase.
Figure 2.
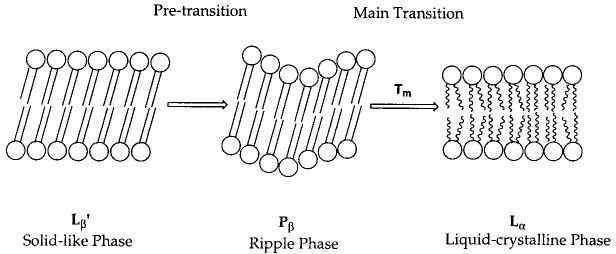
Schematic of the transformation of a solidlike bilayer, where the lipid tails are in the all-trans extended confomation, to the liquid-crystalline phase, where gauche conformers are abundant in the lipid tails. The intervening ripple phase is found in some pure lipid systems.
Figure 5.
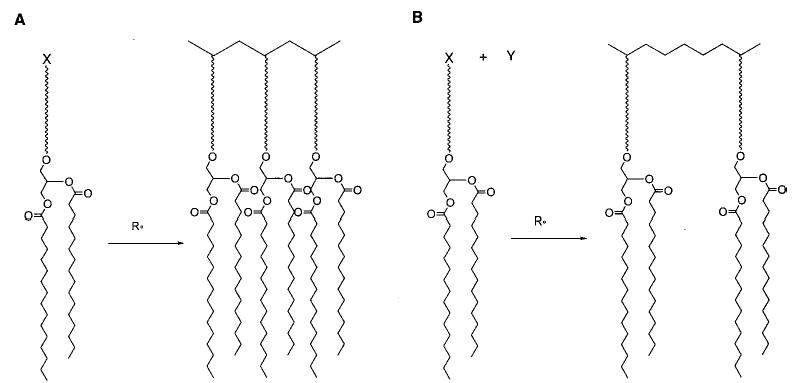
Representation of reactive lipids that incorporate a hydrophilic spacer group between the lipid headgroup and the reactive group (X), where (A) a homopolymer of the lipid is formed and (B) the lipid is copolymerized with a hydrophilic comonomer (Y), such as hydroxyethylacrylate (ref 13).
Figure 7.
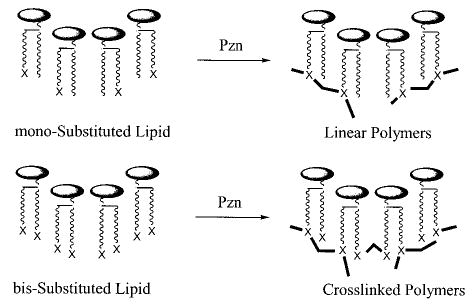
Schematic representation of the linear or cross-linked lipid polymers formed by polymerization (Pzn) of either monosubstituted or bissubstituted lipids, respectively.
Figure 8.
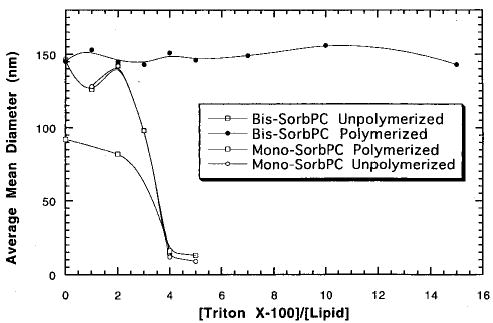
Effect of surfactant (TX-100) on the particle size of LUV before and after polymerization of mono-SorbPC (17) and bis-SorbPC (18). The un-cross-linked LUV were converted to mixed micelles with 4 equiv of TX-100 per lipid (ref 38).
Figure 9.
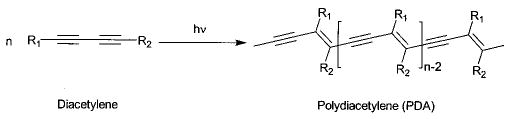
Photochemical conversion of diacetylene to poly-(diacetylene).
Figure 10.
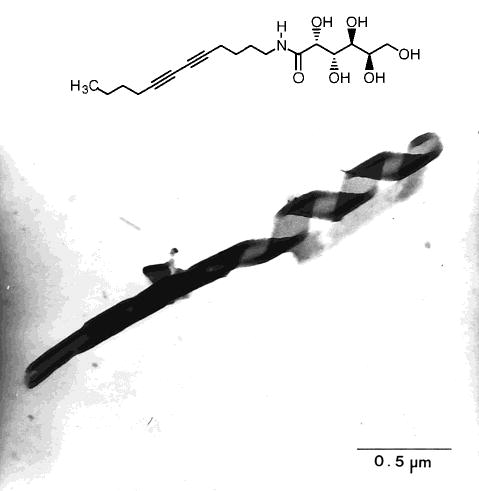
TEM of unstained hydrated N-dodeca-5,7-diyne galactonamide assembly that shows the winding of a bilayer helix and tubule (ref 131).
Figure 11.
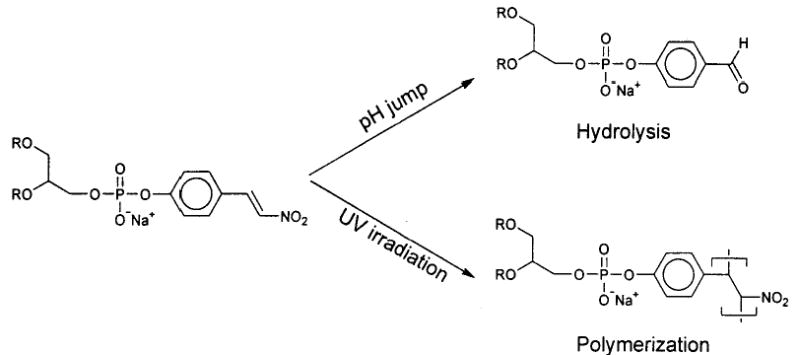
Stategy for separate polymerization of the inner and outer leaflets of a bilayer by taking advantage of the difference in chemical and photochemical reactions of monomer 46.
Figure 12.
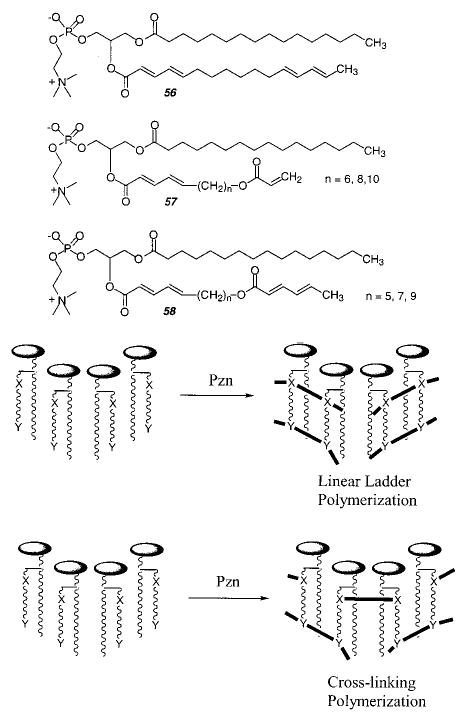
Schematic representation of the polymers formed from dependent (ladder-like) and independent (cross-linked) polymerization (Pzn) paths of two reactive groups tethered via an aliphatic sn-2 chain.
Figure 13.
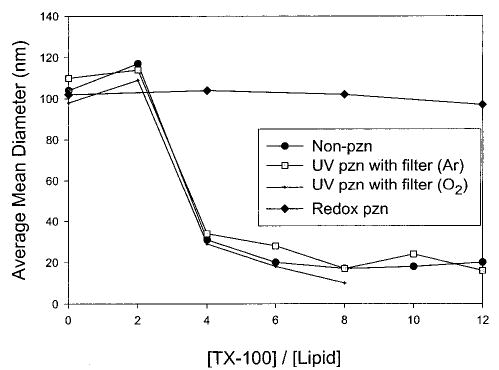
Effect of surfactant on the stability of LUV before and after polymerization of monomer 57 (n = 9). The redox-polymerized LUV were stable to more than 12 equiv of TX-100 per lipid, whereas photopolymerization of the single dienoyl group did not increase the stability beyond that of the unpolymerized LUV (ref 165).
Figure 14.
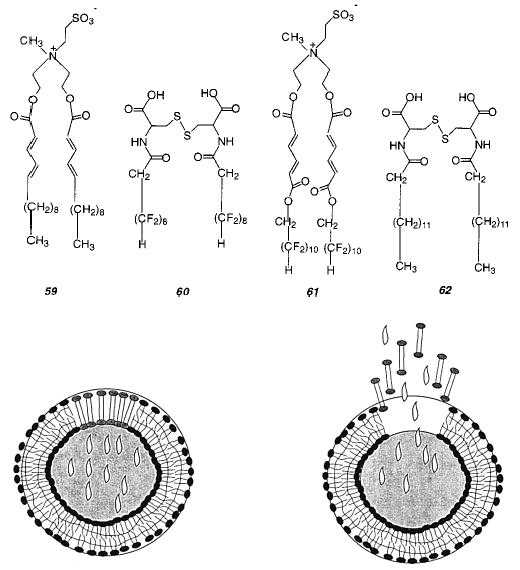
Uncorking of a phase-separated vesicle.
Figure 15.

Electon microscopy of freeze–fracture replicas of rhodopsin-poly-16/DOPC membranes in 30% glycerol–water frozen from room temperature. The particles on the fracture faces are morphological manifestations of the protein embedded in the bilayer.
Figure 16.
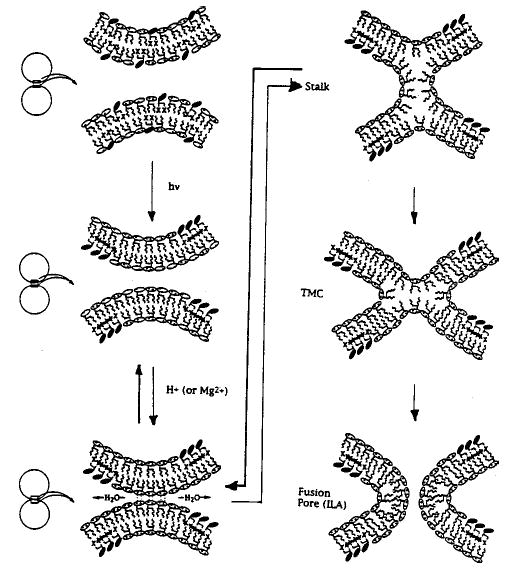
Schematic representation of the polymerization-induced phase separation of DOPE and poly-18 leading to the fusion of bilayer vesicles through contact of the enriched DOPE domains. Bilayer contact is postulated to occur through a stalk intermediate which then transforms into an interlamellar attachment (ILA) (aka fusion pore).
(Reprinted with permission from ref 170. Copyright 1996 Springer-Verlag.)
Figure 19.
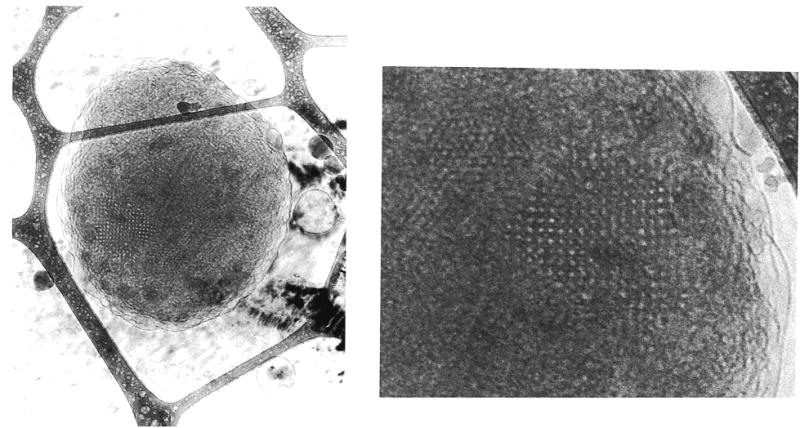
Cryo-transmission electron microscopy of a polymerized QII phase prepared from a 3:1 M mixture of monomers 63 and 64. The particle on the left is approximately 3 micrometers across. The image of the cubic lattice on the right is a 10-fold enlargement (ref 196). The dark border images are due to the lacy carbon grid.
References
- 1.Lindblom G, Rilfors L. Biochim Biophys Acta. 1989;988:221–256. [Google Scholar]
- 2.Gruner SM. J Phys Chem. 1989;93:7562–7570. [Google Scholar]
- 3.Seddon JM. Biochim Biophys Acta. 1990;1031:1–69. doi: 10.1016/0304-4157(90)90002-t. [DOI] [PubMed] [Google Scholar]
- 4.Evans DF, Wennerstrom H. VCH Publishers; New York: 1994. The Colloidal Domain: Where Physics, Chemistry, Biology, and Technology Meet. [Google Scholar]
- 5.Benedicto AD, O’Brien DF. Macromolecules. 1997;30:3395–3402. [Google Scholar]
- 6.Bates FS. Science. 1991;251:898–905. doi: 10.1126/science.251.4996.898. [DOI] [PubMed] [Google Scholar]
- 7.Gruner SM. Proc Natl Acad Sci. 1985;82:3665–3669. doi: 10.1073/pnas.82.11.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lewis RNAH, McElhaney RM. Biophys J. 2000;79:1455–1464. doi: 10.1016/S0006-3495(00)76397-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Epand RM, Epand RF, Decicco A, Schwarz D. Eur J Biochem. 2000;112:2909–2915. doi: 10.1046/j.1432-1327.2000.01304.x. [DOI] [PubMed] [Google Scholar]
- 10.Li XJ, Schick M. J Chem Phys. 2000;112:6063–6072. [Google Scholar]
- 11.Fontell K. Adv Colloid Interface Sci. 1992;41:127–147. [Google Scholar]
- 12.Elbert R, Laschewsky A, Ringsdorf H. J Am Chem Soc. 1985;107:4134–4141. [Google Scholar]
- 13.Laschewsky A, Ringsdorf H, Schmidt G, Schneider J. J Am Chem Soc. 1987;109:788–796. [Google Scholar]
- 14.Ringsdorf H, Schlarb B, Venzmer J. Angew Chem, Int Ed Engl. 1988;27:113–158. [Google Scholar]
- 15.New RRC. Liposomes: A Practical Approach. In: New RRC, editor. IRL Press; Oxford, UK: 1990. [Google Scholar]
- 16.Kaler EW, Murthy AK, Rodriguez BE, Zasadzinski JAN. Science. 1989;245:1371–1374. doi: 10.1126/science.2781283. [DOI] [PubMed] [Google Scholar]
- 17.Fukuda H, Kawata K, Okuda H, Regen SL. J Am Chem Soc. 1990;112:1635–1637. [Google Scholar]
- 18.Fahey PF, Webb WW. Biochemistry. 1978;17:3046–3053. doi: 10.1021/bi00608a016. [DOI] [PubMed] [Google Scholar]
- 19.Gros L, Ringsdorf H, Schupp H. Angew Chem, Int Ed Engl. 1981;20:305–325. [Google Scholar]
- 20.Fendler JH. Science. 1984;223:888–894. doi: 10.1126/science.223.4639.888. [DOI] [PubMed] [Google Scholar]
- 21.Paleos CM. Chem Soc Rev. 1985;45:45–66. [Google Scholar]
- 22.Bader H, Dorn K, Hupfer B, Ringsdorf H. Adv Polym Sci. 1985;64:1–62. [Google Scholar]
- 23.Regen SL. Polymerized Liposomes. In: Regen SL, editor. Marcel Dekker; New York: 1987. pp. 73–108. [Google Scholar]
- 24.O’Brien DF, Ramaswami V. Encycl Polym Sci Eng. 1989;17:108–135. [Google Scholar]
- 25.Singh A, Schnur JM. Polymerizable Phospholipids. In: Singh A, Schnur JM, editors. Marcel Dekker; New York: 1993. pp. 233–291. [Google Scholar]
- 26.O’Brien DF, Armitage B, Benedicto A, Bennett DE, Lamparski HG, Lee YS, Srisiri W, Sisson TM. Acc Chem Res. 1998;31:861–868. [Google Scholar]
- 27.Srisiri W, Lee YS, O’Brien DF. Tetrahedron Lett. 1995;36:8945–8948. [Google Scholar]
- 28.Regen SL, Czech B, Singh A. J Am Chem Soc. 1980;102:6638–6640. [Google Scholar]
- 29.Regen SL, Singh A, Oehme G, Singh M. J Am Chem Soc. 1982;104:791–795. [Google Scholar]
- 30.Kusumi A, Singh M, Tirrell DA, Oehme G, Singh A, Samuel NKP, Hyde JS, Regen SL. J Am Chem Soc. 1983;105:2975–2980. [Google Scholar]
- 31.Meier H, Sprenger I, Bärmann M, Sackmann E. Macromolecules. 1994;27:7581–7588. [Google Scholar]
- 32.Dorn K, Klingbiel RT, Specht DP, Tyminski PN, Ringsdorf H, O’Brien DF. J Am Chem Soc. 1984;106:1627–1633. [Google Scholar]
- 33.Dorn K, Patton EV, Klingbiel RT, O’Brien DF, Ringsdorf H. Makromol Chem, Rapid Commun. 1983;4:513. [Google Scholar]
- 34.Bolikal D, Regen SL. Macromolecules. 1984;17:1287–1289. [Google Scholar]
- 35.Sells TD, O’Brien DF. Macromolecules. 1994;27:226–233. [Google Scholar]
- 36.Lei J, O’Brien DF. Macromolecules. 1994;27:1381–1388. [Google Scholar]
- 37.Lamparski HG, Oblinger E, O’Brien DF. Macromolecules. 1999;32:5450–5452. [Google Scholar]
- 38.Sisson TM, Lamparski HG, Kölchens S, Elyadi A, O’Brien DF. Macromolecules. 1996;29:8321–8329. [Google Scholar]
- 39.Sisson TM, Lamparski H, Kölchens S, Peterson T, Elayadi A, O’Brien DF. Methodologies and Models of Cross-linking Polymerization in Supramolecular Assemblies. In: Sisson TM, Lamparski H, Kölchens S, Peterson T, Elayadi A, O’Brien DF, editors. Vol. 695. American Chemical Society; Washington, DC: 1998. pp. 119–130. [Google Scholar]
- 40.Kölchens S, Lamparski H, O’Brien DF. Macromolecules. 1993;26:398–400. [Google Scholar]
- 41.Lei J, Sisson TM, Lamparski HG, O’Brien DF. Macromolecules. 1999;32:73–78. [Google Scholar]
- 42.Penner TL, Schildkraut JS, Ringsdorf H, Schuster A. Macromolecules. 1991;24:1041–1049. [Google Scholar]
- 43.Kato S, Kunitake T. Polym J. 1991;23:135–146. [Google Scholar]
- 44.Hirano K, Fukuda H, Regen SL. Langmuir. 1991;7:1045–1047. [Google Scholar]
- 45.Regen SL, Shin JS, Yamaguchi K. J Am Chem Soc. 1984;106:2446–2447. [Google Scholar]
- 46.Aliev KV, Ringsdorf H, Schlarb B. Makromol Chem Rapid Commun. 1984;5:345–352. [Google Scholar]
- 47.Fukuda H, Diem T, Stefely J, Kezdy FJ, Regen SL. J Am Chem Soc. 1986;108:2321–2327. doi: 10.1021/ja00269a030. [DOI] [PubMed] [Google Scholar]
- 48.Ringsdorf H, Schlarb B, Tyminski PN, O’Brien DF. Macromolecules. 1988;21:671–677. [Google Scholar]
- 49.Regen SL, Shin JS, Hainfeld JF, Wall JS. J Am Chem Soc. 1984;106:5756–5757. [Google Scholar]
- 50.Everaars MD, Marcelis ATM, Sudhölter EJR. Langmuir. 1996;12:3964–3968. [Google Scholar]
- 51.Marra KG, Winger TM, Hanson ST, Chaikof EL. Macromolecules. 1997;30:6483–6488. [Google Scholar]
- 52.Marra KG, Kidani DDA, Chaikof EL. Langmuir. 1997;13:5697–5701. [Google Scholar]
- 53.Chon JH, Marra KG, Chaikof EL. J Biomater Sci: Polym Ed. 1999;10:95–107. doi: 10.1163/156856299x00306. [DOI] [PubMed] [Google Scholar]
- 54.Orban JM, Faucher KM, Dluhy RA, Chaikof EL. Macromolecules. 2000;33:4205–4212. [Google Scholar]
- 55.Ross EE, Bondurant B, Spratt T, Conboy JC, O’Brien DF, Saavedra SS. Langmuir. 2001;17:2305–2307. [Google Scholar]
- 56.Cho I, Chung KC. Macromolecules. 1984;17:2935–2937. [Google Scholar]
- 57.Abid SK, Sherrington DC. Polymer. 1992;33:175–180. [Google Scholar]
- 58.Merkel R, Simon J, Ringsdorf H, Sackmann E. J Phys II. 1994;4:703–713. [Google Scholar]
- 59.Barraud A, Rosilio C, Ruaudel-Teixier A. Thin Solid Films. 1980;68:7–12. [Google Scholar]
- 60.Ringsdorf H, Schupp H. J Macromol Sci Chem. 1981;A15:1015–1026. [Google Scholar]
- 61.Laschewsky A, Ringsdorf H. Macrmolecules. 1988;21:1936–1941. [Google Scholar]
- 62.Fukuda K, Shibasaki Y, Nakahara H. Thin Solid Films. 1985;113:39–49. [Google Scholar]
- 63.Tieke B. Colloid Polym Sci. 1985;263:965–972. [Google Scholar]
- 64.Tieke B. Polymerization at Interfaces. In: Tieke B, editor. Gordon Breach; Philadelphia: 1992. pp. 105–181. [Google Scholar]
- 65.Matsumoto A, Ishizu Y, Yokoi K. Macromol Chem Phys. 1998;199:2511–2516. [Google Scholar]
- 66.Odani T, Matsumoto A. Macromol Rapid Commun. 2000;21:40–44. [Google Scholar]
- 67.Tyminski PN, Ponticello IS, O’Brien DF. J Am Chem Soc. 1987;109:6541–6542. [Google Scholar]
- 68.Ohno H, Takeoka S, Iwai H, Tsuchida E. Macromolecules. 1989;22:61–66. [Google Scholar]
- 69.Tsuchida E, Hasegawa E, Kimura N, Hatashita M, Makino C. Macromolecules. 1992;25:207–212. [Google Scholar]
- 70.Lamparski H, O’Brien DF. Macromolecules. 1995;28:1786–1794. [Google Scholar]
- 71.Liu S, O’Brien DF. Macromolecules. 1999;32:5519–5524. [Google Scholar]
- 72.Clapp PJ, Armitage BA, O’Brien DF. Macromolecules. 1997;30:32–41. [Google Scholar]
- 73.Ohno H, Ogata Y, Tsuchida E. Macromolecules. 1987;20:929–933. [Google Scholar]
- 74.Ohno H, Takeoka S, Iwai H, Tsuchida E. J Polym Sci. 1987;25:2737–2746. [Google Scholar]
- 75.Lopez E, O’Brien DF, Whitesides TH. J Am Chem Soc. 1982;104:305–307. [Google Scholar]
- 76.Takeoka S, Ohno H, Iwai H, Tsuchida E. J Polym Sci. 1990;28:717–730. [Google Scholar]
- 77.Anikin A, Chupin V, Anikin M, Serebrennikova G, Tarahovsky J. Makromol Chem. 1993;194:2663–2673. [Google Scholar]
- 78.Roks MFM, Visser HGJ, Zwikkrt JW, Verkley AJ, Nolte RJM. J Am Chem Soc. 1983;105:4507–4510. [Google Scholar]
- 79.Binder H, Anikin A, Kohlstrunk B. J Phys Chem B. 1999;103:450–460. [Google Scholar]
- 80.Binder H, Anikin A, Lantzsch G, Klose G. J Phys Chem B. 1999;103:461–471. [Google Scholar]
- 81.Binder H, Dietrich U, Schalke M, Pfieffer H. Langmuir. 1999;15:4857–4866. [Google Scholar]
- 82.Hupfer B, Ringsdorf H. Chem Phys Lipids. 1983;33:263–282. doi: 10.1016/0009-3084(83)90078-6. [DOI] [PubMed] [Google Scholar]
- 83.Day D, Ringsdorf H. J Polym Sci. 1978;16:205–210. [Google Scholar]
- 84.Day DR, Ringsdorf H. Makromol Chem. 1979;180:1059–1063. [Google Scholar]
- 85.Yager P, Schoen PE. Mol Cryst Liq Cryst. 1984;106:371–381. [Google Scholar]
- 86.Georger JH, Singh A, Price RR, Schnur JM, Yager P, Schoen PE. J Am Chem Soc. 1987;109:6169–6175. [Google Scholar]
- 87.Baughman RH, Chance RR. J Polym Sci, Polym Phys Ed. 1976;14:2037–2045. [Google Scholar]
- 88.Chance RR. Macromolecules. 1980;13:396–398. [Google Scholar]
- 89.Rubner MF, Sandman DJ, Velasquez C. Macromolecules. 1987;20:1296–1300. [Google Scholar]
- 90.Kollmar C, Sixl J. J Chem Phys. 1988;88:1343–1358. [Google Scholar]
- 91.Lio A, Reichert A, Ahn DJ, Nagy JO, Salmeron M, Charych DH. Langmuir. 1997;13:6524–6532. [Google Scholar]
- 92.Orchard BJ, Tripathy SK. Macromolecules. 1986;19:1844–1850. [Google Scholar]
- 93.Dobrosavljevic V, Stratt RM. Phys Rev B. 1987;35:2781–2794. doi: 10.1103/physrevb.35.2781. [DOI] [PubMed] [Google Scholar]
- 94.Singh A, Thompson RB, Schnur JM. J Am Chem Soc. 1986;108:2785–2787. [Google Scholar]
- 95.Tanaka H, Gomez MA, Tonelli AE, Thakur M. Macromolecules. 1989;22:1208–1215. [Google Scholar]
- 96.Okada S, Peng S, Spevak W, Charych D. Acc Chem Res. 1998;31:229–239. [Google Scholar]
- 97.Rhodes DG, Hui SW, Xu YH, Byun HS, Singh M, Bittman R. Biochim Biophys Acta. 1994;1215:237–244. doi: 10.1016/0005-2760(94)90048-5. [DOI] [PubMed] [Google Scholar]
- 98.Cheng Q, Stevens RC. Langmuir. 1998;14:1974–1976. [Google Scholar]
- 99.Jonas U, Shah K, Norvez S, Charych D. J Am Chem Soc. 1999;121:4580–3588. [Google Scholar]
- 100.Day D, Hub HH, Ringsdorf H. Isr J Chem. 1979;18:325–329. [Google Scholar]
- 101.Hub HH, Hupfer B, Koch H, Ringsdorf H. Angew Chem, Int Ed Engl. 1980;19:938–940. doi: 10.1002/anie.198009381. [DOI] [PubMed] [Google Scholar]
- 102.Hub HH, Hupfer B, Koch H, Ringsdorf H. J Macromol Sci Chem. 1981;A15:701–715. [Google Scholar]
- 103.O’Brien DF, Whitesides TH, Klingbiel RT. J Polym Sci: Polm Lett Ed. 1981;19:95–101. [Google Scholar]
- 104.Johnston DS, Sanghera S, Pons M, Chapman D. Biochim Biophys Acta. 1980;602:57–69. doi: 10.1016/0005-2736(80)90289-8. [DOI] [PubMed] [Google Scholar]
- 105.Johnston DS, Sanghera S, Manjon-Rubio A, Chapman D. Biochim Biophys Acta. 1980;602:213–216. doi: 10.1016/0005-2736(80)90304-1. [DOI] [PubMed] [Google Scholar]
- 106.Johnston DS, McLean LR, Whittam MA, Clark AD, Chapman D. Biochemistry. 1983;22:3194–3202. doi: 10.1021/bi00282a024. [DOI] [PubMed] [Google Scholar]
- 107.Roy BC, Fazal AM, Arruda A, Mallik S, Campiglia AC. Org Lett. 2000;2:3067–3070. doi: 10.1021/ol006357p. [DOI] [PubMed] [Google Scholar]
- 108.Peek BM, Callahan JH, Namboodiri K, Singh A, Gaber B. Macomolecules. 1994;27:292–297. [Google Scholar]
- 109.Ladika M, Fisk TE, Wu WW, Jons SD. J Am Chem Soc. 1994;116:12093–12094. [Google Scholar]
- 110.Lee YS, O’Brien DF. Chem Phys Lipids. 1992;61:209–218. doi: 10.1016/0009-3084(92)90100-4. [DOI] [PubMed] [Google Scholar]
- 111.Rhodes DG, Xu Z, Bittman R. Biochim Biophys Acta. 1992;1128:93–104. doi: 10.1016/0005-2760(92)90262-t. [DOI] [PubMed] [Google Scholar]
- 112.Albrecht O, Laschewsky A, Ringsdorf H. Macromolecules. 1984;17:937–940. [Google Scholar]
- 113.Tieke B. Adv Polym Sci. 1985;71:79–151. [Google Scholar]
- 114.Fouassier JP, Tieke B, Wegner G. Isr J Chem. 1979;18:22–232. [Google Scholar]
- 115.Mino N, Tamura H, Ogawa K. Langmuir. 1991;7:2336–2341. [Google Scholar]
- 116.Kunitake T, Shimomura M. Thin Solid Films. 1984;121:L89–L91. [Google Scholar]
- 117.Nakashima N, Kunitake M, Kunitake T, Tone S, Kajiyama T. Macromolecules. 1985;18:1515–1516. [Google Scholar]
- 118.Kuo T, O’Brien DF. J Am Chem Soc. 1988;110:7571–7572. [Google Scholar]
- 119.Kuo T, O’Brien DF. Langmuir. 1991;7:584–589. [Google Scholar]
- 120.Leaver J, Alonso A, Durrani AA, Chapman D. Biochim Biophys Acta. 1983;732:210–218. doi: 10.1016/0005-2736(83)90418-2. [DOI] [PubMed] [Google Scholar]
- 121.Papahadjopoulos D, Vail WJ, Jacobson K, Poste G. Biochim Biophys Acta. 1975;394:483–491. doi: 10.1016/0005-2736(75)90299-0. [DOI] [PubMed] [Google Scholar]
- 122.Schnur JM. Science. 1993;262:1669–1676. doi: 10.1126/science.262.5140.1669. [DOI] [PubMed] [Google Scholar]
- 123.Yager P, Schoen PA, Davies C, Price R, Singh A. Biophys J. 1985;48:899–906. doi: 10.1016/S0006-3495(85)83852-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Schoen PE, Yager P. J Polym Sci: Polym Phys. 1985;23:2203–2216. [Google Scholar]
- 125.Yager P, Price RR, Schnur JM, Schoen PE, Singh A, Rhodes DG. Chem Phys Lipids. 1988;46:171–179. [Google Scholar]
- 126.Schnur JM, Ratna BR, Selinger JV, Singh A, Jyothi G, Easwaran KRK. Science. 1994;264:945–948. doi: 10.1126/science.264.5161.945. [DOI] [PubMed] [Google Scholar]
- 127.Thomas BN, Lindemann CM, Clark NA. Phys Rev E. 1999;59:3040–3047. [Google Scholar]
- 128.Markowitz MA, Singh A. Chem Phys Lipid. 1996;84:65–74. [Google Scholar]
- 129.Frankel DA, O’Brien DF. J Am Chem Soc. 1991;113:7436–7437. [Google Scholar]
- 130.Fuhrhop JH, Blumtritt P, Lehmann C, Luger P. J Am Chem Soc. 1991;113:7437–7439. [Google Scholar]
- 131.Frankel DA, O’Brien DF. J Am Chem Soc. 1994;116:10057–10069. [Google Scholar]
- 132.Fuhrhop JH, Svenson S, Boettcher C, Rossler E, Vieth HM. J Am Chem Soc. 1990;112:4307–4312. [Google Scholar]
- 133.Fuhrhop JH, Boettcher C. J Am Chem Soc. 1990;112:1768–1776. [Google Scholar]
- 134.Giulieri F, Guillod F, Greiner J, Krafft MP, Riess JG. Chem-Eur J. 1996;2:1335–1339. [Google Scholar]
- 135.Cheng Q, Yamamoto M, Stevens RC. Langmuir. 2000;16:5333–5342. [Google Scholar]
- 136.Thomas BN, Corcoran RC, Cotant CL, Lindemann CM, Kirsch JE, Persichini PJ. J Am Chem Soc. 1998;120:12178–12186. doi: 10.1021/ja012137a. [DOI] [PubMed] [Google Scholar]
- 137.Svenson S, Messersmith PB. Langmuir. 1999;15:4464–4471. [Google Scholar]
- 138.Singh A, Markowitz MA. New J Chem. 1994;18:377–385. [Google Scholar]
- 139.Selinger JV, Schnur JM. Phys Rev Lett. 1993;71:4091–4094. doi: 10.1103/PhysRevLett.71.4091. [DOI] [PubMed] [Google Scholar]
- 140.Spector MS, Easwaran KRK, Jyothi G, Selinger JV, Singh A, Schnur JM. Proc Natl Acad Sci USA. 1996;93:12943–12946. doi: 10.1073/pnas.93.23.12943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Spector MS, Selinger JV, Singh A, Rodriguez JM, Price RR, Schnur JM. Langmuir. 1998;14:3493–3500. [Google Scholar]
- 142.Ringler P, Muller W, Ringsdorf H, Brisson A. Chem-Eur J. 1997;3:620–625. [Google Scholar]
- 143.Schnur JM, Price R, Schoen P, Yager P, Calvert JM, Georger J, Singh A. Thin Solid Films. 1987;152:181–206. [Google Scholar]
- 144.Markowitz M, Barah S, Brandow S, Singh A. Thin Solid Films. 1993;224:242–247. [Google Scholar]
- 145.Wang GJ, Hollingsworth RI. Langmuir. 1999;15:6135–6138. [Google Scholar]
- 146.Rosenblatt C, Yager P, Schoen PE. Biophys J. 1987;52:295–301. doi: 10.1016/S0006-3495(87)83216-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Archibald DD, Mann S. Nature. 1993;364:430–433. [Google Scholar]
- 148.Lvov YM, Price RR, Selinger JV, Singh A, Spector MS, Schnur JM. Langmuir. 2000;16:5932–5935. [Google Scholar]
- 149.Regen SL, Yamaguchi K, Samuel NKP, Singh M. J Am Chem Soc. 1983;105:6354–6355. [Google Scholar]
- 150.Samuel NKP, Singh M, Yamaguchi K, Regen SL. J Am Chem Soc. 1985;107:42–47. [Google Scholar]
- 151.Sadownik A, Stefely J, Regen SL. J Am Chem Soc. 1986;108:7789–7791. doi: 10.1021/ja00284a050. [DOI] [PubMed] [Google Scholar]
- 152.Stefely J, Markowitz MA, Regen SL. J Am Chem Soc. 1988;110:7463–7469. [Google Scholar]
- 153.Chung YC, Regen SL. Macromolecules. 1991;24:5738–5739. [Google Scholar]
- 154.Ravoo BJ, Weringa WD, Engberts JBFN. Langmuir. 1996;12:5773–5780. [Google Scholar]
- 155.Ravoo BJ, Weringa WD, Engberts JBFN. Biophys J. 1999;76:374–386. doi: 10.1016/S0006-3495(99)77204-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Tundo P, Kippenberger DJ, Klahn PL, Prieto NE, Jao TC, Fendler JH. J Am Chem Soc. 1982;104:456–461. [Google Scholar]
- 157.Kippenberger D, Rosenquist K, Odberg L, Tundo P, Fendler JH. J Am Chem Soc. 1983;105:1129–1135. [Google Scholar]
- 158.Reed W, Guterman L, Tundo P, Fendler JH. J Am Chem Soc. 1984;106:1897–1907. [Google Scholar]
- 159.Serrano J, Mucino S, Millan S, Reynoso R, Fucugauchi LA, Reed W, Nome F, Tundo P, Fendler JH. Macromolecules. 1985;18:1999–2005. [Google Scholar]
- 160.Higashi N, Adachi T, Niwa M. Macromolecules. 1988;21:2297–2299. [Google Scholar]
- 161.Jung M, den Ouden I, Montoya-Gõni A, Hubert DHW, Frederik PM, van Herk AM, German AL. Langmuir. 2000;16:4185–4195. [Google Scholar]
- 162.Nezu S, Lando JB. J Polym Sci, Polym Chem. 1995;33:2455–2461. [Google Scholar]
- 163.Srisiri W, Sisson TM, O’Brien DF. J Am Chem Soc. 1996;118:11327–11328. [Google Scholar]
- 164.Sisson TM, Srisiri W, O’Brien DF. J Am Chem Soc. 1998;120:2322–2329. [Google Scholar]
- 165.Liu S, Sisson TM, O’Brien DF. Macromolecules. 2001;34:465–473. [Google Scholar]
- 166.Möhwald H. Annu Rev Phys Chem. 1990;41:441–476. doi: 10.1146/annurev.pc.41.100190.002301. [DOI] [PubMed] [Google Scholar]
- 167.McConnell H. Annu Rev Phys Chem. 1991;42:171–195. [Google Scholar]
- 168.Klausner RD, Kleinfeld AM. Lipid Domains in Membranes. In: Klausner RD, Kleinfeld AM, editors. Marcel Dekker; New York: 1984. pp. 23–58. [Google Scholar]
- 169.Silvius JR. Biochim Biophys Acta. 1986;857:217–228. doi: 10.1016/0005-2736(86)90350-0. [DOI] [PubMed] [Google Scholar]
- 170.Armitage BA, Bennett DE, Lamparski HG, O’Brien DF. Adv Polym Sci. 1996;126:53–84. [Google Scholar]
- 171.Lopez E, O’Brien DF, Whitesides TH. Biochim Biophys Acta. 1982;693:437–443. doi: 10.1016/0005-2736(82)90451-5. [DOI] [PubMed] [Google Scholar]
- 172.Büschl R, Hupfer B, Ringsdorf H. Makromol Chem, Rapid Commun. 1982;3:589–596. [Google Scholar]
- 173.Hupfer B, Ringsdorf H, Schupp H. Makromol Chem. 1981;182:247–253. [Google Scholar]
- 174.Sackmann E, Eggl P, Fahn C, Bader H, Ringsdorf H, Schollmeier M. Ber Bunsen-Ges Phys Chem. 1985;89:1198–1208. [Google Scholar]
- 175.Elbert R, Folda T, Ringsdorf H. J Am Chem Soc. 1984;106:7687–7692. [Google Scholar]
- 176.Büschl R, Folda T, Ringsdorf H. Makromol Chem Suppl. 1984;6:245–258. [Google Scholar]
- 177.Seki N, Tsuchida E, Ukaji K, Sekiya T, Nozawa Y. Polym Bull. 1985;13:489–492. [Google Scholar]
- 178.Tsuchida E, Seki N, Ohno H. Makromol Chem. 1986;187:1351–1358. [Google Scholar]
- 179.Ohno H, Takeoka S, Tsuchida E. Polym Bull. 1985;14:487–490. [Google Scholar]
- 180.Takeoka S, Sakai H, Ohno H, Tsuchida E. Macromolecules. 1991;24:1279–1283. [Google Scholar]
- 181.Tyminski PN, Latimer LH, O’Brien DF. J Am Chem Soc. 1985;107:7769–7770. [Google Scholar]
- 182.Tyminski PN, Latimer LH, O’Brien DF. Biochemistry. 1988;27:2696–2705. doi: 10.1021/bi00408a009. [DOI] [PubMed] [Google Scholar]
- 183.Frankel DA, Lamparski H, Liman U, O’Brien DF. J Am Chem Soc. 1989;111:9262–9263. [Google Scholar]
- 184.Lamparski H, Liman U, Frankel DA, Barry JA, Ramaswami V, Brown MF, O’Brien DF. Biochemistry. 1992;31:685–694. doi: 10.1021/bi00118a008. [DOI] [PubMed] [Google Scholar]
- 185.Ellens H, Bentz J, Szoka FC. Biochemistry. 1984;23:1532–1538. doi: 10.1021/bi00302a029. [DOI] [PubMed] [Google Scholar]
- 186.Ellens H, Bentz J, Szoka FC. Biochemistry. 1986;25:285–294. doi: 10.1021/bi00350a001. [DOI] [PubMed] [Google Scholar]
- 187.Rand RP, Parsegian VA. Biochim Biophys Acta. 1989;988:351–376. [Google Scholar]
- 188.Siegel DP. Biophys J. 1993;65:2124–2140. doi: 10.1016/S0006-3495(93)81256-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Barry JA, Lamparski H, Shyamsunder E, Osterberg F, Cerne J, Brown MF, O’Brien DF. Biochemistry. 1992;31:10114–10120. doi: 10.1021/bi00156a035. [DOI] [PubMed] [Google Scholar]
- 190.Bennett DE, O’Brien DF. Biochemistry. 1995;34:3102–3113. doi: 10.1021/bi00009a042. [DOI] [PubMed] [Google Scholar]
- 191.Bentz J, Ellens H. Colloids Surf. 1988;30:65–112. [Google Scholar]
- 192.Gaub H, Sackmann E, Buschl R, Ringsdorf H. Biophys J. 1984;45:725–731. doi: 10.1016/S0006-3495(84)84215-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Armitage BA, Klekotka PA, Oblinger E, O’Brien DF. J Am Chem Soc. 1993;115:7920–7921. [Google Scholar]
- 194.Srisiri W, Lee YS, Sisson TM, Bondurant B, O’Brien DF. Tetrahedron. 1997;53:15397–15414. [Google Scholar]
- 195.Sjolund M, Lindblom G, Rilfors L, Arvison G. Biophys J. 1987;52:145–153. doi: 10.1016/S0006-3495(87)83202-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Lee YS, Yang JZ, Sisson TM, Frankel DA, Gleeson JT, Aksay E, Keller SL, Gruner SM, O’Brien DF. J Am Chem Soc. 1995;117:5573–5578. [Google Scholar]
- 197.Schoen AH. NASA Tech Note. 1970;D-5541:1–98. [Google Scholar]
- 198.Tate MW, Eikenberry EF, Turner DC, Shyamsunder E, Gruner SM. Chem Phys Lipids. 1991;57:147–164. doi: 10.1016/0009-3084(91)90073-k. [DOI] [PubMed] [Google Scholar]
- 199.Lindblom G, Larsson K, Johansson L, Fontell K, Forsén S. J Am Chem Soc. 1979;101:5465–5470. [Google Scholar]
- 200.Briggs J, Caffrey M. Biophys J. 1994;66:573–587. doi: 10.1016/s0006-3495(94)80847-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Briggs J, Caffrey M. Biophys J. 1994;67:1594–1602. doi: 10.1016/S0006-3495(94)80632-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Srisiri W, Lamparski H, O’Brien DF. J Org Chem. 1996;61:5911–5915. [Google Scholar]
- 203.Srisiri W, Benedicto A, O’Brien DF, Trouard TP, Orädd G, Persson S, Lindblom G. Langmuir. 1998;14:1921–1926. [Google Scholar]
- 204.Eriksson PO, Lindblom G. Biophys J. 1993;64:129–136. doi: 10.1016/S0006-3495(93)81347-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Smith RC, Fischer WM, Gin DL. J Am Chem Soc. 1997;119:4092–4093. [Google Scholar]
- 206.Gray DH, Hu S, Juang E, Gin DL. Adv Mater. 1997;9:731–736. [Google Scholar]
- 207.Srisiri W, Sisson TM, O’Brien DF, McGrath KM, Han Y, Gruner SM. J Am Chem Soc. 1997;119:4866–4873. [Google Scholar]
- 208.Gin DL, Gray DH, Smith RC. Synlett. 1999;10:1509–1522. [Google Scholar]
- 209.Deng H, Gin DL, Smith RC. J Am Chem Soc. 1998;120:3522–3523. [Google Scholar]
- 210.Jenekhe SA, Osaheni JA. Chem Mater. 1994;6:1906. [Google Scholar]
- 211.Gray DH, Gin DL. Chem Mater. 1998;10:1827–1832. [Google Scholar]
- 212.Miller SA, Kim E, Gray DH, Gin DL. Angew Chem, Int Ed. 1999;38:3022–3026. doi: 10.1002/(sici)1521-3773(19991018)38:20<3021::aid-anie3021>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 213.Hoag BP, Gin DL. Macromolecules. 2000;33:8549–8558. [Google Scholar]
- 214.Lewis RNAH, Mannock DA, McElhaney RN, Turner DC, Gruner SM. Biochemistry. 1989;28:541–548. doi: 10.1021/bi00428a020. [DOI] [PubMed] [Google Scholar]
- 215.Tate MW, Gruner SM. Biochemistry. 1989;28:4245–4253. doi: 10.1021/bi00436a019. [DOI] [PubMed] [Google Scholar]
- 216.Koch H, Ringsdorf H. Makromol Chem Rapid Commun. 1981;182:255–259. [Google Scholar]
- 217.Bader H, Ringsdorf H, Skura J. Angew Chem, Int Ed Engl. 1981;20:91–92. [Google Scholar]
- 218.Charych DH, Nagy JO, Spevak W, Bednarski MD. Science. 1993;261:585–588. doi: 10.1126/science.8342021. [DOI] [PubMed] [Google Scholar]
- 219.Spevak W, Nagy JO, Charych DH, Schaefer ME, Gilbert JH, Bednarski MD. J Am Chem Soc. 1993;115:1146–1147. [Google Scholar]
- 220.Reichert A, Nagy JO, Spevak W, Charych D. J Am Chem Soc. 1995;117:829–830. [Google Scholar]
- 221.Spevak W, Nagy JO, Charych DH. Adv Mater. 1995;7:85–89. [Google Scholar]
- 222.Charych DH, Cheng Q, Reichert A, Kuziemko G, Stroh M, Nagy JO, Spevak W, Stevens RC. Chem Biol. 1996;3:113–120. doi: 10.1016/s1074-5521(96)90287-2. [DOI] [PubMed] [Google Scholar]
- 223.Okada SY, Jelinek R, Charych D. Angew Chem, Int Ed Engl. 1999;38:655–659. doi: 10.1002/(SICI)1521-3773(19990301)38:5<655::AID-ANIE655>3.0.CO;2-4. [DOI] [PubMed] [Google Scholar]
- 224.Cheng Q, Stevens RC. Adv Mater. 1997;9:481–483. [Google Scholar]
- 225.Kolusheva S, Shahal T, Jelinek R. J Am Chem Soc. 2000;122:776–780. [Google Scholar]
- 226.Tilmann RW, Radmacher M, Gaub HE, Kenney P, Ribi HO. J Phys Chem. 1993;97:2928–2932. [Google Scholar]
- 227.Juliano RL, Hsu MJ, Peterson D, Regen SL, Singh A. Exp Cell Res. 1983;146:422–427. doi: 10.1016/0014-4827(83)90144-1. [DOI] [PubMed] [Google Scholar]
- 228.Juliano RL, Hsu MJ, Regen SL. Biochim Biophys Acta. 1985;812:42–48. doi: 10.1016/0005-2736(85)90519-x. [DOI] [PubMed] [Google Scholar]
- 229.Regen S. Ann NY Acad Sci. 1985;446:296–307. doi: 10.1111/j.1749-6632.1985.tb18409.x. [DOI] [PubMed] [Google Scholar]
- 230.Miller CR, Clapp PJ, O’Brien DF. FEBS Lett. 2000;467:52–56. doi: 10.1016/s0014-5793(00)01122-4. [DOI] [PubMed] [Google Scholar]
- 231.Ramakrishnan R, Ahmad N, LaBell R, O’Brien DF. Manuscript in preparation. [Google Scholar]
- 232.Bondurant B, O’Brien DF. J Am Chem Soc. 1998;120:13541–13542. [Google Scholar]
- 233.Mueller A, Bondurant B, O’Brien DF. Macromolecules. 2000;33:4799–4804. [Google Scholar]
- 234.Papahadjopoulos D, Allen TM, Gabison A, Mayhew E, Matthay K, Huang SK, Lee KD, Woodle MC, Lasic DD, Redemann C, Martin FJ. Proc Natl Acad Sci USA. 1991;88:11460–11464. doi: 10.1073/pnas.88.24.11460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Gabizon AA. Cancer Res. 1992;52:891–896. [PubMed] [Google Scholar]
- 236.Huang SK, Lee KD, Hong K, Friend DS, Papahadjopoulos D. Cancer Res. 1992;52:5135–5143. [PubMed] [Google Scholar]
- 237.Wu NZ, Da D, Rudoll TL, Needham D, Whorton AR, Dewhirst MW. Cancer Res. 1993;53:3765–3770. [PubMed] [Google Scholar]
- 238.Cho I, Kim YD. Macromol Symp. 1997;118:631–640. [Google Scholar]
- 239.Fendler JH, Meldrum FC. Adv Mater. 1995;7:607–632. [Google Scholar]
- 240.Ferrar WT, O’Brien DF, Wahrshawsky A, Voycheck CL. J Am Chem Soc. 1988;110:288. [Google Scholar]
- 241.Mann S, Williams RJP. J Chem Soc, Dalton Trans. 1983:311–316. [Google Scholar]
- 242.Tricot YM, Emeren A, Fendler JH. J Phys Chem. 1985;89:4721–4726. [Google Scholar]
- 243.Tricot YM, Fendler JH. J Phys Chem. 1986;90:3369–3374. [Google Scholar]
- 244.Markowitz MA, Chow GM, Singh A. Langmuir. 1994;10:4095–4102. [Google Scholar]
- 245.Markowitz MA, Schnur JM, Singh A. Chem Phys Lipids. 1992;62:193–204. [Google Scholar]