

Abstract
Centrosome- and cilia-associated proteins play crucial roles in establishing polarity and regulating intracellular transport in post-mitotic cells. Using genetic mapping and positional candidate strategy, we have identified an in-frame deletion in a novel centrosomal protein CEP290 (also called NPHP6), leading to early-onset retinal degeneration in a newly identified mouse mutant, rd16. We demonstrate that CEP290 localizes primarily to centrosomes of dividing cells and to the connecting cilium of retinal photoreceptors. We show that, in the retina, CEP290 associates with several microtubule-based transport proteins including RPGR, which is mutated in ~15% of patients with retinitis pigmentosa. A truncated CEP290 protein (ΔCEP290) is detected in the rd16 retina, but in considerably reduced amounts; however, the mutant protein exhibits stronger association with specific RPGR isoform(s). Immunogold labeling studies demonstrate the redistribution of RPGR and of phototransduction proteins in the photoreceptors of rd16 retina. Our findings suggest a critical function for CEP290 in ciliary transport and provide insights into the mechanism of early-onset photoreceptor degeneration.
INTRODUCTION
The centrosome is a highly specialized organelle consisting of a pair of centrioles surrounded by amorphous proteinaceous matrix, the pericentriolar material (PCM) (1). It serves as the primary microtubule organizing center of the cell by orchestrating nucleation, anchoring, and release of micro-tubules during cell division and chromosome segregation (2). In post-mitotic cells, such as photoreceptors, one of the centrioles (basal body) migrates to the base of the cellmembrane, recruits intraflagellar transport proteins and microtubule motors and nucleates the assembly of primary cilia (3). Owing to the involvement of primary cilia in diverse cellular processes, mutations in centrosomal/ciliary proteins result in human disorders with a wide phenotypic spectrum, including randomization of body symmetry, obesity, cystic kidney diseases and retinal degeneration (4).
The photoreceptors are highly polar neurons with a distinct inner and an outer segment; the outer segment is a specialized sensory cilium linked to the inner segment by the connecting cilium. The photoreceptor polarity is established by unique distribution of phototransduction proteins to the outer segments from their site of synthesis in the inner segment (5). Approximately 10% of the outer segments are turned over each day, with new discs being formed proximally and shed distally. The proteins destined for outer segments are believed to dock at the basal bodies to be transported distally via the connecting cilium (6,7). Perturbations in the intersegmental ciliary transport are associated with retinal degeneration (7,8).
Animal models of spontaneous retinal degeneration provide insights into pathological mechanism(s) of disease progression and help in designing therapeutic strategies. Moreover, identification of disease-associated genes in mice has led to the discovery of corresponding genes that cause retinal degeneration in humans. For example, identification of retinal degeneration 1 (rd1), retinal degeneration 7 (rd7) and retinal degeneration slow (rds) mouse models led to the identification of β-phosphodiesterase, photoreceptor-specific nuclear receptor and peripherin-rds mutations, respectively, which are associated with inherited retinopathies (9–14). Additional retinal mutants have now been described by screening genetically diverse but inbred strains of mice at The Jackson Laboratory (15).
Here, we report the identification and characterization of the rd16 mouse, which exhibits early-onset retinal degeneration with autosomal recessive inheritance. We show that the rd16 mouse carries an in-frame deletion in a novel centrosomal protein, CEP290. The CEP290 protein also localizes to the connecting cilium of photoreceptors and associates with several ciliary and centrosomal proteins, including RPGR. In the rd16 retina, we observe altered interaction of RPGR and mutant CEP290 and a redistribution of RPGR and phototransduction proteins. Our findings suggest that CEP290 plays an important role in protein trafficking and shed light on the pathways leading to photoreceptor degeneration, a major cause of inherited blindness in developed countries.
RESULTS
Clinical and histological examination of the rd16 mouse
The phenotype of homozygote rd16 mice can be distinguished from wild-type (WT) animals by the appearance of white retinal vessels at 1 month and large pigment patches at 2 months of age (Fig. 1A). Electroretinograms under dark- and light-adapted conditions indicate a considerable deterioration of rod and cone functions in the rd16 homozygotes compared with the WT as early as postnatal (P) day 18 (Fig. 1B). Light microscopy of the rd16/rd16 retina shows degeneration of outer segments and reduction in the thickness of the outer nuclear layer as early as postnatal day 19 and progresses with age. Little or no change was observed in other cellular layers (Fig. 1C).
Figure 1.
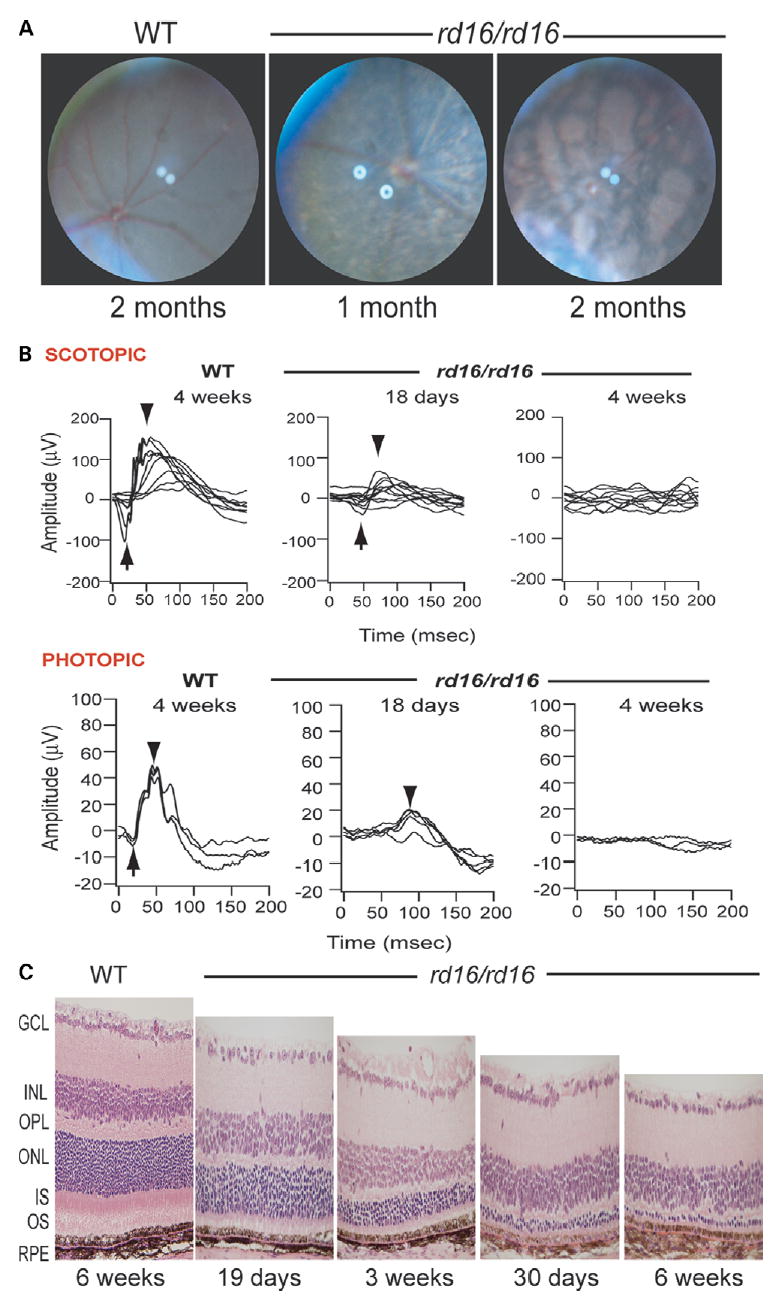
Examination of the rd16 mouse retina. (A) Fundus photographs of WT C57BL/6J mouse and the rd16 homozygote mutants (rd16/rd16) demonstrating retinal degeneration at 1 month of age and at 2 months. (B) ERG responses of WT and mutant (rd16/rd16) mice under dark- (SCOTOPIC) and light- (PHOTOPIC) adapted conditions. Arrows indicate the A-wave and arrowheads the B-wave. (C) Histology of retina ofWT and rd16 homozygotesmice at indicated ages. RPE, retinal pigment epithelium; OS, outer segments; IS, inner segments; ONL, outer nuclear layer; OPL, outer plexiformlayer; INL, inner nuclear layer; GCL, ganglion cell layer.
Cep290 is mutated in the rd16 mouse
By linkage analysis of back-crossed mice, we mapped the causative gene defect in rd16 to chromosome 10 in the genomic region flanked by D10Mit244 (99.4 M) and D10Nds2 (105 M) (Fig. 2A and B). In silico analysis of the critical region revealed over 30 putative expressed sequences, which were then examined for differential expression in mouse photoreceptors using gene expression profiles generated by us (16) or others (17). We found that the expression of one of the hypothetical genes, BC004690, was increased nearly 3-fold during rod maturation (P2–P6). Its expression was dramatically reduced in the Crx−/− mice in which photoreceptors fail to develop (18) and in the rodless, cone-enriched retina of Nrl−/− mice (19) (data not shown). Real-time PCR analysis using primer pair F1–R1 derived from BC004690 (Supplementary Material, Table S1) validated the gene-profiling data (Fig. 2C and D).
Figure 2.
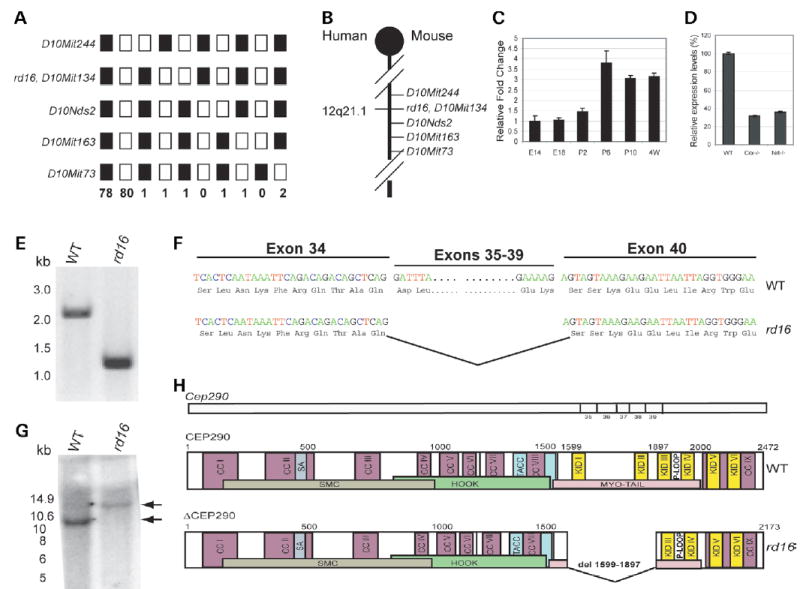
Cep290 mutation in rd16. (A) Linkage cross-data: 165 back-cross progeny from the (rd16 × CAST/EiJ)F1 × rd16/rd16 were phenotyped for ERG phenotype and genotyped for the indicated microsatellite markers. Black boxes represent homozygosity for rd16-derived alleles and white boxes represent heterozygosity for rd16- and CAST-derived alleles. The number of animals sharing the corresponding haplotype is indicated below each column of squares. The order of marker loci was determined by minimizing the number of crossovers. The rd16 locus was inferred from the ERG phenotype of mice showing recombinations. (B) Genetic map of mouse chromosome 10 showing the rd16 critical region, which is syntenic to human chromosome 12q21.1. (C) Real-time RT-PCR analysis of BC004690 (Cep290, exons 27–48) in the retina of WT mice. The expression levels at different developmental stages were calculated as relative fold change with respect to embryonic day, E14, after normalization to Hprt levels. P, postnatal day. Each bar represents the mean ± SE (n = 6). (D) Real-time RT-PCR analysis of BC004690 in the retina of Crx−/− and Nrl−/− versus WT mice. The expression levels in the Crx−/− and Nrl−/− retina were calculated as percentage of the level in the WT mouse retina after normalization to Hprt levels. Each bar represents the mean ± SE (n = 6). (E) RT-PCR analysis (with F2–R2 primer set) of BC004690 using rd16 and WT retinal RNA. A 1.2 kb band is detected in rd16 compared with a 2.1 kb product in WT. DNA size markers are shown on the left (in kb). (F) BC004690 sequence in rd16 showing an in-frame deletion of 897 bp encompassing exons 35–39. Three-letter codes for amino acids were used. (G) Southern analysis of WT and rd16 DNA using an exon 34 probe. DNA was digested with EcoRV, which cuts the WT DNA five times between exons 34 and 40, whereas in the rd16 DNA, only three EcoRV sites remain. WT DNA gave the expected band of 10.6 kb, whereas with the rd16 DNA, a heavier band at ~15 kb (indicated by arrows) is seen. Molecular weight markers are in kilobases. (H) Schematic representation of the Cep290 gene and the CEP290 and ΔCEP290 proteins showing putative domains and motifs. CC, coiled-coil; KID, RepA/Re+ protein KID; P-loop, ATP-GTP-binding site motif A; spindle association (SA) domain; MYO-Tail, myosin tail homology domain.
Further in silico analysis revealed that BC004690 is part of the mouse Cep290 gene (exons 27–48), which encodes a protein similar to human centrosomal protein CEP290 (20). Given that mutations in certain centrosomal proteins may result in retinal degeneration owing to ciliary dysfunction in photoreceptors (4), we screened the Cep290 gene for possible mutations in the rd16 mouse. Initial RT-PCR analysis using the F1–R1 primer pair did not amplify any product (data not shown); yet another primer set (F2–R2; Supplementary Material, Table S1) encompassing the complete BC004690 sequence detected a 1.2 kb product with the rd16 retinal cDNA compared with the expected 2.1 kb product in WT cDNA (Fig. 2E). Sequence analysis of the RT-PCR products identified an in-frame deletion of 897 bp (5073–5969 bp in cDNA), which corresponded to CEP290 amino acid residues 1599–1897 (Fig. 2F showing the junction sequence). The truncated CEP290 protein was designated ΔCEP290. No other sequence alteration was detected. Southern analysis of the WT and rd16 homozygote genomic DNAs confirmed a deletion (from exon 35 to 39) within the Cep290 gene (Fig. 2G).
Domain composition of CEP290
The Cep290 gene, spanning over 85 kb and 52 exons, encodes a putative protein of 2472 amino acids (apparent molecular weight 290 kDa). To investigate the domain structure of CEP290, we scanned the MotifScan and SMART protein databases (www.expasy.org) and identified at least nine coiled-coil domains and a C-terminal myosin-tail homology domain, which provides a structural backbone to the myosin motor (Fig. 2H). Moreover, CEP290 exhibits significant similarity to SMC (structural maintenance of chromosomes) chromosomal segregation ATPases (21), six RepA/Rep+ protein motifs KID, glycine-rich ATP/GTP-binding site motif (P-loop) involved in the binding of motor proteins to the nucleotides and the transforming acidic coiled-coil (TACC) domain involved in microtubule organization by centrosomal proteins. A majority of the myosin-tail homology region is deleted in rd16 mouse (see Fig. 2H and blue region in Supplementary Material, Fig. S1). CLUSTALW analysis shows strong evolutionary conservation of the CEP290 protein, with orthologs in Danio rerio and Anopheles gambiae (Supplementary Material, Fig. S1).
Expression and localization of CEP290 in mouse retina
A monoclonal antibody, 3G4 (22), against CEP290 recognized a band at ~290 kDa in protein extracts from different tissues of WT mice as well as in COS1 cells transfected with a full-length myc-tagged CEP290 construct (data not shown). We also generated a polyclonal antibody against two peptides corresponding to the mouse CEP290 protein; this antibody also recognized the CEP290 protein in transfected COS-1 cells (Fig. 3A). Immunoblot analysis revealed a fainter band of faster mobility (ΔCEP290) in retinal extracts from the rd16 mouse compared with the 290 kDa band in WT (Fig. 3B). Additional bands of low molecular mass are also observed in bovine retina extracts (our unpublished data). On the basis of this and in silico analysis, we suggest that these bands represent alternately spliced isoforms of CEP290.
Figure 3.
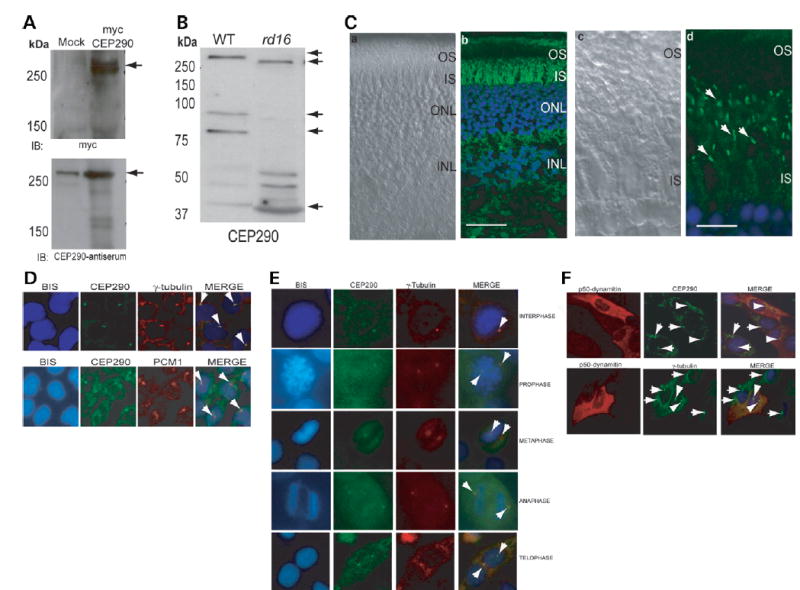
Expression and localization of CEP290. (A) COS-1 cells were transfected with empty vector (mock) or a vector encoding full-length human CEP290 protein fused to a myc-tag. Cells were lysed and analyzed by immunoblotting (IB), using anti-myc (upper panel) or anti-CEP290 antiserum (lower panel). Arrows indicate specific bands. The immunoreactive band in the mock transfected lane (lower panel) is endogenous CEP290 protein. Pre-immune serum shows no signal (data not shown). (B) Immunoblots of protein extracts from WT (20 μg) and rd16 (200μg) retina were analyzed using CEP290 antibody. Arrows indicate the full length and predicted alternatively spliced products of CEP290. (C) Immunohistochemical analysis of WT mouse retina. The sections were incubated with the CEP290 antibody followed by secondary antibody incubation. (a) and (c) Nomarski image of the retinal sections. (b) and (d) Staining with the CEP290 antibody (green) reveals intense labeling of the connecting cilium (indicated by arrows). Labeling in the IS is also observed. Scale bar: 40 μm for (a), (b); 10 μm for (c), (d). (D) CEP290 (green) co-localizes with γ-tubulin (red; upper panel) and PCM1 (red; lower panel) at the centrosomes (arrows; merge) in IMCD-3 cells. Bisbenzimide (BIS) was used to stain the DNA. (E) CEP290 is associated with centrosomes during cell cycle. Synchronized HeLa cells were co-stained with antibodies against γ-tubulin (red) and CEP290 (green) and analyzed by confocal microscopy. Arrows indicate the centrosomal staining of CEP290 (merge) at all indicated stages of cell division. (F) IMCD-3 cells were transfected with p50-dynamitin expression construct. Cells were stained with p50 (red), CEP290 or γ-tubulin (green) antibodies. Arrows denote centrosomal CEP290 and γ-tubulin in untransfected cells, whereas arrowheads denote the localization of CEP290 and γ-tubulin to centrosomes in p50-overexpressing cells. Merge image shows blue nuclear staining.
We then investigated the localization of CEP290 in mouse retina by immunofluorescence and immunogold microscopy. CEP290 is localized primarily to the connecting cilium of mouse photoreceptors, although some labeling is detected in the inner segments (Fig. 3C; Supplementary Material, Fig. S2). Connecting cilium staining of CEP290 was also observed in dissociated rod photoreceptors of mouse retina, as determined by co-localization with acetylated alpha-tubulin (data not shown).
CEP290 localizes to centrosomes in a dynein-independent manner
Immunocytochemical analysis using the CEP290 antibody revealed that CEP290 co-localizes with the centrosomal and pericentriolar matrix markers γ-tubulin and PCM1 (2) at the centrosomes of mouse kidney inner medullary collecting duct (IMCD-3) (Fig. 3D). Co-localization with PCM1 is reminiscent of the staining pattern of BBS4, a ciliary/centrosomal protein involved in microtubule dynamics (23). We also find consistent co-labeling of CEP290 with γ-tubulin through different stages of cell cycle (Fig. 3E).
We next queried how CEP290 is recruited or assembled at the centrosomes. Previous studies have shown that microtubule depolymerization using nocodazole does not alter centrosomal localization of CEP290 (20). Given that a number of centrosomal proteins, including RPGR-ORF15 and PCM1, are anchored at the centrosomes via the functional dynein–dynac-tin molecular motor, whereas others such as γ-tubulin and BBS6 are not (24,25), we examined whether localization of CEP290 depends on dynein–dynactin motor by overexpressing the p50-dynamitin subunit of the dynactin complex (26). Like γ-tubulin, the localization of CEP290 at centrosomes is not altered in cells overexpressing p50-dynamitin (Fig. 3F). Our data suggest that functional microtubule motor or polymerized microtubules are not necessary to maintain CEP290 at the centrosomes; however, we cannot rule out their requirement for the recruitment of newly synthesized CEP290 to the centrosomes.
CEP290 associates with RPGR in mammalian retina
Given that RPGR, a ciliary/centrosomal protein (27–29) mutations in which are detected in retinitis pigmentosa (30–32), interacts with centrosomal disease-associated proteins (29,33–35), we hypothesized that CEP290 may also associate with RPGR and its interacting proteins and participate in common functional pathways. The ORF15CP antibody against the retina-enriched RPGR-ORF15 isoform(s) (28,29,34) was able to precipitate very low amounts of CEP290 from WT mouse retinal extracts (Fig. 4A). Reverse co-immunoprecipitation using the 3G4 antibody detected RPGR-ORF15 upon immunoblotting (Fig. 4B). Notably, yeast two-hybrid experiments do not appear to reveal a direct interaction of CEP290 with RPGR (data not shown).
Figure 4.
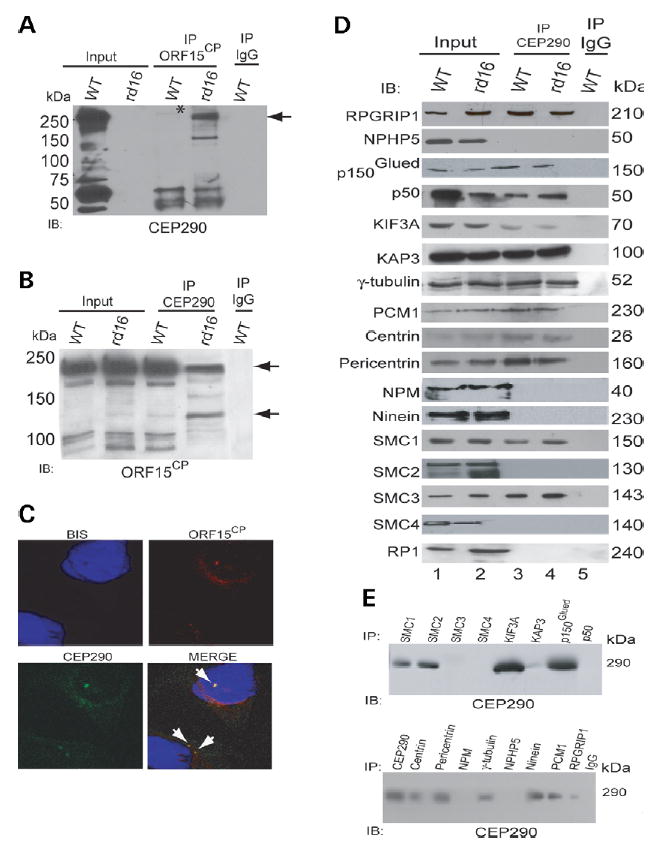
CEP290 and Δ CEP290 associate with RPGR-ORF15 and other centrosomal/microtubule-associated proteins in the retina. (A, B) IP was performed using ORF15CP (A), CEP290 (B) antibodies or normal IgG from WT and rd16 retinal extract (200 μg each). The immunoprecipitated proteins were analyzed by IB using CEP290 (A) or ORF15CP (B) antibodies. Input lane contains 20% of the protein extract used for IP. Longer exposure of the blot in (A) shows an immunoreactive band for ΔCEP290 in rd16 input lane (data not shown). Molecular weight markers are shown in kilo Dalton. Asterisk indicates the faint full-length CEP290-immunoreactive band (290 kDa) immunoprecipitated from the WT retina using the ORF15CP antibody. Arrow in (A) points to the ΔCEP290 protein immunoprecipitated from rd16 retina using ORF15CP. Arrows in (B) indicate multiple RPGR-ORF15 isoforms recognized by the ORF15CP antibody (29). Less high molecular weight (120–220 kDa) RPGR-ORF15 isoforms are immunoprecipitated by the CEP290 antibody in rd16. (C) Immunocytochemistry using the CEP290 (green) and ORF15CP (red) antibodies shows co-localization of endogenous CEP290 and RPGR-ORF15 in IMCD-3 cells. Arrows indicate co-localization (Merge). (D) WT and rd16 retinal extracts were subjected to IP using the CEP290 antibody and analyzed by IB using indicated antibodies. Input lane represents 5% of the total protein extract used for IP. Molecular weight markers are shown in kilo Dalton. Lanes 1 and 2: input from WT and rd16 retinal extracts; 3 and 4: IP using the CEP290 antibody from WT and rd16, respectively; 5: IP with normal IgG from WT retina. (E) Reverse IP was performed by incubating protein extracts of WT retina with indicated antibodies for IP followed by IB using the CEP290 antibody. Molecular weight markers are shown in kilo Dalton.
We next performed co-immunoprecipitation experiments using rd16 retinal extracts. RPGR-ORF15 could recruit over 50 times higher levels of the ΔCEP290 protein from rd16 retina compared with the WT protein (Fig. 4A). Reverse immunoprecipitation pulled down a few, but not all, RPGR-ORF15 isoforms from the rd16 retina (Fig. 4B). Consistent with this, the endogenous CEP290 co-localizes with RPGR-ORF15 in IMCD-3 cells (Fig. 4C) and dissociated mouse rod photoreceptors (data not shown).
CEP290 is part of selected centrosomal and microtubule-associated protein complex(es)
To evaluate whether CEP290 and ΔCEP290 are part of multi-protein complex(es) with other centrosomal and microtubule-associated motor assemblies, some of which may also overlap with RPGR-ORF15-containing complexes, we performed additional co-immunoprecipitation experiments using mouse or bovine retinal extracts. Our data show that CEP290 is present in complex with RPGR-interacting protein 1 (RPGRIP1), dynactin subunits p150Glued and p50-dynamitin, kinesin subunit KIF3A, kinesin-associated protein (KAP3), γ-tubulin, PCM1, centrin, pericentrin and ninein, but not with nucleophosmin (NPM), or Nephrocystin-5 (NPHP5) (Fig. 4D and E). Dynein subunits are not detectable probably because of the low abundance or instability of the dynein–dynactin interaction. As RPGR-ORF15, CEP290 also interacts with SMC1 and 3. Varying degree of association with SMC proteins and p50-dynamitin may be due to relative abundance of the proteins. CEP290 is not associated with RP1, another ciliary protein mutated in retinopathies (36) (Fig. 4D). Similar results are obtained with rd16 as well as bovine retinal extracts (data not shown). No immunoreactive bands are detected when normal IgG is used for IP. Notably, RPGR-ORF15 interacts with NPM (28) and NPHP5 (34) but not with centrin and pericentrin (29). Our results therefore indicate that CEP290 and RPGR may perform multiple overlapping yet distinct microtubule-based transport functions in the retina.
Perturbed localization of RPGR and opsin in the rd16 retina
We next examined whether increased association of ΔCEP290 affects the localization of RPGR-ORF15 in the rd16 retina. ImmunoEM experiments revealed that RPGR-ORF15 aggregates in the inner segments of P12 rd16 retina, indicating a trafficking defect, whereas, as shown previously (29), the axoneme and basal bodies in photoreceptors of normal retinas are strongly labeled with the ORF15CP antibody (Fig. 5A–C). However, we did not observe any obvious structural defects in the connecting cilium of the rd16 retina.
Figure 5.
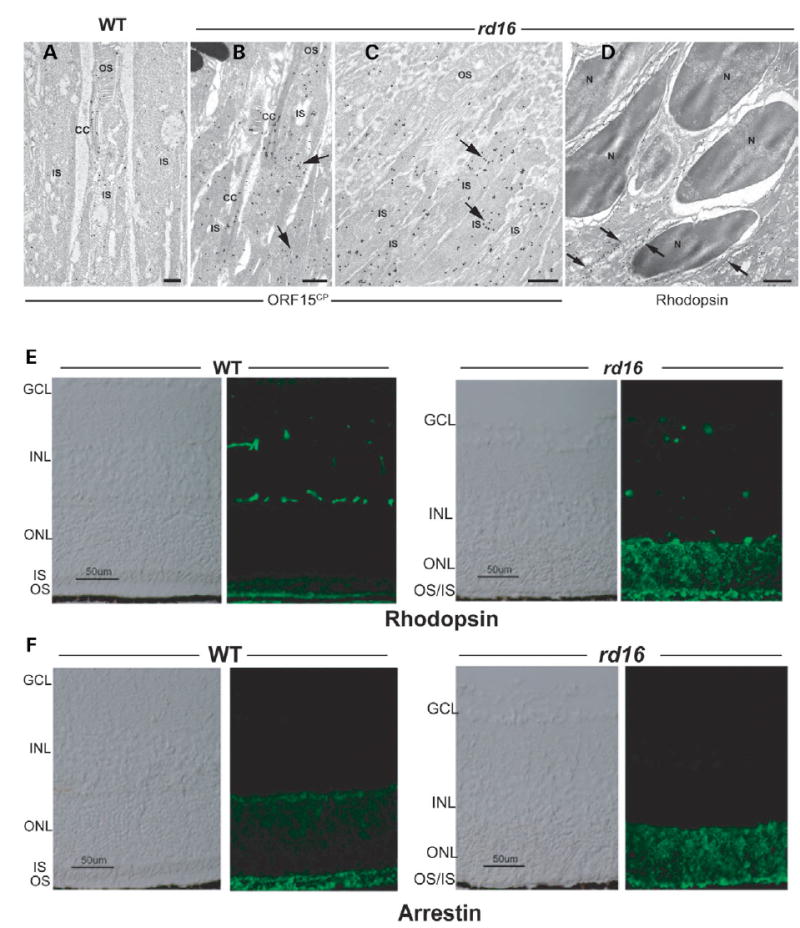
Localization of RPGR-ORF15, rhodopsin and arrestin in rd16 retinas. (A–D) Immunogold EM of WT or rd16 retinas with indicated antibodies. Labeling with ORF15CP antibody showed a predominant connecting cilium (CC) staining of RPGR-ORF15 (A) as opposed to abnormal extensive labeling throughout the photoreceptor IS in the rd16 retina (B, C). Arrows indicate clusters of immunogold particles. Labeling of rhodopsin in the rd16 retina (D) is evident around the photoreceptor cell bodies (indicated by arrows) with no exclusive OS localization; N, nucleus. (E, F) Immunohistochemical analysis of the WT and rd16 retinas at P12, dissected under normal light/dark cycle, with antibodies against rhodopsin (E) or arrestin (F). As shown, both rhodopsin and arrestin are localized primarily in the OS of WT retina, whereas in rd16, rhodopsin and arrestin are also detected in the ONL and ISs of photoreceptors. OS in the rd16 retina degenerate at P12 and therefore are represented in conjunction with the inner segments (OS/IS). Scale bar: 50 μm.
Given the involvement of RPGR-ORF15 in regulating intracellular trafficking in photoreceptors (29,37), we sought to examine the effect of CEP290 mutation and/or RPGR mislocalization on the trafficking of phototransduction proteins in the retina. Immunogold EM and immunofluorescence analyses revealed redistribution of rhodopsin and arrestin throughout the plasma membrane of rd16 photoreceptors when compared with the normal outer segment localization in WT photoreceptors (Fig. 5D–F).
DISCUSSION
Photoreceptor degeneration is associated with a number of syndromic diseases, such as Senior–Loken syndrome and Bardet–Biedl syndrome (BBS) (4,34), as well as with non-syndromic inherited retinal degenerative diseases. Over 100 genetic loci for inherited retinal degenerations have been identified (see RetNet: http://www.sph.uth.tmc.edu/Retnet/). Mouse models of retinal degeneration provide candidate genes for human retinopathies and offer critical insights into disease pathogenesis. Here, we report that early-onset retinal degeneration in the rd16 mouse is associated with an in-frame deletion in a novel centrosomal protein CEP290. Our data suggest that CEP290 regulates intracellular protein trafficking and that perturbation of its function leads to mislocalization of ciliary and phototransduction proteins resulting in photoreceptor degeneration.
The defects observed in the rd16 retina are consistent with a role of CEP290 in ciliary transport processes. Because the association of ΔCEP290 with microtubule motors (dynein and kinesin-II) remains unaltered, we conclude that abnormal transport of RPGR and opsins in rd16 is not a direct effect of impaired or defective association of ΔCEP290 with the motor proteins. An intriguing finding in this study is that the reduced amount of intact ΔCEP290 is able to associate with considerably higher amounts of specific RPGR-ORF15 isoforms. Such higher association may recruit RPGR from the connecting cilium and redistribute it in the inner segments. Consistent with the involvement of RPGR in intracellular protein trafficking, we observe redistribution of phototransduction proteins, which are potential ‘cargo’ proteins (8) being transported via the connecting cilium to the outer segments, in the rd16 retina. However, we do not rule out the possibility of a direct association of CEP290 (or ΔCEP290) with rhodopsin and/or arrrestin. Attempts to directly identify associations of cargo proteins with the ciliary transport assemblies have been unsuccessful so far perhaps because of weak or transient interactions (29,38,39).
Our findings suggest that misrouting of phototransduction proteins is the underlying cause of photoreceptor degeneration in the rd16 retina. Such redistribution of opsins in photoreceptor inner segments owing to impaired ciliary transport has been detected previously in Kif3a conditional mutants, Tg737orpk, Bbs2−/− and Bbs4−/− mice (7,40–42) as well as some other types of retinal degenerations (e.g. rd1, rds/peripherin and opsin mutants, Q334ter and P347S, but not P23H) (43,44). The mislocalization of opsins is possibly due to redistribution of RPGR-ORF15 to the inner segments. The support for this hypothesis comes from a previous observation that the connecting cilium staining of RPGR is impaired in the Rpgrip1−/− mice, which also develop early-onset photoreceptor degeneration owing to abrogated opsin trafficking (45). Notably, the Rpgr−/− mice show considerably slower retinal degeneration with only minor redistribution of rhodopsin (37) compared with the rd16 mouse. This may be due to the presence of a few RPGR-ORF15 isoforms that are still expressed in the Rpgr−/− mice and localize to the connecting cilium (29).
CEP290 associates predominantly with centrosomes throughout the cell cycle, suggesting its role in microtubule nucleation, originating from the centrioles in dividing cells. However, the delineation of its precise role in cell division will require further investigation. Intriguingly, CEP290 shows homology to and associates with SMC proteins, which in addition to regulating chromosomal dynamics during cell division, also localize to primary cilia of photoreceptors and are postulated to be involved in ciliary transport in photoreceptors by interacting with RPGR (29). Several ciliary and centrosomal proteins contain SMC-like domains and are involved in microtubule dynamics during retrograde and anterograde protein trafficking (20,46,47). Hence, the SMC-like domains can be hypothesized as signature sequences that are recognized during these transport processes.
While this study was in progress, mutations in the CEP290 gene (also called NPHP6) were identified in patients with Joubert syndrome, which includes cystic kidney disease (nephronophthisis) and cerebellar defects in addition to early-onset retinal degeneration or coloboma (48). Extensive evaluation of 6-month-old rd16 homozygotes did not detect any gross brain or kidney pathology (Supplementary Material, Table S2), indicating that the ΔCEP290 protein in rd16 mouse retains at least partial function. Notably, only nonsense or frame-shift mutations (probably, loss of function) in the CEP290 gene have been identified in the Joubert syndrome patients. These observations suggest that the myosin-tail homology domain of CEP290 performs a retina-specific function. However, in the absence of the CEP290 protein, a pleiotropic phenotype is observed in Joubert syndrome patients.
In conclusion, our study shows that CEP290 participates in regulating intracellular protein-trafficking probably by organizing distinct multi-protein complexes for microtubule-based transport. Our findings provide a direct link between microtubule organization, intracellular transport and ciliary function in neurons and may facilitate functional dissection of centrosomal disease-associated proteins using retinal photoreceptors as a paradigm.
MATERIALS AND METHODS
Animal studies
The mice were bred and maintained in standardized conditions at The Jackson Laboratory and Kellogg Eye Center. The use of mice was approved by the Institutional Animal Care and Use Committee. The rd16 mouse was discovered in strain BXD-24/Ty at about F140 generation and the mutation was fixed in this strain, but all BXD-24/Ty mice recovered from the embryo freezer at about F84 generation had normal retinas. Detailed methods for retinal examination, histology and electroretinography have been described (49). We mated BXD-24/Ty-rd16 mice with CAST/EiJ mice. The F1 mice, which exhibit no retinal abnormalities, were backcrossed (BC) to BXD-24/Ty-rd16 mice. DNAs from 165 BC offspring were genotyped using microsatellite markers to develop a structure map; detailed methods for mapping and mutation screening have been reported (49).
DNA, RNA and protein analyses
DNA and RNA analysis methods have been described (19). Primer pairs for RT-PCR are given in Supplementary Material, Table S1. The co-IP experiments with retinal extracts were performed as described (29). The rabbit polyclonal CEP290 peptide antibody was generated (Invitrogen) against the mouse sequence 517SKRLKQQQYRAENQ530 and 2457SEHS-EDGESPHSFPIY2472. Rabbit polyclonal antibodies to RPGR, RPGRIP1 and NPHP5 have been described previously (29,34). Antibodies against acetylated α-tubulin, γ-tubulin, p50-dynamitin, SMC1 and SMC3 were purchased from Chemicon (Temeculla, CA). Mouse anti-p150Glued antibody was obtained from BD Transduction Labs (San Jose, CA); anti-KIF3A, anti-KAP3, anti-centrin and anti-pericentrin antibodies were obtained from Sigma and anti-ninein was from BioLegend (San Diego, CA). Anti-RP1 antibody was generously provided by Dr Eric A. Pierce, anti-NPM antibody by Dr Alan F. Wright and anti-PCM1 by Dr A. Merdes.
Cell culture and immunolocalization
Kidney m-IMCD-3 cells (American Type Culture Collection, Manassas, VA; CRL 2123) were grown in six well plates and transfected with p50-dynamitin expression construct (a gift from Dr Trina A. Schroer) using Fugene-6 reagent (Roche). Experimental details about immunocytochemistry and immunogold EM procedures are as described (29). Immunofluorescence microscopy of retinal sections for rhodopsin and arrestin was performed using published procedures (50). For immunolabeling of CEP290, eyes were fixed in methanol, and sections were labeled with 3G4, followed by goat anti-mouse conjugated to AlexaFluor 488.
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG Online.
Supplementary Material
Acknowledgments
We are grateful to Muriel T. Davisson for guidance, Masayuki Akimoto for the Nrl-p-GFP mice, Stephen Lentz for confocal microscopy, Robbie Duerr and Steve Merino for technical assistance, Edwin Oh and other members of the Swaroop Lab for discussions and Sharyn Ferrara for administrative assistance. This research was supported by grants from the National Institutes of Health (EY07961, EY07003, EY13408, EY12598, EY07758, RR01183, DK1069274, DK1068306, DK064614 and DK20572), the Foundation Fighting Blindness, Research to Prevent Blindness and George M. O’Brien Kidney Research Center. Funding to pay the Open Access Publication charges for this article was provided by NIH grant EY07961.
Footnotes
The mouse Cep290 cDNA and gene sequence correspond to GenBank accession no. XM_618806.
Conflict of Interest statement. The authors declare no conflict of interest.
References
- 1.Nigg EA. Centrosomes in Development and Disease. Wiley-VCH Verlag GmbH & Co; KGaA: 2004. [Google Scholar]
- 2.Doxsey S. Re-evaluating centrosome function. Nat Rev Mol Cell Biol. 2001;2:688–698. doi: 10.1038/35089575. [DOI] [PubMed] [Google Scholar]
- 3.Beisson J, Wright M. Basal body/centriole assembly and continuity. Curr Opin Cell Biol. 2003;15:96–104. doi: 10.1016/s0955-0674(02)00017-0. [DOI] [PubMed] [Google Scholar]
- 4.Badano JL, Teslovich TM, Katsanis N. The centrosome in human genetic disease. Nat Rev Genet. 2005;6:194–205. doi: 10.1038/nrg1557. [DOI] [PubMed] [Google Scholar]
- 5.Young RW. Passage of newly formed protein through the connecting cilium of retina rods in the frog. J Ultrastruct Res. 1968;23:462–473. doi: 10.1016/s0022-5320(68)80111-x. [DOI] [PubMed] [Google Scholar]
- 6.Liu X, Udovichenko IP, Brown SD, Steel KP, Williams DS. Myosin VIIa participates in opsin transport through the photoreceptor cilium. J Neurosci. 1999;19:6267–6274. doi: 10.1523/JNEUROSCI.19-15-06267.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Marszalek JR, Liu X, Roberts EA, Chui D, Marth JD, Williams DS, Goldstein LS. Genetic evidence for selective transport of opsin and arrestin by kinesin-II in mammalian photoreceptors. Cell. 2000;102:175–187. doi: 10.1016/s0092-8674(00)00023-4. [DOI] [PubMed] [Google Scholar]
- 8.Besharse JC, Baker SA, Luby-Phelps K, Pazour GJ. Photoreceptor intersegmental transport and retinal degeneration: a conserved pathway common to motile and sensory cilia. Adv Exp Med Biol. 2003;533:157–164. [PubMed] [Google Scholar]
- 9.Bowes C, Li T, Danciger M, Baxter LC, Applebury ML, Farber DB. Retinal degeneration in the rd mouse is caused by a defect in the beta subunit of rod cGMP-phosphodiesterase. Nature. 1990;347:677–680. doi: 10.1038/347677a0. [DOI] [PubMed] [Google Scholar]
- 10.Akhmedov NB, Piriev NI, Chang B, Rapoport AL, Hawes NL, Nishina PM, Nusinowitz S, Heckenlively JR, Roderick TH, Kozak CA, et al. A deletion in a photoreceptor-specific nuclear receptor mRNA causes retinal degeneration in the rd7 mouse. Proc Natl Acad Sci USA. 2000;97:5551–5556. doi: 10.1073/pnas.97.10.5551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Travis GH, Brennan MB, Danielson PE, Kozak CA, Sutcliffe JG. Identification of a photoreceptor-specific mRNA encoded by the gene responsible for retinal degeneration slow (rds) Nature. 1989;338:70–73. doi: 10.1038/338070a0. [DOI] [PubMed] [Google Scholar]
- 12.McLaughlin ME, Sandberg MA, Berson EL, Dryja TP. Recessive mutations in the gene encoding the beta-subunit of rod phosphodiesterase in patients with retinitis pigmentosa. Nat Genet. 1993;4:130–134. doi: 10.1038/ng0693-130. [DOI] [PubMed] [Google Scholar]
- 13.Haider NB, Jacobson SG, Cideciyan AV, Swiderski R, Streb LM, Searby C, Beck G, Hockey R, Hanna DB, Gorman S, et al. Mutation of a nuclear receptor gene, NR2E3, causes enhanced S cone syndrome, a disorder of retinal cell fate. Nat Genet. 2000;24:127–131. doi: 10.1038/72777. [DOI] [PubMed] [Google Scholar]
- 14.Farrar GJ, Kenna P, Jordan SA, Kumar-Singh R, Humphries MM, Sharp EM, Sheils DM, Humphries P. A three-base-pair deletion in the peripherin-RDS gene in one form of retinitis pigmentosa. Nature. 1991;354:478–480. doi: 10.1038/354478a0. [DOI] [PubMed] [Google Scholar]
- 15.Chang B, Hawes NL, Hurd RE, Davisson MT, Nusinowitz S, Heckenlively JR. Retinal degeneration mutants in the mouse. Vision Res. 2002;42:517–525. doi: 10.1016/s0042-6989(01)00146-8. [DOI] [PubMed] [Google Scholar]
- 16.Akimoto M, Cheng H, Zhu D, Brzezinski JA, Khanna R, Filippova E, Oh EC, Jing Y, Linares JL, Brooks M, et al. Targeting of green fluorescent protein to new-born rods by Nrl promoter and temporal expression profiling of flow-sorted photoreceptors. Proc Natl Acad Sci USA. 2006;103:3890–3895. doi: 10.1073/pnas.0508214103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Blackshaw S, Harpavat S, Trimarchi J, Cai L, Huang H, Kuo WP, Weber G, Lee K, Fraioli RE, Cho SH, et al. Genomic analysis of mouse retinal development. PLoS Biol. 2004;2:E247. doi: 10.1371/journal.pbio.0020247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Furukawa T, Morrow EM, Li T, Davis FC, Cepko CL. Retinopathy and attenuated circadian entrainment in Crx-deficient mice. Nat Genet. 1999;23:466–470. doi: 10.1038/70591. [DOI] [PubMed] [Google Scholar]
- 19.Mears AJ, Kondo M, Swain PK, Takada Y, Bush RA, Saunders TL, Sieving PA, Swaroop A. Nrl is required for rod photoreceptor development. Nat Genet. 2001;29:447–452. doi: 10.1038/ng774. [DOI] [PubMed] [Google Scholar]
- 20.Andersen JS, Wilkinson CJ, Mayor T, Mortensen P, Nigg EA, Mann M. Proteomic characterization of the human centrosome by protein correlation profiling. Nature. 2003;426:570–574. doi: 10.1038/nature02166. [DOI] [PubMed] [Google Scholar]
- 21.Nasmyth K, Haering CH. The structure and function of SMC and kleisin complexes. Annu Rev Biochem. 2005;74:595–648. doi: 10.1146/annurev.biochem.74.082803.133219. [DOI] [PubMed] [Google Scholar]
- 22.Guo J, Jin G, Meng L, Ma H, Nie D, Wu J, Yuan L, Shou C. Subcellullar localization of tumor-associated antigen 3H11Ag. Biochem Biophys Res Commun. 2004;324:922–930. doi: 10.1016/j.bbrc.2004.09.133. [DOI] [PubMed] [Google Scholar]
- 23.Kim JC, Badano JL, Sibold S, Esmail MA, Hill J, Hoskins BE, Leitch CC, Venner K, Ansley SJ, Ross AJ, et al. The Bardet–Biedl protein BBS4 targets cargo to the pericentriolar region and is required for microtubule anchoring and cell cycle progression. Nat Genet. 2004;36:462–470. doi: 10.1038/ng1352. [DOI] [PubMed] [Google Scholar]
- 24.Dammermann A, Merdes A. Assembly of centrosomal proteins and microtubule organization depends on PCM-1. J Cell Biol. 2002;159:255–266. doi: 10.1083/jcb.200204023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JC, Ou YY, Badano JL, Esmail MA, Leitch CC, Fiedrich E, Beales PL, Archibald JM, Katsanis N, Rattner JB, et al. MKKS/BBS6, a divergent chaperonin-like protein linked to the obesity disorder Bardet–Biedl syndrome, is a novel centrosomal component required for cytokinesis. J Cell Sci. 2005;118:1007–1020. doi: 10.1242/jcs.01676. [DOI] [PubMed] [Google Scholar]
- 26.Vaughan KT, Vallee RB. Cytoplasmic dynein binds dynactin through a direct interaction between the intermediate chains and p150Glued. J Cell Biol. 1995;131:1507–1516. doi: 10.1083/jcb.131.6.1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hong DH, Pawlyk B, Sokolov M, Strissel KJ, Yang J, Tulloch B, Wright AF, Arshavsky VY, Li T. RPGR isoforms in photoreceptor connecting cilia and the transitional zone of motile cilia. Invest Ophthalmol Vis Sci. 2003;44:2413–2421. doi: 10.1167/iovs.02-1206. [DOI] [PubMed] [Google Scholar]
- 28.Shu X, Fry AM, Tulloch B, Manson FD, Crabb JW, Khanna H, Faragher AJ, Lennon A, He S, Trojan P, et al. RPGR ORF15 isoform co-localizes with RPGRIP1 at centrioles and basal bodies and interacts with nucleophosmin. Hum Mol Genet. 2005;14:1183–1197. doi: 10.1093/hmg/ddi129. [DOI] [PubMed] [Google Scholar]
- 29.Khanna H, Hurd TW, Lillo C, Shu X, Parapuram SK, He S, Akimoto M, Wright AF, Margolis B, Williams DS, et al. RPGR-ORF15, which is mutated in retinitis pigmentosa, associates with SMC1, SMC3, and microtubule transport proteins. J Biol Chem. 2005;280:33580–33587. doi: 10.1074/jbc.M505827200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vervoort R, Lennon A, Bird AC, Tulloch B, Axton R, Miano MG, Meindl A, Meitinger T, Ciccodicola A, Wright AF. Mutational hot spot within a new RPGR exon in X-linked retinitis pigmentosa. Nat Genet. 2000;25:462–466. doi: 10.1038/78182. [DOI] [PubMed] [Google Scholar]
- 31.Breuer DK, Yashar BM, Filippova E, Hiriyanna S, Lyons RH, Mears AJ, Asaye B, Acar C, Vervoort R, Wright AF, et al. A comprehensive mutation analysis of RP2 and RPGR in a North American cohort of families with X-linked retinitis pigmentosa. Am J Hum Genet. 2002;70:1545–1554. doi: 10.1086/340848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sharon D, Sandberg MA, Rabe VW, Stillberger M, Dryja TP, Berson EL. RP2 and RPGR mutations and clinical correlations in patients with X-linked retinitis pigmentosa. Am J Hum Genet. 2003;73:1131–1146. doi: 10.1086/379379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dryja TP, Adams SM, Grimsby JL, McGee TL, Hong DH, Li T, Andreasson S, Berson EL. Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am J Hum Genet. 2001;68:1295–1298. doi: 10.1086/320113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Otto EA, Loeys B, Khanna H, Hellemans J, Sudbrak R, Fan S, Muerb U, O’Toole JF, Helou J, Attanasio M, et al. Nephrocystin-5, a ciliary IQ domain protein, is mutated in Senior–Loken syndrome and interacts with RPGR and calmodulin. Nat Genet. 2005;37:282–288. doi: 10.1038/ng1520. [DOI] [PubMed] [Google Scholar]
- 35.Hong DH, Yue G, Adamian M, Li T. Retinitis pigmentosa GTPase regulator (RPGRr)-interacting protein is stably associated with the photoreceptor ciliary axoneme and anchors RPGR to the connecting cilium. J Biol Chem. 2001;276:12091–12099. doi: 10.1074/jbc.M009351200. [DOI] [PubMed] [Google Scholar]
- 36.Liu Q, Zuo J, Pierce EA. The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J Neurosci. 2004;24:6427–6436. doi: 10.1523/JNEUROSCI.1335-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hong DH, Pawlyk BS, Shang J, Sandberg MA, Berson EL, Li T. A retinitis pigmentosa GTPase regulator (RPGR)-deficient mouse model for X-linked retinitis pigmentosa (RP3) Proc Natl Acad Sci USA. 2000;97:3649–3654. doi: 10.1073/pnas.060037497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cole DG, Diener DR, Himelblau AL, Beech PL, Fuster JC, Rosenbaum JL. Chlamydomonas kinesin-II-dependent intraflagellar transport (IFT): IFT particles contain proteins required for ciliary assembly in Caenorhabditis elegans sensory neurons. J Cell Biol. 1998;141:993–1008. doi: 10.1083/jcb.141.4.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Piperno G, Siuda E, Henderson S, Segil M, Vaananen H, Sassaroli M. Distinct mutants of retrograde intraflagellar transport (IFT) share similar morphological and molecular defects. J Cell Biol. 1998;143:1591–1601. doi: 10.1083/jcb.143.6.1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Pazour GJ, Baker SA, Deane JA, Cole DG, Dickert BL, Rosenbaum JL, Witman GB, Besharse JC. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol. 2002;157:103–113. doi: 10.1083/jcb.200107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nishimura DY, Fath M, Mullins RF, Searby C, Andrews M, Davis R, Andorf JL, Mykytyn K, Swiderski RE, Yang B, et al. Bbs2-null mice have neurosensory deficits, a defect in social dominance, and retinopathy associated with mislocalization of rhodopsin. Proc Natl Acad Sci USA. 2004;101:16588–16593. doi: 10.1073/pnas.0405496101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mykytyn K, Mullins RF, Andrews M, Chiang AP, Swiderski RE, Yang B, Braun T, Casavant T, Stone EM, Sheffield VC. Bardet–Biedl syndrome type 4 (BBS4)-null mice implicate Bbs4 in flagella formation but not global cilia assembly. Proc Natl Acad Sci USA. 2004;101:8664–8669. doi: 10.1073/pnas.0402354101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nir I, Papermaster DS. Immunocytochemical localization of opsin in degenerating photoreceptors of RCS rats and rd and rds mice. Prog Clin Biol Res. 1989;314:251–264. [PubMed] [Google Scholar]
- 44.Li T, Snyder WK, Olsson JE, Dryja TP. Transgenic mice carrying the dominant rhodopsin mutation P347S: evidence for defective vectorial transport of rhodopsin to the outer segments. Proc Natl Acad Sci USA. 1996;93:14176–14181. doi: 10.1073/pnas.93.24.14176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhao Y, Hong DH, Pawlyk B, Yue G, Adamian M, Grynberg M, Godzik A, Li T. The retinitis pigmentosa GTPase regulator (RPGR)-interacting protein: subserving RPGR function and participating in disk morphogenesis. Proc Natl Acad Sci USA. 2003;100:3965–3970. doi: 10.1073/pnas.0637349100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, Subramaniam S, Zuker CS. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004;117:527–539. doi: 10.1016/s0092-8674(04)00412-x. [DOI] [PubMed] [Google Scholar]
- 47.Li JB, Gerdes JM, Haycraft CJ, Fan Y, Teslovich TM, May-Simera H, Li H, Blacque OE, Li L, Leitch CC, et al. Comparative genomics identifies a flagellar and basal body proteome that includes the BBS5 human disease gene. Cell. 2004;117:541–552. doi: 10.1016/s0092-8674(04)00450-7. [DOI] [PubMed] [Google Scholar]
- 48.Sayer JA, Otto EA, O’Toole JF, Nurnberg G, Kennedy MA, Becker C, Hennies HC, Helou J, Attanasio M, Fausett B, et al. A novel centrosomal protein nephrocystin-6 is mutated in Joubert syndrome and activates transcription factor ATF4/CREB2. Nat Genet. 2006 doi: 10.1038/ng1786. in press. [DOI] [PubMed] [Google Scholar]
- 49.Pang JJ, Chang B, Hawes NL, Hurd RE, Davisson MT, Li J, Noorwez SM, Malhotra R, McDowell JH, Kaushal S, et al. Retinal degeneration 12 (rd12): a new, spontaneously arising mouse model for human Leber congenital amaurosis (LCA) Mol Vis. 2005;11:152–162. [PubMed] [Google Scholar]
- 50.Cheng H, Khanna H, Oh EC, Hicks D, Mitton KP, Swaroop A. Photoreceptor-specific nuclear receptor NR2E3 functions as a transcriptional activator in rod photoreceptors. Hum Mol Genet. 2004;13:1563–1575. doi: 10.1093/hmg/ddh173. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.