

Abstract
The activin receptor-like kinase 1 (ALK1) is a type I receptor for transforming growth factor-β (TGF-β) family proteins. Expression of ALK1 in blood vessels and mutations of the ALK1 gene in human type II hereditary hemorrhagic telangiectasia patients suggest that ALK1 may have an important role during vascular development. To define the function of ALK1 during development, we inactivated the ALK1 gene in mice by gene targeting. The ALK1 homozygous embryos die at midgestation, exhibiting severe vascular abnormalities characterized by excessive fusion of capillary plexes into cavernous vessels and hyperdilation of large vessels. These vascular defects are associated with enhanced expression of angiogenic factors and proteases and are characterized by deficient differentiation and recruitment of vascular smooth muscle cells. The blood vessel defects in ALK1-deficient mice are reminiscent of mice lacking TGF-β1, TGF-β type II receptor (TβR-II), or endoglin, suggesting that ALK1 may mediate TGF-β1 signal in endothelial cells. Consistent with this hypothesis, we demonstrate that ALK1 in endothelial cells binds to TGF-β1 and TβR-II. Furthermore, the ALK1 signaling pathway can inhibit TGF-β1-dependent transcriptional activation mediated by the known TGF-β1 type I receptor, ALK5. Taken together, our results suggest that the balance between the ALK1 and ALK5 signaling pathways in endothelial cells plays a crucial role in determining vascular endothelial properties during angiogenesis.
Transforming growth factor-β (TGF-β) is a multifunction protein and is involved in diverse biological processes, including growth, differentiation, and inflammation (1). It is generally thought that TGF-β1 plays an important role in vascular remodeling (2, 3). TGF-β1 can inhibit the activities of other angiogenic factors in endothelial cell (EC) proliferation and migration under most cell-culture conditions and can stimulate the production of extracellular matrix proteins and proteinase inhibitors (3). However, TGF-β1 displays a biphasic effect on angiogenesis. Low concentrations of TGF-β1 synergistically enhance, whereas high concentrations of TGF-β1 decrease, the vascular invasion of cultured ECs induced by angiogenic factors such as vascular endothelial growth factor (VEGF) or basic fibroblast growth factor (4, 5). Although TGF-β1 appears to stimulate neovascularization in vivo, such stimulatory activity may be caused by an indirect effect through recruiting inflammatory cells (6).
TGF-β family cytokines exert their effects by binding to heteromeric complexes of type I and type II transmembrane serine/threonine kinase receptors (7). On binding to the ligands, type II receptors recruit and phosphorylate the type I receptors, which in turn activate the downstream signaling mediators Smads (7). To date, seven type I receptors have been identified and designated as activin receptor-like kinase (ALK) 1 to 7 (8–11, 39). The ligand specificity of these ALKs has been determined primarily by their ability to bind to a given ligand and to activate specific downstream genes in the presence of corresponding type II receptors. ALK1 is able to bind to TGF-β1 or activins in the presence of either TβR-II or activin type II receptors, respectively. However, ALK1 does not elicit a specific transcriptional response (7, 12). Thus, ALK1 has been considered an “orphan” receptor.
Several observations have prompted the suggestion that ALK1 may mediate signals important for the formation or remodeling of blood vessels. ALK1 is expressed in blood vessels during embryogenesis (13) and adult stages (14). In addition, mutations of the ALK1 gene have been linked to the type II hereditary hemorrhagic telangiectasia (15), also known as Osler—Rendu–Weber syndrome, an autosomal dominant disorder characterized by multisystemic vascular dysplasia and recurrent hemorrhage (16).
In this paper, we present genetic and biochemical evidence indicating that a TGF-β1 signal may be mediated by two distinct type I receptors, ALK1 and ALK5, in ECs, and that the balance between these two TGF-β1 signaling pathways plays an important role in determining the properties of the endothelium during angiogenesis.
Materials and Methods
Construction of the Targeting Vector and Generation of Mice Carrying Targeted Mutations.
A 12-kb XbaI fragment corresponding to the ALK1 locus was isolated from 129/Sv genomic library (Stratagene). A PGK-neo-poly(A) cassette (the XhoI-SalI fragment from pKJ-1) was inserted into the XhoI site in exon 7-encoding kinase subdomain V in the opposite transcriptional orientation as the ALK1 gene (Fig. 1A; ref. 10). The genotype of G418r clones was analyzed by Southern blot hybridization by using the BamHI-XbaI genomic fragment as a probe (Fig. 1 A and B). Of 188 G418-resistant embryonic stem cell clones analyzed, 13 contained the corrected targeting event. Two independent clones were injected into C57BL/6J blastocysts to obtain chimeric mice.
Figure 1.

Targeted disruption of the mouse ALK1 gene results in embryonic lethality. (A) Schematic diagram (from top to bottom) of the wild-type allele, the knockout (KO) vector, and the recombinant mutant allele. Exons are indicated by rectangles and roman numerals. The PGK-neo cassette was inserted into the XhoI site of exon VII encoding kinase subdomain V (10). An EcoRI site was introduced into the target ALK1 locus, thus the 9.8-kb and 7.2-kb EcoRI fragments indicated by lines represent wild-type and mutant alleles, respectively. PCR primers (arrowheads) and the 5′ external probe (B-X) for Southern hybridization are indicated. B, BamHI; E, EcoRI; S, SalI; X, XbaI; Xh, XhoI. (B) Southern blot analysis of tail DNA of a litter of newborns from the ALK1+/− intercross. (C-F) Gross morphology of yolk sac (C, D) and embryo proper (E, F) of wild-type (Left) and mutant embryos (Right) at E9.5 (C, E) and E10.5 (D, F). Arrows with asterisks (C) indicate the yolk sac blood vessels. Arrows (F) indicate clumps of blood cells in the mutant embryos. pc, pericardium. (G, H) Histological analysis of E9.5 wild type (G) and mutant (H) embryos in decidua. In the mutant yolk sac, blood, and ECs are present, but the vessels are enlarged (H). bc, blood cells; d, decidua; en, endothelium.
Histology and 5-Bromo-4-chloro-3-indolyl β-d-galactoside (X-Gal) Staining.
Histological analysis was carried out by using standard methods. At embryonic day (E)9.5, embryos in decidua were fixed with Bouin's fixative (Polysciences) for 2–4 hr, sectioned at 7 μm thickness, and stained with hematoxylin and eosin. The X-Gal staining was performed as described (17). The stained embryos were postfixed in 4% paraformaldehyde and embedded for plastic section JB-4 plus solution (Polysciences). The embryo sections were cut at 3 μm and counterstained with Nuclear Fast red for 30 sec.
RNA Extraction and Quantitative Reverse Transcription—PCR (RT-PCR).
Total RNA was isolated from E9.5 embryos as described (18), followed by DNase I treatment. First strand cDNA was synthesized by using Superscript kit (GIBCO/BRL). A murine β-actin primer set was used to normalize the amount of total cDNA in each genotype by ABI PRISM 7700 sequence detection system (Applied Biosystems) and SYBR green PCR kit (Perkin–Elmer). Various concentrations of wild-type cDNA (10×, 1×, 0.1×, and 0.01×) were used for the calibration curve for each primer set. The primer sequences are shown in supplemental Table 1 (published on the PNAS web site, www.pnas.org). After 40 cycles of PCR reaction, the relative amount of gene transcripts was calibrated by the comparison with wild-type curve (supplemental Fig. 6, published on the PNAS web site). In addition, the PCR products were separated on the agarose gel and visualized with ethidium bromide (Fig. 3A). In most cases, the ethidium bromide staining pattern reflected the quantitative difference measured by the real-time PCR amplification curve.
Figure 3.
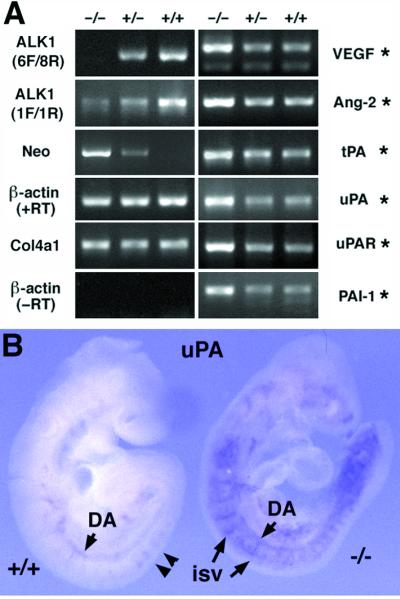
Elevated expression of angiogenic factors and plasminogen activators in the ALK1−/− embryos. (A) Visualized amplified PCR products by the ethidium bromide staining. The same amount of cDNA and methods were used for PCR amplification of each gene. After 40 cycles of amplification and quantitative analyses, PCR products were separated on an agarose gel. All the ethidium bromide staining patterns reflecting amplification plots as represented in supplemental Fig. 6 (www.pnas.org) are shown. ALK1 primers (6F/8R) spanning the neor insertion site did not amplify a DNA band from −/− embryos. ALK1 primers (1F/1R) upstream of the neor insertion site amplified a DNA band from −/− embryos in a greatly diminished level, indicating that a reduced level of transcripts is made in the mutant allele. Conversely, neo gene primers amplified cDNA from −/− and +/− embryos but not from the wild-type embryos. Note that the intensity of the amplified signal for ALK1 and Neo was gene-dosage dependent. The transcript level of the α1 type IV collagen (Col4a1) gene was unaltered, regardless of the genotypes of embryos, whereas, transcript levels of VEGF, Ang-2, tissue-type PA, uPA, uPA-receptor, and PAI-1 were significantly elevated in the ALK1−/− embryos (indicated by asterisks). (B) Whole-mount in situ hybridization of the uPA gene in the E9.5 wild-type (Left) and ALK1−/− (Right) embryos. uPA was detected in the branching points (arrow heads) of the dorsal aorta and ISV of wild-type embryo, whereas overexpressed uPA expression was detected in the various developing blood vessels in the mutant embryos.
Affinity Crosslinking and Immunoprecipitation.
Human umbilical-vein ECs (HUVEC) were affinity labeled with 125I-TGF-β1 followed by immunoprecipitation by using the specific antisera to ALK1 or ALK5. COS7 cells were transfected with hemagluttinin-tagged constitutively active forms of type I receptors, (ALK5/T204D or ALK1/Q201D) (19) and Flag-tagged Smad1, Smad2, or Smad5 by FuGENE6 (Boehringer Mannheim). Smads were immunoprecipitated by Flag antibody (Aves Labs, Tigard, OR), and phosphorylated Smads were detected by immunoblotting by using the antiphosphoserine antibody (Zymed) (20).
Transfection and Luciferase Assays.
For the transient expression assays, the cells were seeded at a density of 2 × 105 cells/well in six-well dishes. They were then cotransfected by using lipofectin (Life Technologies, Grand Island, NY) with a promoter construct, p3TP-Lux, which contains multiple copies of the TGF-β response elements (21) and either ALK5 alone or ALK5 with ALK1 (or ALK2) expression constructs. After 5 hr, complete medium was added. The cells were incubated for 24 hr and then treated with 5 ng/ml of TGF-β1 and incubated for an additional 24 hr. Luciferase activity was measured by using an assay kit (Analytical Luminescence Laboratory, San Diego) and a Dynatech ML3000 luminometer. Results from the luciferase assay were normalized on the basis of β-galactosidase expression from pSV-β-galactosidase in all luciferase reporter experiments. All experiments were repeated at least three times, and similar results were obtained each time.
Results
Targeted Disruption of the ALK1 Gene Results in Embryonic Lethality.
To elucidate the function of ALK1 during mouse development, we generated ALK1-deficient mice by targeted disruption of the ALK1 gene in embryonic stem cells (Fig. 1A) (10). Mice heterozygous for the ALK1 mutation (ALK1+/−) were normal and fertile, suggesting that the ALK1 mutation did not exhibit dominant effects. Of 352 offspring from ALK1+/− crosses, 226 (64%) were heterozygous, and 126 (36%) were wild-type mice. However, no viable ALK1 homozygous (ALK1−/−) mice were recovered at the weaning age (Fig. 1B). ALK1−/− embryos were morphologically indistinguishable from their wild-type littermates at E8.5 but were readily identifiable at E9.5 by their smaller head size, retarded posterior development, and absence of mature blood vessels in the yolk sac (Fig. 1 C and E). ALK1−/− embryos became severely distorted at E10.5, as shown by their severe growth retardation, avascular yolk sac, clustered blood cells, and enlarged pericardium (Fig. 1 D and F) and were resorbed by E11.5. Histological analysis of E9.5 ALK1−/− embryos revealed that the yolk sac vasculature was highly dilated (Fig. 1 G and H).
Angiogenesis Defects in ALK1 Mutant Embryos.
To visualize ECs in the ALK1 mutant embryos, we introduced an EC-specific lacZ transgene into the ALK1−/− mutant mice by crossing ALK1+/− mice with the Flk1 knockout mice in which a lacZ transgene was inserted into the Flk1 gene locus (17). We found that the compound heterozygous embryos [ALK1+/−, Flk1+/−] were viable and normal, and [ALK1−/−, Flk1+/−] embryos were phenotypically indistinguishable from the ALK1−/− embryos, indicating no cooperative effect between the Flk1 and the ALK1 mutations. At E8.5, the primary capillary plexus in the yolk sac and embryo proper was indistinguishable between normal and ALK1−/− embryos (Fig. 2 A, B, D, and E). Histological analyses confirmed that the formation of the primary capillary plexus or vasculogenesis was normal in ALK1−/− embryos (data not shown).
Figure 2.

Dilation of major vessels and fusion of capillaries in ALK1−/− embryos. Developing blood vessels of [ALK1+/+, Flk1+/−] (A–C, G, I) and [ALK1−/−, Flk1+/−] (D–F, H, J) embryos are visualized by X-Gal staining (blue). (A, B, D, and E) Lateral (A) and posterior (B) views of a normal E8.5 embryo and lateral (D) and posterior (E) views of an ALK1−/− littermate. Note the vascular patterns, in particular the primary capillary plexus (pcp) in head and the yolk sac indicated by arrows, are essentially indistinguishable between normal and mutant embryos. (C and F) Gross vascular morphology of the head region in E9.5 embryos. Note the absence of a capillary network in the mutant embryo indicated by an arrow with an asterisk (F). (G–J) Histological sections of E9.5 [ALK1+/+, Flk1+/−] (G, I) and [ALK1−/−, Flk1+/−] (H, J) embryos after whole-mount X-Gal staining followed by plastic embedding. (G) Transverse sections of a normal embryo at otic vesicle (ov) region show Flk+ cells in the endothelium of the dorsal aorta (da), branchial arch arteries (baa), and peripheral head capillaries (phc). (H) Transverse sections of a mutant embryo in corresponding to G. Note that dorsal aorta and branchial arch arteries were greatly dilated and fused with surrounding capillaries, forming large cavernous vessels. The endothelial linings of the mutant blood vessels were intact, suggesting that the clumps of blood cells seen in the mutant embryos (Fig. 1F) result not from hemorrhages but probably from impaired circulation of blood in malformed vessels. (I) A normal embryo section showing neural tube (nt), somites (s), and intersomitic vessels (isv). (J) Sections of mutant embryos corresponding to I. Note the dilation and fusion of intersomitic vessels in the mutant embryos. a, allantois; bc, blood cells; fbv, forebrain vesicle; hbv, hind brain vesicle.
The presence of major blood vessels as well as a capillary network was apparent in E9.5 normal embryos (Fig. 2C). In contrast, very few defined capillary vessels were visible in ALK1−/− embryos (Fig. 2F). Serial sections of embryos after whole-mount X-Gal staining revealed that the Flk-expressing (Flk+) cells were confined to the endothelium lining the blood vessels in both normal and mutant embryos (Fig. 2 G and H). However, the lumen of major blood vessels including dorsal aorta and branchial arch arteries was highly dilated in ALK1−/− embryos (Fig. 2H). Furthermore, very few capillary vessels were found in the ALK1−/− embryos (Fig. 2H), probably because of excessive fusion of capillaries including the intersomitic vessels in the mutant embryos (Fig. 2 I and J). The endocardium and myocardium were present in the mutant embryos but appeared to be immature as compared with the wild-type heart (data not shown).
ALK1 Deficiency Leads to Up-Regulation of Angiogenic Factors and Plasminogen Activators.
To investigate the molecular basis underlying the vascular defects in ALK1 mutant embryos, we analyzed the expression of a set of genes known to be involved in the blood vessel formation by a quantitative RT-PCR method (supplemental Fig. 6; www.pnas.org). The plasminogen–plasmin system has been implicated in proteolysis of perivascular matrix during angiogenesis (22, 23). We found that the transcript levels of tissue-type plasminogen activator (PA), urokinase-type PA (uPA), uPA receptor, and PA inhibitor-1 (PAI-1) were significantly elevated in the ALK1−/− embryos (Fig. 3A). Consistent with the RT-PCR result, analysis by whole-mount in situ hybridization further showed that uPA expression was dramatically increased throughout the entire vasculature in mutant embryos (Fig. 3B), in contrast to its expression in the branching points of intersomitic vessels and dorsal aorta in normal embryos (Fig. 3B). Although PAs are not essential for angiogenesis (24), high expression of uPA has been shown to be a hallmark of actively migrating ECs in vitro (25) and in vivo (26).
Analysis of expression of angiogenic factors and their receptors revealed that levels of VEGF and Ang-2 transcripts were significantly elevated in the mutant embryos (Fig. 3A). It has been shown that VEGF increases the expression of uPA and uPA receptor in ECs, thus increasing the vascular permeability during angiogenesis (27). In addition, Ang-2, a natural antagonist of Ang-1, shows synergistic effects on vascular induction when it is applied with VEGF (28). Therefore, up-regulation of angiogenic factors and PAs in the developing blood vessels may increase perivascular proteolysis, leading to excessive growth of ECs and fusion of capillary vessels in the ALK1 mutant embryos.
ALK1 Is Required for the Differentiation and Recruitment of Vascular Smooth Muscle Cells.
To determine whether differentiation or recruitment of VSMC is affected in mutant embryos, we examined the expression of a smooth muscle cell marker SM22α by crossing ALK1+/− mice with transgenic mice containing a lacZ transgene (tg-SM) driven by the SM22α promoter (29). At E9.5, the tg-SM is expressed in the heart, dorsal aorta, and the myotome of the somites, recapitulating the endogenous SM22α gene expression pattern (Fig. 4A) (29). In E9.5 ALK1−/− embryos, the tg-SM expression was detected in the heart tube and weakly in the somites, but was undetected in the dorsal aorta (Fig. 4B). In E10.5 ALK1−/− embryos, the tg-SM expression was weakly detectable in the dorsal aorta region of the ALK1−/− embryos (Fig. 4C). However, histological analysis of the stained embryos revealed that the lacZ-positive cells in the mutants did not localize to the perivascular regions surrounding the endothelium (Fig. 4E) as in the wild type (Fig. 4D).
Figure 4.
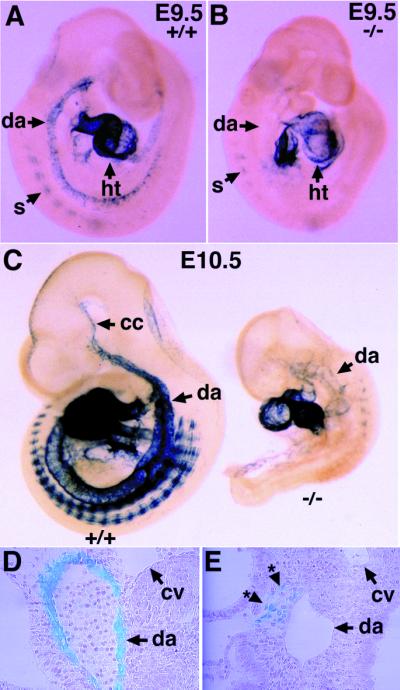
Delayed differentiation and improper localization of vascular smooth muscle cells in the ALK1−/− embryos. (A and B) Expression of tg-SM in E9.5 wild-type (A) and ALK1−/− mutant (B) embryos. Note the absence of tg-SM expression in the dorsal aorta (arrow with asterisk) of the mutant embryos (B). (C) Expression of the tg-SM in E10.5 wild-type (Left) and ALK1−/− mutant (Right) embryos. The mutant embryo is severely growth retarded and expresses tg-SM in the anterior part of the dorsal aorta and somites. (D and E) Plastic sections of embryos after X-Gal staining shown in C. Note the dorsal aorta in the wild-type embryo is surrounded by lacZ-positive vascular smooth muscle cells (D), whereas the lacZ-positive cells (*) in the mutant embryo are localized near the dorsal aorta but fail to encircle the vessel (E). cc, common carotid; cv, cardinal vein; da, dorsal aorta; ht, heart; s, somite.
These results indicate that the ALK1 signaling is required for differentiation and proper localization of VSMC to the perivascular region. Because perivascular cells exert an antagonistic effect on proliferation of ECs on contact (30), the delayed differentiation and improper localization of VSMC may partly contribute to the dilation of the dorsal aorta and branchial arch arteries in the ALK1 mutants. However, we could not determine whether the excessive fusion of capillaries results from abnormal pericyte differentiation, because SM22α is not expressed in pericytes surrounding the capillary vessels.
ALK1 Mediates TGF-β1 Signals Through Smad1/Smad5.
It has been shown that ALK1 is able to bind to TGF-β1 when coexpressed with TβR-II in transfected cells (12). The dilation of the yolk sac blood vessels in the ALK1 homozygotes is reminiscent of that in TGF-β1 (31), TβR-II (32), or endoglin (33) knockout embryos. This phenotypic similarity strongly suggests that TGF-β1 is the major ligand for ALK1 for vascular development. To test whether endogenous ALK1 in ECs can bind to TGF-β1, we performed a binding assay with 125I-labeled TGF- β1 in HUVEC that express both ALK1 and ALK5 (also termed TβR-I), the known TGF-β1 type I receptor (8). In the HUVEC, antisera specific to ALK1 and ALK5 immunoprecipitated ALK1- and ALK5-TβR-II crosslinked complexes, respectively (Fig. 5A), indicating that both type I receptors bind TGF-β1 in conjunction with TβR-II in ECs.
Figure 5.

ALK1 binds to TGF-β1 in ECs, activates Smad1 and Smad5, and can inhibit the TGF-β1-responsive 3TP-Lux induction. (A) TGF-β1 binds to ALK1 as well as ALK5 (TβR-I) in HUVEC. HUVEC were affinity crosslinked with 125I-TGF-β1 and immunoprecipitated by using antibodies specific to either ALK1 or ALK5. Blocking of ALK1 antiserum was done in the presence of the peptide used for immunization of the rabbit (15). The ALK1 band migrated slightly slower than ALK5, in agreement with the difference in the sizes of ALK1 and ALK5. (B) ALK1 phosphorylates Smad1 and Smad5 but not Smad2. COS cells were transfected with Flag-tagged (F) Smad1, Smad5, or Smad2, and constitutively active (c.a.) forms of ALK1 (A1) or ALK5 (A5). (Top). Phosphorylation of Smads was detected by immunoprecipitation of each Smad with a Flag antibody followed by immunoblotting by using an antiphosphoserine antiserum. (Center and Bottom). Expression of Smads and ALKs was determined by anti-Flag and antihemagglutinin antibodies, respectively. (C) ALK1 inhibits TGF-β1-dependent 3TP-Lux induction mediated by ALK5 in HepG2 cells. A TGF-β responsive element linked to the luciferase gene (3TP-Lux) was transfected into HepG2 cells together with increasing amounts of ALK1 or ALK2 in the presence of ALK5. After transfection, cells were treated with (solid bars) or without (open bars) TGF-β1 (5 ng/ml) for 24 hr. Luciferase activities were then measured and plotted. In all assays, luciferase activities are plotted in arbitrary units. (D) The balance model for TGF-β1 signaling in regulation of angiogenesis. Explanation is in the text.
Smad family proteins are directly phosphorylated by activated ALKs and mediate signaling of TGF-β superfamily members. Recent studies have shown that Smad1/Smad5 transduce signals for bone morphogenetic proteins, whereas Smad2/Smad3 transduce signals for TGF-β/activin (7). To investigate which Smads are activated by ALK1, we transfected cDNAs encoding constitutively active forms of ALK1 or ALK5 (ALK1/Q201D or ALK5/T204D, respectively) with various Smads into COS cells and analyzed the phosphorylation of Smads. We found that ALK5/T204D could phosphorylate Smad2 but Smad1 only very weakly (Fig. 5B). In contrast, ALK1/Q201D could phosphorylate Smad1 and Smad5 but not Smad2 (Fig. 5B) and Smad3 (data not shown), suggesting that ALK1 transduces signals through Smad1 and/or Smad5.
These results are consistent with recent findings that ALK1 and ALK2 can be classified into a specific subgroup of type I receptors that interact with the TβR-II or activin type II receptors, respectively, but phosphorylate Smad1 instead of Smad2 (34, 35). Furthermore, remarkably similar vascular phenotypes between ALK1 and Smad5 knockout embryos support the notion that ALK1 transduces signals through Smad5 in vivo (36). Taken together, our data suggest that TGF-β1 may signal through different type I receptors and downstream Smads in ECs, i.e., TGF-β1 > TβR-II > ALK1 > Smad1/5, and TGF-β1 > TβR-II > ALK5 > Smad2/3.
ALK1 Inhibits TGF-β1-Induced PAI-1 Expression.
Because ALK1 activation on TGF-β1 treatment could not activate the PAI-1 promoter (8, 12), and inactivation of ALK1 resulted in elevated expression of PAI-1 (Fig. 3A), we speculated that the ALK1 pathway may inhibit the activation of PAI-1 mediated by the TGF-β1 > ALK5 signaling pathway (9). To test this hypothesis, we analyzed TGF-β1-induced PAI-1 promoter activity in the human hepatoblastoma cell line, HepG2, which expresses endogenous ALK5 but not ALK1 (8) and responds to TGF-β1 treatment. We transiently transfected the reporter construct, p3TP-Lux, into HepG2 cells. Treatment of transfected HepG2 cells with TGF-β1 induced luciferase activity 70-fold. However, this TGF-β1-dependent induction of 3TP-Lux activity was reduced to 35-fold (50% reduction) in HepG2 cells cotransfected with ALK1, indicating that ALK1 inhibits the TGF-β1 response (data not shown).
To determine whether this inhibitory effect of ALK1 on the TGF-β1 response is because of inhibition of the ALK5 pathway, we cotransfected p3TP-Lux together with either ALK5 alone or both ALK5 and ALK1. ALK5 alone could further induce TGF-β1-dependent p3TP-Lux activity by about 300-fold. However, this ALK5-mediated TGF-β1 response was reduced in a dose-dependent manner when ALK1 was cotransfected (Fig. 5C). In a control experiment, cotransfection of ALK2, which was shown to phosphorylate Smad1 when it interacts with activin type II receptors with bone morphogenetic protein 7 (34), did not affect the TGF-β1-dependent 3TP-Lux induction (Fig. 5C). These results suggest that the ALK1 signaling specifically and negatively regulates TGF-β1 signalings by ALK5. In addition, we found that a kinase-deficient form of ALK1 could not inhibit the TGF-β1-dependent 3TP-Lux induction (data not shown), implying that phosphorylation of Smad proteins by ALK1 is important for the inhibitory effect of ALK1 on TGF-β1 signaling.
Discussion
ECs are in either the activation phase or the resolution phase during angiogenesis (3). In the activation phase, ECs degrade perivascular basement membrane, invade and migrate into the extracellular space, proliferate, and form capillary lumen. In the resolution phase, ECs cease migration and proliferation and reconstitute basement membrane. The in vivo mechanism by which these two phases are regulated has not been clearly defined. ALK1−/− embryos exhibited hyperfusion and hyperdilation of blood vessels. Molecular studies in ALK1−/− embryos indicate that the ALK1 signaling represses angiogenic factors and plasminogen activators, which are marker genes for the activation phase of angiogenesis. Furthermore, we show that ALK1 is also required for the differentiation and recruitment of vascular smooth muscle cells. These results suggest that the vascular abnormalities of ALK1−/− embryos may result from the persistence of the activation phase of angiogenesis, and that ALK1 may regulate the transition of ECs from the activation phase to the resolution phase of angiogenesis.
In addition, we demonstrate that ALK1 binds to TGF-β1 and phosphorylates Smad1 and Smad5. Overexpression of ALK1 in HepG2 cells inhibits the ALK5-mediated TGF-β1 response. On the basis of these findings, we propose a balance model for the role of TGF-β1 in angiogenesis (Fig. 5D): the TGF-β1 signal is mediated by two signaling pathways, i.e., ALK1 and ALK5, in ECs. The TGF-β1 signal mediated by ALK1 is essential for the transition of ECs from the activation phase to the resolution phase by repressing expression of angiogenic factors and proteases, whereas TGF-β1 signaling via ALK5, together with other angiogenic factors, induces secretion of proteinases and promotes the activation phase of angiogenesis. Therefore, the balance between ALK1 and ALK5 may be crucial for controlling the properties of endothelium during angiogenesis.
The growth-inhibiting activity of TGF-β1 can be abolished in ECs expressing a dominant negative form of TβR-II but maintained in ECs expressing a dominant negative form of ALK5 (36). This result indicates that signaling pathways other than ALK5 mediate TGF-β signals for growth inhibition in ECs. Therefore, this is consistent with our hypothesis that ALK1 mediates a TGF-β1 signal, and that ALK1 signaling is required for the resolution phase of angiogenesis, during which ECs cease migration and proliferation. This balance model may offer an explanation for the biphasic effects of TGF-β1 on ECs (4, 5). If ALK5 has a higher sensitivity to TGF-β1 than ALK1, then low concentrations of TGF-β1 may activate only the ALK5 pathway favoring the activation phase. On the other hand, high concentrations of TGF-β1 would activate the ALK1 pathway, which in turn inhibits the ALK5 pathway, leading to the resolution phase of angiogenesis.
It is interesting to compare the phenotypes of two groups of knockout mice, the ALK1 and Smad5 knockout mice (ALK1 group; ref. 37) and the TGF-β1, TβR-II, and endoglin (ENG) knockout mice (ENG group; 31–33). Vascular phenotypes appear to be identical within each group but different, although overlapping, between these two groups. All these mutant mice exhibit normal EC differentiation and vasculogenesis at E8.5, distended blood yolk sac blood vessels at E9.5, and lethality at around E10.5-E11.5 with severe growth retardation. Both groups show similar defects in the differentiation and recruitment of VSMCs (Fig. 4; ref. 33). A major difference between the two groups is the vascular morphogenesis of the embryo proper at E9.5. The ENG group embryos have relatively normal branching capillary network and lumen size at E9.5 (31–33), whereas the capillaries of the ALK1 group embryos fail to form a branching capillary network, and the blood vessels are greatly dilated at E9.5 (Fig. 2 F and H; ref. 37). The differences between these two groups can probably be attributed to the fact that mutations in the ENG group disrupt both ALK1 and ALK5 pathways, whereas mutations of the ALK1 group disrupt only the ALK1 pathway, leading to dominance of the ALK5 signaling pathway.
Hereditary hemorrhagic telangiectasia (HHT) is a genetically heterogeneous disorder, because it has been linked to two major genetic loci. HHT-1 is mapped to ENG on chromosome 9 (38), whereas HHT-2 has been linked to ALK1 on chromosome 12 (15). It is paradoxical that mutation of one allele of either ALK1 or ENG results in an identical phenotype in humans (i.e., HHT), whereas inactivation of both alleles of these genes produces overlapping yet distinct phenotypes in knockout mice. Our model provides an intriguing explanation of this paradox. Because ENG probably binds and enriches TGF-β1 near the cell surface, a reduced dosage of ENG in HHT-1 patients may decrease the functional TGF-β1 concentration. A significant decrease in TGF-β1 concentration may block signaling through ALK1, while leaving the higher sensitivity ALK5 pathway unaffected. Thus, mutation of one ENG allele would produce effects similar to mutations of one ALK1 allele. The dominance of the ALK5 pathway for TGF-β1 signaling would then lead to increased migratory properties of the capillary ECs, resulting in arteriovenous connections between dilated venules and arteriols with no intervening capillary bed (24).
Another possible explanation for the difference is that ALK1-KO could be a “gain-of-function” mutation rather than a “null” mutation. Because the insertion of the neor cassette disrupts the kinase function of the ALK1 gene but leaves the ligand-binding domain and transmembrane domain intact, the presumptive truncated protein may interfere with the normal function of other subfamily member receptors. However, since the quantitative RT-PCR analysis using primers from upstream of the neor insertion site detects only minimal expression of aberrant ALK1 transcripts from ALK1−/− embryos (Fig. 3A), it seems unlikely that this exceedingly low level of truncated protein could cause such dramatic effects. Currently we are generating the second mutant allele in which whole transmembrane domain of the ALK1 gene is deleted, to clarify further this “null”-mutation issue.
Supplementary Material
Acknowledgments
We thank Drs. J. M. Gimble and X. Wu for mouse ALK1 cDNA (Oklahoma Medical Research Foundation, Oklahoma City, OK), Hong Lei for technical assistance, and Drs. C.–H. Heldin, B. Olsen, M. Pepper, M. Kawabata, J. Resnick, and B. Ticho for comments on the manuscript. This work was supported by a grant from Bristol–Myers/Squibb (E. L.), National Institutes of Health (NIH) HD32112 (P.K.D.), Netherlands Heart Foundation (P.td.), and Howard Hughes Research Resources Program and American Heart Association (S.P.O.). S.P.O. was the recipient of National Research Service Award (NIH).
Abbreviations
- TGF-β
transforming growth factor-β
- EC
endothelial cell
- VEGF
vascular endothelial growth factor
- ALK
activin receptor-like kinase
- HHT
hereditary hemorrhagic telangiectasia
- RT-PCR
reverse transcription—PCR
- X-Gal
5-bromo-4-chloro-3-indolyl β-d-galactoside
- E
embryonic day
- PA
plasminogen activator
- uPA
urokinase-type PA
- PAI-1
PA inhibitor-1
- HUVEC
human umbilical-vein EC
- ENG
endoglin
References
- 1.Hoodless P A, Wrana J L. Curr Top Microbial Immunol. 1998;228:235–272. doi: 10.1007/978-3-642-80481-6_10. [DOI] [PubMed] [Google Scholar]
- 2.Risau W. Nature (London) 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 3.Pepper M S. Cytokines Growth Factor Rev. 1997;8:21–43. doi: 10.1016/s1359-6101(96)00048-2. [DOI] [PubMed] [Google Scholar]
- 4.Gajdusek C M, Luo Z, Mayberg M R. J Cell Physiol. 1993;157:133–144. doi: 10.1002/jcp.1041570118. [DOI] [PubMed] [Google Scholar]
- 5.Pepper M S, Vassalli J-D, Orci L, Montesano R. Exp Cell Res. 1993;204:356–363. doi: 10.1006/excr.1993.1043. [DOI] [PubMed] [Google Scholar]
- 6.Roberts A B, Sporn M B. Am Rev Respir Dis. 1989;140:1126–1128. doi: 10.1164/ajrccm/140.4.1126. [DOI] [PubMed] [Google Scholar]
- 7.Kretzschmar M, Massagué J. Curr Opin Genet Dev. 1998;8:103–111. doi: 10.1016/s0959-437x(98)80069-5. [DOI] [PubMed] [Google Scholar]
- 8.Attisano L, Cárcamo J, Ventura F, Weis F M B, Massagué J, Wrana J L. Cell. 1993;75:671–680. doi: 10.1016/0092-8674(93)90488-c. [DOI] [PubMed] [Google Scholar]
- 9.Franzen P, ten Dijke P, Ichijo H, Yamashita H, Schulz P, Heldin C-H, Miyazono K. Cell. 1993;75:681–692. doi: 10.1016/0092-8674(93)90489-d. [DOI] [PubMed] [Google Scholar]
- 10.ten Dijke P, Ichijo H, Franzen P, Schulz P, Saras J, Toyoshima H, Heldin C-H, Miyazono K. Oncogene. 1993;8:2879–2887. [PubMed] [Google Scholar]
- 11.Ryden M, Imamura T, Jornval H, Belluardo N, Nevue I, Trupp M, Okadome T, ten Dijke P, Ibanez C F. J Biol Chem. 1996;271:30603–30609. doi: 10.1074/jbc.271.48.30603. [DOI] [PubMed] [Google Scholar]
- 12.Bassing C H, Yingling J M, Howe D W, Wang T, He W W, Gustafson M L, Shah P, Donahoe P K, Wang X-F. Science. 1994;263:87–89. doi: 10.1126/science.8272871. [DOI] [PubMed] [Google Scholar]
- 13.Roelen B A J, Rooijen M A V, Mummery C L. Dev Dyn. 1997;209:418–430. doi: 10.1002/(SICI)1097-0177(199708)209:4<418::AID-AJA9>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 14.Panchenko M P, Williams M C, Brody J S, Yu Q. Am J Physiol. 1996;270:L574–558. doi: 10.1152/ajplung.1996.270.4.L547. [DOI] [PubMed] [Google Scholar]
- 15.Johnson D W, Berg J N, Baldwin M A, Gallione C J, Marondel I, Yoon S J, Stenzel T T, Speer M, Pericak-Vance M A, Diamond A, et al. Nat Genet. 1996;13:189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- 16.Guttmacher A, Marchuk D A, White R I. New Engl J Med. 1995;333:918–924. doi: 10.1056/NEJM199510053331407. [DOI] [PubMed] [Google Scholar]
- 17.Shalaby F, Rossant J, Yamaguchi T P, Gertenstein M, Wu X-F, Bretman M L, Schuh A C. Nature (London) 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 18.Chomczynski P, Sacchi N. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- 19.Wieser R, Wrana J L, Massagué J. EMBO J. 1995;14:2199–2208. doi: 10.1002/j.1460-2075.1995.tb07214.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawabata M, Inoue H, Hanyu A, Imamura T, Miyazono K. EMBO J. 1998;17:4056–4065. doi: 10.1093/emboj/17.14.4056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wrana J L, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, Wang X F, Massagué J. Cell. 1992;71:1003–1014. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- 22.Tkachuk V, Stepanova V, Little P J, Bobik A. Clin Exp Pharmacol Physiol. 1996;23:759–765. doi: 10.1111/j.1440-1681.1996.tb01177.x. [DOI] [PubMed] [Google Scholar]
- 23.Saksela O, Rifkin D B. Annu Rev Cell Biol. 1988;4:93–126. doi: 10.1146/annurev.cb.04.110188.000521. [DOI] [PubMed] [Google Scholar]
- 24.Carmeliet P, Schoonjans L, Kieckens L, Ream B, Degen J, Bronson R, De Vos R, van den Oord J J, Collen D, Mulligan R C. Nature (London) 1994;368:419–424. doi: 10.1038/368419a0. [DOI] [PubMed] [Google Scholar]
- 25.Pepper M S, Vassalli J-D, Montesano R, Orci L. J Cell Biol. 1987;105:2535–2541. doi: 10.1083/jcb.105.6.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bacharach E, Itin A, Keshet E. Proc Natl Acad Sci USA. 1992;89:10686–10690. doi: 10.1073/pnas.89.22.10686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mandriota S J, Seghezz G, Vassalli J-D, Ferrara N, Wasi S, Mazzier R, Mignatti P, Pepper M S. J Biol Chem. 1995;270:9709–9716. doi: 10.1074/jbc.270.17.9709. [DOI] [PubMed] [Google Scholar]
- 28.Asahara T, Chen D, Takahashi T, Fujikawa K, Kearney M, Magner M, Yancopoulos G D, Isner J M. Circ Res. 1998;83:233–240. doi: 10.1161/01.res.83.3.233. [DOI] [PubMed] [Google Scholar]
- 29.Li L, Miano J M, Mercer B, Olson E N. J Cell Biol. 1996;132:849–859. doi: 10.1083/jcb.132.5.849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Orlidge A, D'Amore P A. J Cell Biol. 1987;105:155–1462. doi: 10.1083/jcb.105.3.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dickson M C, Martin J S, Cousins F M, Kulkarni A B, Karlsson S, Akhurst R J. Development (Cambridge, U K) 1995;121:1846–1854. doi: 10.1242/dev.121.6.1845. [DOI] [PubMed] [Google Scholar]
- 32.Oshima M, Oshima H, Taketo M M. Dev Biol. 1996;179:297–302. doi: 10.1006/dbio.1996.0259. [DOI] [PubMed] [Google Scholar]
- 33.Li D Y, Sorensen L K, Brooke B S, Urness L D, Davis E C, Taylor D G, Boak B B, Wendel D P. Science. 1999;284:1534–1537. doi: 10.1126/science.284.5419.1534. [DOI] [PubMed] [Google Scholar]
- 34.Macias-Silva M, Hoodless P A, Tang S J, Buchwald M, Wrana J L. J Biol Chem. 1998;273:25628–25636. doi: 10.1074/jbc.273.40.25628. [DOI] [PubMed] [Google Scholar]
- 35.Chen Y G, Massagué J. J Biol Chem. 1999;274:3672–3677. doi: 10.1074/jbc.274.6.3672. [DOI] [PubMed] [Google Scholar]
- 36.Sankar S, Mahooti-Brooks N, Bensen L, McCarthy T L, Centrella M, Madri J A. J Clin Invest. 1996;97:1436–1446. doi: 10.1172/JCI118565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yang, X., Castilla, L. H., Xu, X., Li, C., Gotay. J., Weinstein, M., Liu, P. P. & Deng, C. X. Development (Cambridge, U. K.)126, 1571–1580. [DOI] [PubMed]
- 38.McAllister K A, Grogg K M, Johnson D W, Gallione C J, Baldwin M A, Jackson C E, Helmbold E A, Markel D S, McKinnon W C, Murrell J. Nat Genet. 1994;8:345–351. doi: 10.1038/ng1294-345. [DOI] [PubMed] [Google Scholar]
- 39.He W W, Gustafson M L, Hirobe S, Donahoe P K. Dev Dyn. 1993;196:133–142. doi: 10.1002/aja.1001960207. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}