

Abstract
‘Suture zones’ are areas where hybrid and contact zones of multiple taxa are clustered. Such zones have been regarded as strong evidence for allopatric divergence by proponents of the Pleistocene forest refugia theory, a vicariance hypothesis frequently used to explain diversification in the Amazon basin. A central prediction of the refugia and other vicariance theories is that the taxa should have a common history so that divergence times should be coincident among taxa. A suture zone for Ithomiinae butterflies near Tarapoto, NE Peru, was therefore studied to examine divergence times of taxa in contact across the zone. We sequenced 1619 bp of the mitochondrial COI/COII region in 172 individuals of 31 species from across the suture zone. Inferred divergence times differed remarkably, with divergence between some pairs of widespread species (each of which may have two or more subspecies interacting in the zone, as in the genus Melinaea) being considerably less than that between hybridizing subspecies in other genera (for instance in Oleria). Our data therefore strongly refute a simple hypothesis of simultaneous vicariance and suggest that ongoing parapatric or other modes of differentiation in continuous forest may be important in driving diversification in Amazonia.
Keywords: Lepidoptera, mitochondrial DNA, phylogeography, speciation, Pleistocene refuge hypothesis, DNA barcodes
1. Introduction
Various hypotheses have been proposed to explain the record richness of Amazonian biodiversity, of which the Pleistocene refuge hypothesis has received particular attention. The refuge hypothesis (Haffer 1969; Simpson & Haffer 1978; Haffer 1997; Haffer & Prance 2001) contends that dry intervals associated with climatic oscillations of the Tertiary and Quaternary reduced forest habitats to fragments isolated by expanses of open vegetation. Within such fragments evolutionary innovations accumulated, splitting phyletic lineages. By the time refuges re-expanded and brought forest biotas back into contact, the diverged taxa were hypothesized to have become partially or completely reproductively isolated.
Temperate glacial refuges are well documented and widely accepted (Hewitt 2000) and genetic studies based on Northern Hemisphere taxa have provided support for a temperate zone refuge hypothesis (Jaarola & Searle 2002). Pleistocene rainforest refuges have also been implicated in the historical isolation of tropical taxa (Schneider & Moritz 1999). However, a study which identified genetic evidence of population growth associated with habitat expansions after the late Pleistocene in North American mammals, failed to do so for Amazonian small mammals (Lessa et al. 2003). In another recent study, Flanagan et al. (2004) reported that the radiation of wing patterning in the Neotropical Heliconius erato and Heliconius melpomene had not been coincident, and that these butterflies did not share historical demographies, findings which were also inconsistent with hypotheses of Pleistocene population fragmentation. Such reports are in accordance with recent paleoecological data from the Amazon basin (Colinvaux 1997; Colinvaux & De Oliveira 2000, 2001; Colinvaux et al. 2000), which suggest that the climatic oscillations of the Pleistocene did not have a dramatic effect on forest cover. Using palynological analyses, Colinvaux et al. (Colinvaux & De Oliveira 2001; Colinvaux et al. 2001) identified species turnover in the flora, but not the biome replacements required to support historical forest fragmentation. These geological findings place Pleistocene refuge theory as an explanation for Neotropical biodiversity under strong attack.
Here, we test a key biological prediction of the Pleistocene refuge theory. Implicit in the theory, as well as in other vicariance theories, is that fragmentation would have had a simultaneous impact across the whole biota. Thus, lineage splits caused by forest fragmentation should have occurred at the same time in co-distributed taxa, and, in the case of the Pleistocene refuge theory, commensurate with Pleistocene climatic fluctuations. Clustered divergences due to vicariant events have previously been demonstrated, for example, in marine species pairs found across the Isthmus of Panama (Knowlton et al. 1993; Marko 2002). In accordance with predictions of vicariance theories, lineage splits should be most strikingly correlated in pairs of closely related taxa, which share natural history traits and therefore can be expected to respond similarly to common environmentally induced vicariance.
We here focus on the subfamily Ithomiinae (Lepidoptera: Nymphalidae). The group contains about 355 species (Lamas et al. 2004), each of which may consist of multiple strongly differentiated subspecies, providing an opportunity to study multiple, co-distributed and closely related taxa. Each subspecies is typically involved in Müllerian mimicry ‘rings’ with sympatric taxa from many other species (Brown 1979; Joron & Mallet 1998). Additionally, the distribution of ithomiine subspecies has been used as key biogeographic evidence for the refuge hypothesis (Brown 1979, 1982, 1987b; Haffer 1969; Turner & Mallet 1996).
We sampled from two adjacent centres of endemism in NE Peru separated by the Cerros Escalera mountain range (ranging up to ∼1600 m above sea level): the Río Mayo/upper Río Huallaga valley, and the lower Río Huallaga basin (figure 1). These centres of endemism (Lamas 1982) were interpreted by Brown (1987a,b) as evidence for the Huallaga (Andean valley) and Ucayali (Amazon basin) refuges, respectively. About 40 species of ithomiines show morphologically differentiated pairs of subspecies between these two endemic areas; their strikingly correlated contact and hybrid zones form a very well-defined ‘suture zone’ (sensu Remington 1968). Additionally, approximately 40 species are widespread and monomorphic, and found in both areas, while an additional approximately 40 monomorphic species are distributed only in the Huallaga centre, and approximately 20 are restricted to the Ucayali centre.
Figure 1.
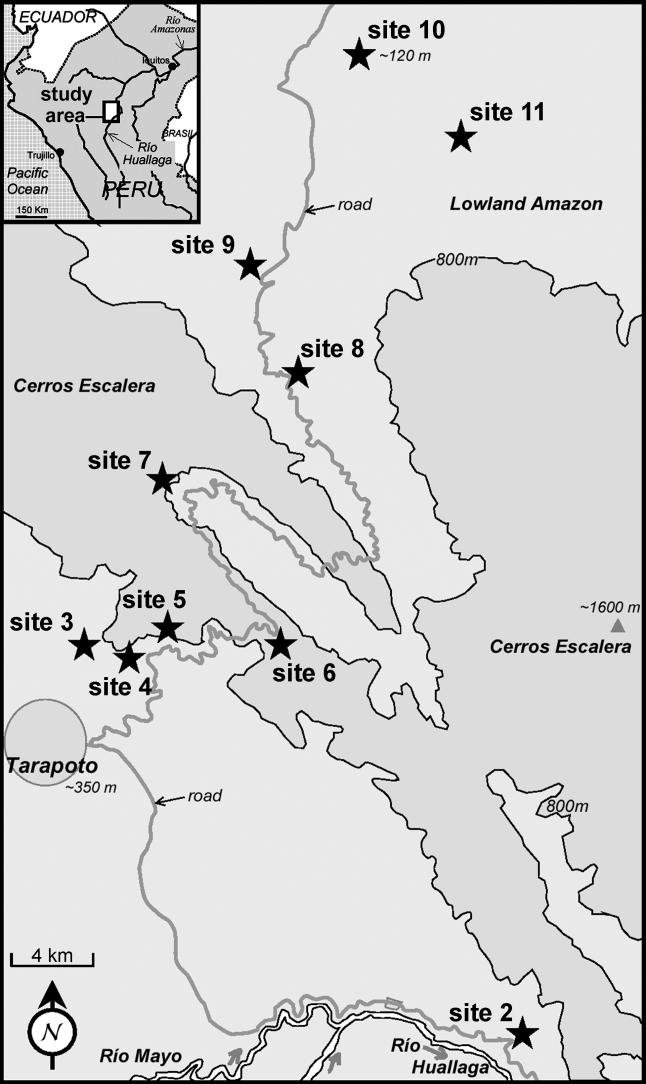
Map of the study area (Joron et al. 1999). Study site 12 is approximately 70 km NW from the top left-hand side corner of the map.
We investigate questions of ithomiine diversification using a 1619 bp aligned COI-tRNA-COII mitochondrial region as it has a high substitution rate, and is expected to show marked differences between even recently diverged taxa. We use a ‘rate-smoothing’ method to investigate the relative chronology of diversifications.
2. Material and methods
Ithomiine samples for DNA analysis were collected and are retained at UCL in 20% dimethylsulphoxide, 0.25 M EDTA and saturated NaCl solution. Taxa were collected in 2002 from study localities described by Joron et al. (1999), with the addition of site 12 (see figure 1 and table 1). Samples were identified by KRW and GL. Study taxa were selected at a range of taxonomic levels to obtain information spanning the whole process of differentiation. Where possible, at least three representatives from each centre of endemism, for each species, were sampled; for example, 3 Hyposcada anchiala interrupta from site 11 and 3 H. anchiala mendax from site 12; and 3 Ithomia agnosia agnosia from each of sites 2 and 12.
Table 1.
Study site details.
| site number | local name | position | zone of endemism | ||
|---|---|---|---|---|---|
| latitude | longitude | altitude (m) | |||
| 12 | Serranoyacu + Pte. Aguas Verdes | 5°67′49″S | 77°67′97″W | 980–1150 | Huallaga (Andean) |
| 2 | Quebrada Pucayaquillo | 6°35′10″S | 76°13′05″W | 300–450 | Huallaga (Andean) |
| 3 | Río Shilcayo + Boca | 6°27′20″S | 76°20′40″W | 350–500 | Huallaga (Andean) |
| 4 | Km 8–9. Sector Uruhuasha | 6°28′00″S | 76°20′05″W | 700–850 | Huallaga (Andean) |
| 6 | Km 15–19. Ahuashiyacu-Túnel | 6°27′25″S | 76°18′00″W | 800–1000 | Huallaga (Andean) |
| 7 | Km 28–30. Tarapoto-Yurimaguas | 6°24′30″S | 76°19′30″W | 750–900 | suture zone |
| 8 | Km 42–52. Tarapoto-Yurimaguas | 6°22′10″S | 76°16′45″W | 450 | suture zone |
| 10 | Km 68–72. Santa Rosa de Davidcillo + Km 26 Yurimaguas-Tarapoto | 6°14′55″S | 76°15′50″W | 120 | Ucayali (Amazonian) |
| 11 | Km 4–8. Carretera Pongo-Barranquita | 6°17′15″S | 76°13′40″W | 150 | Ucayali (Amazonian) |
One-third of the thorax of each newly collected specimen was extracted using the DNA easy kit (Qiagen) according to the manufacturer's instructions, with an initial incubation of 1.5–3 h at 55 °C, and into 300 μl elution buffer. Samples are preserved at −20 °C. Dried wings are retained as vouchers at UCL. Lab protocols closely follow those of Mallarino et al. (2005) using primers Jerry and Pat for COI amplification and sequencing, George and Imelda for COII amplification, and George and Imelda, or Phyllis, Imelda and an additional primer, Romeo, (5′-TAATATGACAGATTATATGTAATGGA-3′), for COII sequencing. Some Pseudoscada individuals would not amplify for COI, so a new specific primer, Talma (5′-AATCAGAATAACGACGAGG-3′) was developed, which successfully paired with Jerry, following the above protocol. Sequence data have been lodged at Genbank (Genbank accession numbers DQ078312-473 and DQ078478-479). Sequence data for eight specimens of Ithomia (Genbank accession numbers AY713067-8, AY713040-1, and AY713075-8, Mallarino et al. 2005) and a danaine outgroup Anetia briarea (Genbank accession number DQ071866, Brower et al. 2005) were also used. Vouchers for these nine specimens are retained by C. Jiggins at The University of Edinburgh (Ithomia) and A. V. Z. Brower at Oregon State University (Anetia).
PAUP* v. 4.0b 10 (Swofford 2000) was used to calculate the number of variable sites. Bayesian analysis was performed using MrBayes v. 3.0 (Huelsenbeck & Ronquist 2001), with nst=6 and a gamma rates heterogeneity model, using four simultaneous chains run for 1 000 000 generations, sampling a tree every 100 generations. The tree with maximum posterior probability was assessed using a consensus of the final 9000 trees (representing the final 900 000 generations), after confirmation that likelihood values had stabilized following 100 000 generations. Pairwise divergences between taxa, corrected for multiple hits, were calculated in PAUP using maximum likelihood parameters identified using PAUP* with a variant of the MODELTEST script (Posada & Crandall 1998); however, the Bayesian consensus tree was used, rather than a NJ tree as in the usual MODELTEST script. Model parameters found and used subsequently were; base frequencies, A: 0.3537, C: 0.0788, G: 0.0911; relative transition rates, A–C: 9.4363, A–G: 26.4378, A–T: 5.5769, C–G: 4.7678 C–T: 109.8022, and G–T: 1.0000; gamma-distributed rates with shape parameter: 0.6605; and fraction of invariable sites: 0.5626 (corresponding to a General Time Reversible, GTR+I+G model). We also used a PAUP* likelihood ratio method to test constancy of evolutionary rates (i.e. the molecular clock null hypothesis) across the Bayesian consensus tree.
Because the molecular clock was rejected, we used Yang's AHRS likelihood method in PAML (using the BASEML subprogram; see Yoder & Yang (2000), Yang & Yoder (2003), Yang (2004)) to obtain branch lengths by fitting a discrete model of rate variation, implemented with four different rates chosen via a clustering analysis (Yang 2004), to the Bayesian consensus tree. There are no fossil data for Ithomiinae, so times of divergence were calculated relative to the Ithomiinae+Anetia briarea (Danainae) root, which was set to 1.0. Relative divergence times were then calibrated to molecular divergence by performing a linear regression of selected, well-supported deeper nodes of species within genera against GTR+I+G distances obtained in PAUP*. As the fit was good (r2=0.926), we used node-depths instead of the more usual averaged pairwise divergences between taxa as estimators of the times of divergence between taxa. The resultant tree, therefore, gives relative estimates of times of divergence in terms of fitted GTR+I+G distances.
3. Results
Sequence data were obtained for 173 individuals representing 31 species from 10 genera, including an Anetia outgroup. The final 1619 bp alignment spanned 829 bp of the 3′ end of COI (alignment positions 1–829), 64 bp tRNA (830–893) and 726 bp COII (894–1619). Excluding Anetia, 532 sites were variable, of which 511 were parsimony-informative. Likelihood ratio tests were used to test the molecular clock by investigating rate heterogeneity across the Bayesian consensus tree. Substitution rates were heterogeneous (2Δln L=−205.95, 171 d.f., p=0.031), implying a poor fit to the molecular clock.
The rate-smoothed tree obtained via MrBayes and PAML is shown in figure 2. Intergeneric relationships were not as predicted from morphology or molecular phylogenetic analyses using a panel of mitochondrial and nuclear genes (Brower et al. 2005). This is attributable to mitochondrial data saturation and resultant weak support for deep branches in comparisons among phylogenetically distant taxa. However, all genera formed very strongly supported monophyletic clades (posterior probabilities of 1), with the exception of Hypothyris, which contained the genus Hyalyris due to the basal position of Hypothyris cantobrica, as well as due to the closer relationship of Hypothyris sp. nov. to Hyalyris oulita than to other Hypothyris. H. cantobrica was formerly placed in the monotypic genus Rhodussa and is notably divergent from remaining Hypothyris+Hyalyris, suggesting that Rhodussa might be a valid taxon if Hyalyris is to be kept separate. Apart from these, only Oleria was recovered as a monophyletic genus with a weaker posterior probability, 0.87, probably due to the effects of saturation on this deep clade.
Figure 2.

Phylogenetic hypothesis based on mitochondrial nucleotide data for Dircenna (D), Hyalyris (Hl), Brevioleria (B), Hyposcada (Hs), Hypothyris (Ht), Ithomia (I), Melinaea (M), Napeogenes (N), Oleria (O) and Pseudoscada (P). Tree topology is consensus of the last 9000 trees inferred using Bayesian methods; branch lengths shown are obtained by the AHRS rate-smoothing procedure of Yang (2004), calibrated by a least-squares fit to average GTR+I+G distances.
Within genera, haplotypes from the same species tended to cluster together. A striking exception is in the genus Melinaea. M. marsaeus, M. menophilus, M. satevis and M. isocomma haplotypes are clearly not clustered by species. In addition, subspecies of O. onega and Brevioleria aelia do not group together, instead of clustering most closely with haplotypes from a different species. For the purposes of the current paper, we therefore consider the subspecies of B. aelia as separate species. However, the subspecies of O. onega, which produce morphologically identifiable hybrids in contact zones (such as specimen 02–1958; see also Gallusser et al. 2004), are still considered to belong to O. onega, in spite of the evidence for paraphyly using this mitochondrial DNA data.
Many subspecies clusters are clearly recovered, as exemplified by the strong differentiation of both subspecies pairs in the genus Hyposcada. However, some well-supported groupings of haplotypes conflict with their taxonomic assignment. It is possible that the position of P. florula gracilis 1914 is the result of hybridization and introgression, as wild hybrids are known between P. florula gracilis and P. florula aureola. Additional cases of individuals not clustering with other representatives from the same subspecies are; I. salapia aquinia 72, Napeogenes inachia ssp. nov. 1188, B. arzalia arzalia 1203 and O. gunilla lota 209.
We found a remarkable range of pairwise comparisons between taxa, over the whole range of taxonomic levels (see table 2, figures 2 and 3). Between-subspecies smoothed pairwise divergences ranged from 0.06% between M. menophilus subspecies, and 6.40% between O. onega subspecies. Between-species divergences within genera varied by more than an order of magnitude, from 0.23% between M. menophilus and M. isocomma, to higher values such as 13.28% between H. cantobrica and the rest of Hypothyris, while divergences among genera within the Ithomiinae varied between 13.24% between Pseudoscada and Brevioleria and 17.74% at the base of the Ithomiinae.
Table 2.
Table showing fitted between-species pairwise percentage divergences within genera, and fitted between-subspecies pairwise percentage divergences within species, assuming a GTR+I+G model. Pairwise divergences were calculated as twice the distance to the deepest node separating the two taxa.
| genus | species | percentage pairwise divergences | percentage pairwise divergences | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | |||||
| Dircenna | adina | 1 | X | ||||||||||
| dero | 2 | 1.65 | X | ||||||||||
| Brevioleria | aelia orolina | 1 | X | ||||||||||
| aelia cf. pachiteae | 2 | 1.75 | X | ||||||||||
| arzalia | 3 | 1.75 | 1.23 | X | B. a. arzalia + ssp. n. | 1.23 | |||||||
| Hyposcada | anchiala | 1 | X | H. a. interrupta + mendax | 0.68 | ||||||||
| zarepha | 2 | 4.61 | X | H. z. flexibilis + ssp. n. | 2.43 | ||||||||
| Hypothyris | anastasia | 1 | X | ||||||||||
| cantobrica | 2 | 13.28 | X | ||||||||||
| euclea | 3 | 7.24 | 13.28 | X | |||||||||
| fluonia | 4 | 7.24 | 13.28 | 7.24 | X | ||||||||
| semifulva cf. pallisteri | 5 | 7.24 | 13.28 | 7.24 | 6.86 | X | |||||||
| mansuetus. meterus | 6 | 5.93 | 13.28 | 3.40 | 7.24 | 7.24 | X | ||||||
| ninonia | 7 | 7.24 | 13.28 | 2.52 | 7.24 | 7.24 | 3.40 | X | |||||
| sp. n. | 8 | 8.25 | 13.28 | 8.25 | 8.25 | 8.25 | 8.25 | 8.25 | X | ||||
| Hyalyris | oulita | 9 | 8.25 | 13.28 | 8.25 | 8.25 | 8.25 | 8.25 | 8.25 | 5.74 | X | ||
| Ithomia | agnosia | 1 | X | ||||||||||
| lagusa | 2 | 12.08 | X | ||||||||||
| salapia | 3 | 12.08 | 8.29 | X | I. s. aquinia + derasa | 0.23 | |||||||
| Melinaea | isocomma | 1 | X | ||||||||||
| ludovica | 2 | 4.97 | X | ||||||||||
| marsaeus | 3 | 0.94 | 4.97 | X | M. m. mothone +. phasiana | 0.94 | |||||||
| M. m. mothone + rileyi | 0.94 | ||||||||||||
| M. m. phasiana +. rileyi | 0.31 | ||||||||||||
| menophilus | 4 | 0.23 | 4.97 | 0.94 | X | M. m. hicetas + ssp. n. | 0.06 | ||||||
| satevis | 5 | 0.94 | 4.97 | 0.94 | 0.94 | X | M. s. cydon +. tarapotensis | 0.94 | |||||
| Napeogenes | inachia | 1 | X | N. i. pozziana + ssp. n. | 0.46 | ||||||||
| larina | 2 | 12.77 | X | ||||||||||
| pharo | 3 | 12.77 | 10.99 | X | |||||||||
| sylphis | 4 | 5.76 | 12.77 | 12.77 | X | N. s. corena +. rindgei | 0.39 | ||||||
| Oleria | agarista | 1 | X | ||||||||||
| gunilla | 2 | 10.68 | X | O. g. lota + serdolis | 1.25 | ||||||||
| ilerdina | 3 | 6.40 | 10.68 | X | |||||||||
| onega | 4 | 6.40 | 10.68 | 6.40 | X | O. o. janarilla + ssp. n. | 6.40 | ||||||
| Pseudoscada | florula | 1 | X | P. f. gracilis + aureola | 2.11 | ||||||||
Figure 3.
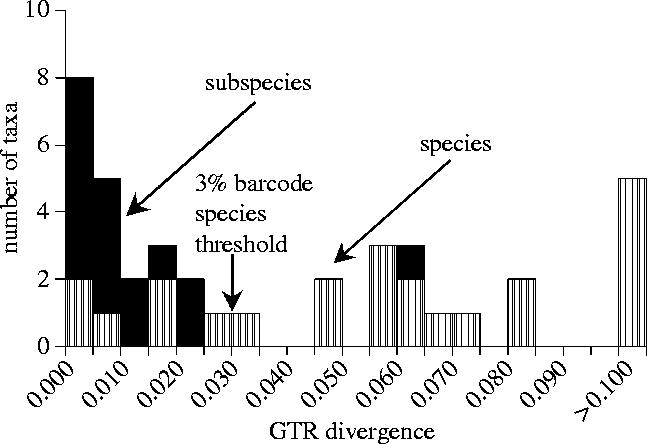
Histogram of fitted GTR+I+G distances across nodes between subspecies and species in the rate-smoothed tree (figure 2). These distances are similar to (and calibrated using) traditionally measured average pairwise between taxon GTR+I+G distances, except that each node is only recorded once to avoid duplication of information.
4. Discussion
Perhaps the major findings of this work are the highly variable levels of divergence in mtDNA between taxa of the same rank. Molecular divergence is an estimator of relative time of divergence, so these results seem to imply that times of divergence are also highly scattered. Our data did not conform to clock-like molecular evolution, so we estimated instead rate-smoothed percentage divergences as a metric of time. (Forcing our tree to obey a molecular clock, however, made little difference to the overall finding of variability in times of divergence—data not shown.) Applying Brower's (1994) 2.3% sequence divergence at COI and COII per million years to the rate-smoothed divergence values, subspecies within species, species with genera, and species among genera (including Hyalyris and Hypothyris in the same genus) appear to have diverged 0.03–2.78, 0.10–5.77, and 5.76–7.71 Myr ago, respectively. Zakharov et al. (2004) recently suggested substitution rates for COI and COII to be 0.78–1.02% sequence divergence per million years—much less than those calculated by Brower. Approximating this to 0.90% would increase our respective estimated times of divergence to 0.07–7.11, 0.26–14.76, and 14.71–19.71 Myr ago.
This variability of taxon divergences challenges assumptions underlying DNA ‘barcoding’, which aims to categorize taxa as species on the basis of mtDNA divergence. Proponents of DNA barcoding have suggested a 3% threshold at the COI mitochondrial locus for Lepidoptera species (Hebert et al. 2003). However, six of our 23 species divergence nodes (within genera) had smoothed GTR distance values less than 3% (figure 3, table 2), and one divergence node out of 16 between conspecific subspecies gave a smoothed distance value of greater than 3%. Somewhat later, Hebert et al. (2004) identified 10 cryptic species from within a single taxonomically recognized species of Astraptes (Hesperiidae), 8 of which had divergences of <3%, some of which were only 0.2–0.3% divergent (comparable with our closest Melinaea). Heliconius may have virtually no mtDNA divergence (0–2%) between subspecies, while values of >∼2% were reported for closest species (Brower 1994; Brower & Egan 1997; Flanagan et al. 2004). Although the use of DNA as a tool in taxonomy is well established, DNA taxonomy alone would have failed to recognize many of these species and/or would have recognized too many, if the threshold was lowered to say 1 or 2%. Our results strongly argue against use of a 3% COI mtDNA ‘barcode’ (or any other threshold divergence value) as a protocol to distinguish species without supplementation by other methods.
The concordant distributions of subspecies differing in mimetic colour pattern demonstrate that the Cerros Escalera is a suture zone for many ithomiine and heliconiine taxa. By recovering repeated genealogical splits between multiple pairs of taxa distributed across this divide, our work shows the same pattern for molecular data. Coyne & Orr (2004) recently identified three requirements by which molecular data could provide evidence for vicariance (for example, a Pleistocene refuge hypothesis). (i) Vicariant events should affect co-distributed taxa simultaneously, so that a main prediction of vicariance hypotheses is that divergence times should be similar and should correlate with the timing of relevant geological events, in this case in the Pleistocene. (ii) Each interacting pair should consist of sister taxa. (iii) There should be evidence of reproductive isolation between members of each pair.
Our results were in striking contrast to the first of these requirements. We found remarkable variability in pairwise divergences between different groups of taxa in comparisons at each taxonomic level. A number of hypotheses might explain the observed variance in sequence divergence values. The first is variability of mutation rates and the controversial reliability of molecular clocks (Graur & Martin 2004). However, we believe that the method of rate-smoothing used here (Yang 2004) accommodates such rate variation to calibrate the divergence to time as well as or better than any other existing method.
It is also possible that the mitochondrial genealogies do not accurately reflect organismal tree topologies, due to confounding factors such as selection, nuclear copies or cytoplasmic factors (Hurst & Jiggins 2005), or because coalescence time variance is itself large (e.g. Hudson & Turelli 2003). To investigate this further, we have sequenced the nuclear loci triose phosphate isomerase, wingless and elongation factor 1-α. Preliminary comparisons between two of the most strongly contrasting genera, Oleria and Melinaea, show molecular divergence in these nuclear regions that correlate with the strongly divergent patterns obtained using mtDNA data.
Furthermore, although some diversification seems to have occurred during the Pleistocene, molecular clock dating also shows genetic breaks both before and after this period. Therefore, ithomiine taxa do not at all seem to share the same clear genetic signature of subspecific or specific diversification due to one or a few historical events during the Pleistocene.
In their second requirement, Coyne & Orr (2004) argue that members of each pair should be sister taxa. In our data, it is very likely that at least some of our pairs of subspecies are not sister taxa, but determining this is beyond the scope of this study, since it would involve the collection and sequencing of as many as 30 closely related subspecies per species from across the Neotropics. However, whether or not the pairs of subspecies are sister taxa should not affect the central prediction of vicariance for the suture zone under study here. All pairs of taxa across the suture zone should have the same (non-sister) relationships if generated by the same sequence of vicariance events, so that their branching pattern should be similar. Their divergence times should likewise be comparable. This prediction is clearly rejected. An alternative hypothesis, therefore, seems much more likely; the clustering of multiple contact zones in the suture zone must have been caused by range changes and/or independent evolution, rather than primarily by the same vicariance events in each group.
Finally, Coyne & Orr (2004) highlight the importance for evidence of reproductive isolation between members of each taxon pair. Past and present gene exchange are hard to distinguish from persistent ancestral polymorphisms; these effects can affect deductions about the history of taxa. Here, the geographically differentiated ithomiines across the suture zone are classified as subspecies and hybridize to a greater or lesser extent. Putative hybrids between subspecies, based on intermediacy of morphological characters, are readily found, for example, O. onega ssp. nov. × O. onega janarilla (02-1958), and M. marsaeus phasiana × M. marsaeus rileyi (02-234). However, despite commonly hybridizing, the O. onega subspecies remain markedly genetically distinct. Melinaea are in stark contrast, with no genetic differences evident between subspecies. The species M. isocomma, M. marsaeus, M. satevis and M. menophilus also share identical or very similar haplotypes (mean between-species divergences were all 0.9% or less), even though we found no evidence of hybridization between species. Nuclear data show a similar pattern (M. Zimmermann, unpublished data). This is quite unexpected as experts have long recognized the distinctness of these Melinaea species (Brown 1979, Lamas et al. 2004), although there have been some revisions, name changes and reallocations of subspecies among species.
Morphologically distinct taxa showing high levels of gene exchange at many loci are predicted to be rare, so whilst we can expect some ancestral polymorphism and introgression between very closely related species, we hardly expect such effects among a group of four highly distinct and relatively widespread species (Brown 1977; Lamas et al. 2004), as found here. The four closely related Melinaea species comprise, between them, 34 morphologically recognizable subspecies, each of which typically has a divergent colour pattern affecting Müllerian mimicry (Brown 1977; Lamas et al. 2004). Our genetic data provide evidence for very recent diversification for the four closely related Melinaea species and even more recent diversification of their subspecies, coupled with incomplete lineage sorting. All of this diversification of Melinaea has apparently occurred well within the time that it took co-distributed pairs of subspecies in O. onega, O. gunilla, B. arzalia, H. zarepha, or P. florula to diverge.
In conclusion, we infer strikingly varied divergence dates between pairs of taxa interacting in a single suture zone. This variability must be due to processes acting independently in different species and at different times, rather than due to a single or a few vicariant events. Our results imply that diversification of subspecies pairs across a single region are due neither to concordant evolutionary histories nor to simultaneous divergence times. We postulate an alternative: that ithomiine biogeographic patterns are explained by ongoing diversification through repeated and idiosyncratic parapatric or allopatric speciation in nearly continuous forest, interspersed by rapid range changes (Benson 1982; Knapp & Mallet 2003; Mallet 1993). The well-defined Escalera suture zone we see today may then be explained by redistribution of subspecific differentiation across ecotones and by partial environmental barriers to the movement of colour pattern and molecular genes. These current or recent factors must affect multiple pairs of taxa, in order for hybrid zones to collect together in the same suture zone (see also Mallet 1993). Our comparative data are, therefore, among the first to demonstrate that Amazonian diversification is relatively continuous over time, as would be likely if speciation took place via parapatric evolution in continuous forest.
Acknowledgements
We thank Maribel González, Carlos Vergara and Oris Sanjur at The Smithsonian Tropical Research Institute for expert laboratory assistance. We are grateful to Ziheng Yang for many useful discussions, and for help in implementing the rate-smoothing method in PAML. We also thank Andrew Brower for providing sequence data prior to publication.
Footnotes
These authors contributed equally.
References
- Benson W.W. Alternative models for infrageneric diversification in the humid tropics: tests with passion vine butterflies. In: Prance G.T, editor. Biological diversification in the tropics. Columbia University Press; New York: 1982. pp. 608–640. [Google Scholar]
- Brower A.V.Z. Rapid morphological radiation and convergence among races of the butterfly Heliconius erato inferred from patterns of mitochondrial DNA evolution. Proc. Natl Acad. Sci. USA. 1994;91:6491–6495. doi: 10.1073/pnas.91.14.6491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brower A.V.Z, Egan M.G. Cladistic analysis of Heliconius butterflies and relatives (Nymphalidae: Heliconiiti): a revised phylogenetic position for Eueides based on sequences from mitochondrial DNA and a nuclear gene. Proc. R. Soc. B. 1997;264:969–977. 10.1098/rspb.1997.0134 [Google Scholar]
- Brower A.V.Z, Freitas A.V.L, Lee M.-M, Silva Brandão K.L, Whinnett A, Willmott K.R. Phylogenetic relationships among the Ithomiini (Lepidoptera: Nymphalidae) inferred from one mitochondrial and two nuclear gene regions. Syst. Entomol. 2005 (In press.). [Google Scholar]
- Brown K.S. Geographical patterns of evolution in neotropical Lepidoptera: differentiation of the species of Melinaea and Mechanitis (Nymphalidae: Ithomiinae) Syst. Entomol. 1977;2:161–197. [Google Scholar]
- Brown, K. S. 1979 Ecologia Geográfica e Evolução nas Florestas Neotropicais Campinas, Brazil: Universidade Estadual de Campinas.
- Brown K.S. Historical and ecological factors in the biogeography of aposematic Neotropical butterflies. Am. Zool. 1982;22:453–471. [Google Scholar]
- Brown K.S. Areas where humid tropical forest probably persisted. In: Whitmore T.C, Prance G.T, editors. Biogeography and quaternary history in tropical America. Oxford University Press; Oxford, UK: 1987a. p. 45. [Google Scholar]
- Brown K.S. Biogeography and evolution of neotropical butterflies. In: Whitmore T.C, Prance G.T, editors. Biogeography and quaternary history in tropical America. Oxford University Press; Oxford, UK: 1987. pp. 66–104. [Google Scholar]
- Colinvaux P.A. An arid Amazon? Trends Ecol. Evol. 1997;12:318–319. doi: 10.1016/S0169-5347(97)89916-8. 10.1016/S0169-5347(97)89916-8 [DOI] [PubMed] [Google Scholar]
- Colinvaux P.A, De Oliveira P.E. Palaeoecology and climate of the Amazon basin during the last glacial cycle. J. Q. Sci. 2000;15:347–356. 10.1002/1099-1417(200005)15:4%3C347::AID-JQS537%3E3.0.CO;2-A [Google Scholar]
- Colinvaux P.A, De Oliveira P.E. Amazon plant diversity and climate through the Cenozoic. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2001;166:51–63. 10.1016/S0031-0182(00)00201-7 [Google Scholar]
- Colinvaux P.A, De Oliveira P.E, Bush M.B. Amazonian and neotropical plant communities on glacial time-scales: the failure of the aridity and refuge hypotheses. Q. Sci. Rev. 2000;19:141–169. 10.1016/S0277-3791(99)00059-1 [Google Scholar]
- Colinvaux P.A, Irion G, Rasanen M.E, Bush M.B, De Mello J.A.S.N. A paradigm to be discarded: geological and paleoecological data falsify the Haffer & Prance refuge hypothesis of Amazonian speciation. Amazoniana. 2001;16:606–607. [Google Scholar]
- Coyne J.A, Orr H.A. Sinauer Associates; Sunderland, MA: 2004. Speciation. [Google Scholar]
- Flanagan N.S, Tobler A, Davison A, Pybus O.G, Kapan D.D, Planas S, Linares M, Heckel D, McMillan W.O. The historical demography of Müllerian mimicry in the neotropical Heliconius butterflies. Proc. Natl Acad. Sci. USA. 2004;101:9704–9709. doi: 10.1073/pnas.0306243101. 10.1073/pnas.0306243101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallusser S, Guadagnuolo R, Rahier M. Genetic (RAPD) diversity between Oleria onega agarista and Oleria onega ssp. (Ithomiinae, Nymphalidae, Lepidoptera) in north-eastern Peru. Genetica. 2004;121:65–74. doi: 10.1023/b:gene.0000019930.74692.bb. 10.1023/B:GENE.0000019930.74692.bb [DOI] [PubMed] [Google Scholar]
- Graur D, Martin W. Reading the entrails of chickens: molecular timescales of evolution and the illusion of precision. Trends. Genet. 2004;20:80–86. doi: 10.1016/j.tig.2003.12.003. 10.1016/j.tig.2003.12.003 [DOI] [PubMed] [Google Scholar]
- Haffer J. Speciation in amazonian forest birds. Science. 1969;165:131–137. doi: 10.1126/science.165.3889.131. [DOI] [PubMed] [Google Scholar]
- Haffer J. Alternative models of vertebrate speciation in Amazonia: a review. Biodivers. Conserv. 1997;6:451–476. 10.1023/A:1018320925954 [Google Scholar]
- Haffer J, Prance G.T. Climatic forcing of evolution in Amazonia during the Cenozoic: on the refuge theory of biotic differentiation. Amazoniana. 2001;16:579–607. [Google Scholar]
- Hebert P.D.N, Cywinska A, Ball S.L, deWaard J.R. Biological identifications through DNA barcodes. Proc. R. Soc. B. 2003;270:313–321. doi: 10.1098/rspb.2002.2218. 10.1098/rspb.2002.2218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert P.D.N, Penton E.H, Burns J.M, Janzen D.H, Hallwachs W. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl Acad. Sci. USA. 2004;101:14812–14817. doi: 10.1073/pnas.0406166101. 10.1073/pnas.0406166101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hewitt G. The genetic legacy of the Quaternary ice ages. Nature. 2000;405:907–913. doi: 10.1038/35016000. 10.1038/35016000 [DOI] [PubMed] [Google Scholar]
- Hudson R.R, Turelli M. Stochasticity overrules the ‘three-times rule’: genetic drift, genetic draft, and coalescence times for nuclear loci versus mitochondrial DNA. Evolution. 2003;57:182–190. doi: 10.1111/j.0014-3820.2003.tb00229.x. [DOI] [PubMed] [Google Scholar]
- Huelsenbeck J.P, Ronquist F. MrBayes: Bayesian inference of phylogenetic trees. Bioinformatics. 2001;17:754–755. doi: 10.1093/bioinformatics/17.8.754. 10.1093/bioinformatics/17.8.754 [DOI] [PubMed] [Google Scholar]
- Hurst G.D.D, Jiggins F.M. Problems with mitochondrial DNA as a marker in population, phylogeographic, and phylogenetic studies: the effects of inherited symbionts. Proc. R. Soc. B. 2005;272:1525–1534. doi: 10.1098/rspb.2005.3056. 10.1098/rspb.2005.3056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaarola M, Searle J.B. Phylogeography of field voles (Microtus agrestis) in Eurasia inferred from mitochondrial DNA sequences. Mol. Ecol. 2002;11:2613–2621. doi: 10.1046/j.1365-294x.2002.01639.x. 10.1046/j.1365-294X.2002.01639.x [DOI] [PubMed] [Google Scholar]
- Joron M, Mallet J. Diversity in mimicry: paradox or paradigm? Trends Ecol. Evol. 1998;13:461–466. doi: 10.1016/s0169-5347(98)01483-9. 10.1016/S0169-5347(98)01483-9 [DOI] [PubMed] [Google Scholar]
- Joron M, Wynne I.R, Lamas G, Mallet J. Variable selection and the coexistence of multiple mimetic forms of the butterfly Heliconius numata. Evol. Ecol. 1999;13:721–754. 10.1023/A:1010875213123 [Google Scholar]
- Knapp S, Mallet J. Refuting refugia. Science. 2003;300:71–72. doi: 10.1126/science.1083007. 10.1126/science.1083007 [DOI] [PubMed] [Google Scholar]
- Knowlton N, Weigt L.A, Solorzano L.A, Mills D.K, Bermingham E. Divergence in proteins, mitochondrial DNA, and reproductive compatibility across the Isthmus of Panama. Science. 1993;260:1629–1632. doi: 10.1126/science.8503007. [DOI] [PubMed] [Google Scholar]
- Lamas G. A preliminary zoogeographical division of Peru based on butterfly distributions (Lepidoptera, Papilionoidea) In: Prance G.T, editor. Biological diversification in the tropics. Columbia University Press; New York: 1982. pp. 336–357. [Google Scholar]
- Lamas G, Callaghan C, Casagrande M.M, Mielke O, Pyrcz T, Robbins R, Viloria A. Hesperioidea and Papilionoidea. In: Heppner J.B, editor. Atlas of neotropical Lepidoptera, checklist. Association for Tropical Lepidoptera/Scientific Publishers; Gainesville, FL: 2004. [Google Scholar]
- Lessa E.P, Cook J.A, Patton J.L. Genetic footprints of demographic expansion in North America, but not Amazonia, during the late Quaternary. Proc. Natl Acad. Sci. USA. 2003;100:10331–10334. doi: 10.1073/pnas.1730921100. 10.1073/pnas.1730921100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallarino R, Bermingham E, Willmott K.R, Whinnett A, Jiggins C.D. Molecular systematics of the butterfly genus Ithomia (Lepidoptera: Ithomiinae): a composite phylogenetic hypothesis based on seven genes. Mol. Phylogenet. Evol. 2005;34:625–644. doi: 10.1016/j.ympev.2004.10.021. 10.1016/j.ympev.2004.10.021 [DOI] [PubMed] [Google Scholar]
- Mallet J. Speciation, raciation, and color pattern evolution in Heliconius butterflies: evidence from hybrid zones. In: Harrison R.G, editor. Hybrid zones and the evolutionary process. Oxford University Press; New York: 1993. pp. 226–260. [Google Scholar]
- Marko P.B. Fossil calibration of molecular clocks and the divergence times of geminate species pairs separated by the Isthmus of Panama. Mol. Biol. Evol. 2002;19:2005–2021. doi: 10.1093/oxfordjournals.molbev.a004024. [DOI] [PubMed] [Google Scholar]
- Posada N.M, Crandall K.A. MODELTEST: testing the model of DNA substitution. Bioinformatics. 1998;14:817–818. doi: 10.1093/bioinformatics/14.9.817. 10.1093/bioinformatics/14.9.817 [DOI] [PubMed] [Google Scholar]
- Remington C.L. Suture-zones of hybrid interaction between recently joined biotas. Evol. Biol. 1968;1:321–428. [Google Scholar]
- Schneider C, Moritz C. Rainforest refugia and evolution in Australia's wet tropics. Proc. R. Soc. B. 1999;266:191–196. 10.1098/rspb.1999.0621 [Google Scholar]
- Simpson B.B, Haffer J. Speciation patterns in the Amazonian forest biota. Ann. Rev. Ecol. Syst. 1978;9:497–518. 10.1146/annurev.es.09.110178.002433 [Google Scholar]
- Swofford D.L. Sinauer Associates; Sunderland, MA: 2000. PAUP*: phylogenetic analysis using parsiomony (*and other methods) [Google Scholar]
- Turner J.R.G, Mallet J.L.B. Did forest islands drive the diversity of warningly coloured butterflies? Biotic drift and the shifting balance. Phil. Trans. R. Soc. B. 1996;351:835–845. [Google Scholar]
- Yang Z. A heuristic rate smoothing procedure for maximum likelihood estimation of species divergence times. Acta Zool. Sinica. 2004;50:645–656. [Google Scholar]
- Yang Z, Yoder A.D. Comparison of likelihood and Bayesian methods for estimating divergence times using multiple gene loci and calibration points, with application to a radiation of cute-looking mouse lemurspecies. Syst. Biol. 2003;52:705–716. doi: 10.1080/10635150390235557. 10.1080/10635150390235557 [DOI] [PubMed] [Google Scholar]
- Yoder A.D, Yang Z. Estimation of primate speciation dates using local molecular clocks. Mol. Biol. Evol. 2000;17:1081–1090. doi: 10.1093/oxfordjournals.molbev.a026389. [DOI] [PubMed] [Google Scholar]
- Zakharov E.V, Caterino M.S, Sperling F.A.H. Molecular phylogeny, historical biogeography, and divergence time estimates for swallowtail butterflies of the genus Papilio (Lepidoptera: Papilionidae) Syst. Biol. 2004;53:193–215. doi: 10.1080/10635150490423403. 10.1080/10635150490423403 [DOI] [PubMed] [Google Scholar]