

Abstract
In the 14 years since the first higher-level bird phylogenies based on DNA sequence data, avian phylogenetics has witnessed the advent and maturation of the genomics era, the completion of the chicken genome and a suite of technologies that promise to add considerably to the agenda of avian phylogenetics. In this review, we summarize current approaches and data characteristics of recent higher-level bird studies and suggest a number of as yet untested molecular and analytical approaches for the unfolding tree of life for birds. A variety of comparative genomics strategies, including adoption of objective quality scores for sequence data, analysis of contiguous DNA sequences provided by large-insert genomic libraries, and the systematic use of retroposon insertions and other rare genomic changes all promise an integrated phylogenetics that is solidly grounded in genome evolution. The avian genome is an excellent testing ground for such approaches because of the more balanced representation of single-copy and repetitive DNA regions than in mammals. Although comparative genomics has a number of obvious uses in avian phylogenetics, its application to large numbers of taxa poses a number of methodological and infrastructural challenges, and can be greatly facilitated by a ‘community genomics’ approach in which the modest sequencing throughputs of single PI laboratories are pooled to produce larger, complementary datasets. Although the polymerase chain reaction era of avian phylogenetics is far from complete, the comparative genomics era—with its ability to vastly increase the number and type of molecular characters and to provide a genomic context for these characters—will usher in a host of new perspectives and opportunities for integrating genome evolution and avian phylogenetics.
Keywords: community genomics, BAC libraries, bird phylogeny, retroelement, concatenation
1. Introduction
The first phylogenetic analysis of higher categories of birds based on DNA sequence data appeared in 1991 (Edwards et al. 1991), soon after the completion of the chicken mitochondrial genome (Desjardins & Morais 1990) and the first applications of polymerase chain reaction (PCR) to ornithology (Kocher et al. 1989; Edwards & Wilson 1990). Building on these and the comprehensive yet controversial ‘tapestry’ provided by Charles Sibley's and Jon Ahlquist's DNA hybridization studies of the 1980s (Sibley & Ahlquist 1990), the number and size of DNA sequence datasets in avian phylogenetics have steadily increased (figure 1a–c). In the late 1990s the ultimate limit for comparative mitochondrial DNA (mtDNA) sequencing was achieved with the first analyses of complete mitochondrial (mt) genomes of ostrich and rhea (ratites; Härlid et al. 1997, 1998; Mindell et al. 1999). Although comparative protein sequencing had previously been employed in avian phylogenetics (Stapel et al. 1984), the threshold for application of nuclear DNA sequences to avian systematics was crossed with Hedges' analyses of 18srRNA (Hedges 1994), and later with studies by Cooper & Penny (1997) and Prychitko & Moore (1997) using β-fibrinogen intron 7 and c-mos, respectively. The possibility of comparing distantly related birds using conserved nuclear DNA sequences has reinvigorated higher-level phylogenetics, with a number of recent studies spanning all modern birds (Neornithes) and notoriously problematic groups, such as the passerine birds (Groth & Barrowclough 1999; van Tuinen et al. 2000; Irestedt et al. 2001; Ericson et al. 2002; Ericson & Johansson 2003; Barker et al. 2004). In 2003, avian molecular phylogenetics entered a new era with the initiation of two NSF-funded Tree of Life initiatives, centred at the American and Field Museums of Natural History in the US. Additional work in Europe stemming largely from the Swedish Museum of Natural History, Stockholm, is similarly increasing the extent of its approach to the avian tree. Although initial reports from these large-scale collaborative projects suggest that the task of resolving the avian tree with DNA sequence data will be daunting, nearly all of the exciting datasets are still to be published and there is every reason to imagine a host of noteworthy findings over the next few years. The steady advances in avian phylogenetics over the past 15 years are all the more remarkable given the profound scepticism of new synthesis systematists towards the possibility of resolving the higher-level genealogy for birds (reviewed in Cracraft et al. 2004).
Figure 1.
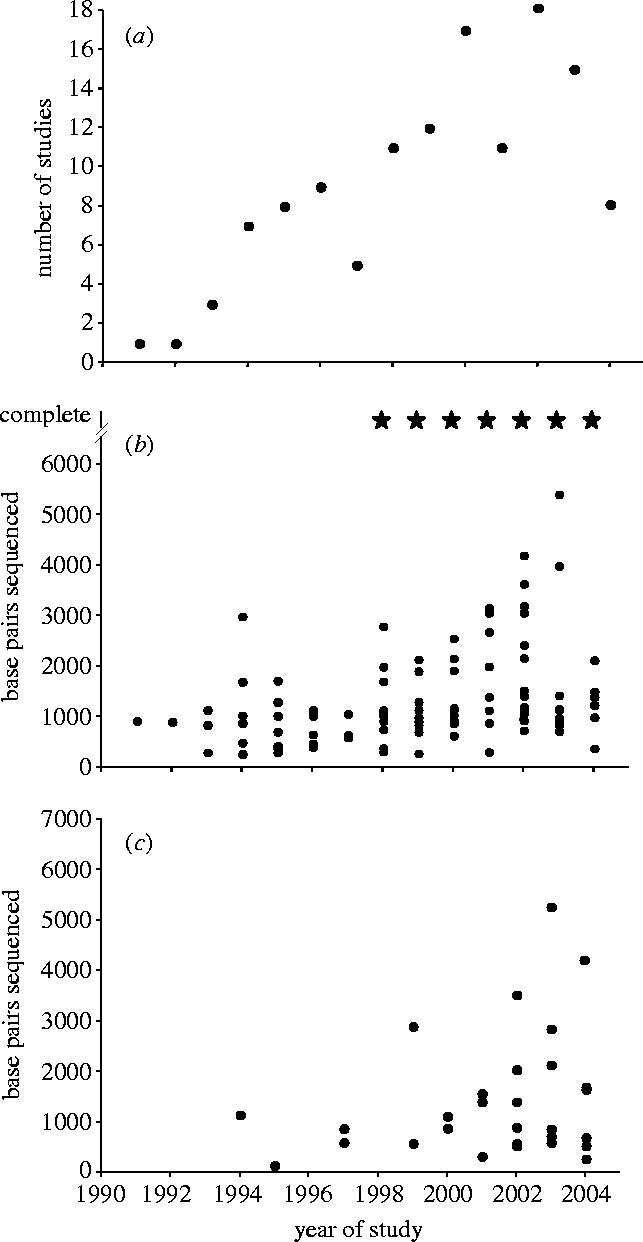
Trends in publication and sequencing for avian higher-level phylogenetics, 1991 to July 2004. The plots are based on 122 publications sampled from the Biosis literature database using keywords ‘bird’, ‘DNA’ and ‘phylogeny’ and which also fulfilled the following criteria: (i) focus on the paper was on a question of higher‐level systematics; (ii) an original phylogenetic analysis was done (as opposed to using a tree solely for comparative purposes; see Electronic Appendix). In addition, we added several key studies that were missed by this search approach. The list is not exhaustive but includes most major studies fulfilling these criteria. (a) Number of publications per year. (b) Length of mitochondrial DNA sequence analysed/produced per publication per year; stars in the upper right of this graph indicate the completion of one or more complete mitochondrial genomes in that year. (c) Length of nuclear DNA sequence analysed/produced per publication per year.
As comparative avian sequences are being accumulated ever more rapidly by PCR approaches, qualitatively new comparative genomics resources and approaches are making their entrance into ornithology, and have already contributed to phylogenetic studies in mammals and other groups (Thomas et al. 2003; Sasaki et al. 2004a). The rate of nucleotide sampling and the signal in currently sampled DNA sequences are probably sufficient to resolve a robust avian tree. However, we suggest that embracing modern genomics will provide a rich portrait of avian evolution at the genomic level, as well as exposure to a host of novel questions that current protocols cannot address. Gathering data applicable to comparative genomics is a worthy subsidiary goal of any systematics endeavour, and genomics and systematics are widely appreciated to be reciprocally illuminating (Matthee et al. 2001). Several recent studies demonstrate the feasibility of these approaches in birds, albeit on a limited number of taxa and genomically idiosyncratic regions such as the MHC (Gasper et al. 2001; Raudsepp et al. 2002; Shiina et al. 2004). Thus, the major issue is not the feasibility of these approaches but whether they can be employed on a taxonomically satisfying scale, thereby substantively furthering the programme of avian phylogenetics (Pollock et al. 2000; Thomas et al. 2003). We also point out some conceptual and analytical issues relevant to the use of the genomic approaches in birds that when addressed will hopefully improve efforts to build a robust species tree for birds.
2. The state of the tree
The current state of knowledge of the phylogenetic relationships of modern birds will not be reviewed here, since a very recent and comprehensive review has just appeared (Cracraft et al. 2004), which concluded that
…research on the higher level relationships of birds has made significant progress over the last decade, yet it is obvious…that compelling evidence for relationships among most major clades is still lacking.
(Cracraft et al. 2004, p. 483).
We agree wholeheartedly with this statement but emphasize with Cracraft et al. that significant amounts of new data will be appearing imminently that will undoubtedly add significant new insight into avian relationships. To briefly reiterate recent major findings, Cracraft et al. (2004) suggest a number of emerging syntheses, several of which have long histories of corroboration from morphology and other markers: (i) that Paleognaths (flightless ratites and tinamous) are the sister group of all other birds (Neognathae), but that relationships within the ratites, including the position of tinamous (Tinamidae) are unclear; (ii) that Galloanserae is the sister group to all other non-ratite birds (Neoaves); (iii) that there is some signal for a large and heterogeneous ‘waterbird assemblage’ including traditional Pelecaniformes and the shoebill (Balaeniceps), as well as other small groupings, such as grebes with flamingos and penguins with tubenosed seabirds; (iv) that passerines may be embedded within, rather than sister to, a ‘higher land bird’ assemblage including Coraciiformes, trogons and woodpeckers and allies; (v) that Passeriformes are strongly monophyletic, with the endemic New Zealand wrens (Acanthisitta) comprising the sister group to all other passeriforms and with New and Old World suboscines as the monophyletic sister group of the oscines; and (vi) lyrebirds (Menura) comprise the sister group to all other oscines, followed by many Corvoidean lineages found primarily in eastern Gondwana. The ‘core Corvida’ has embedded within it a monophyletic Passerida, a subtly but profoundly different arrangement from the sister group relationship of Passerida and Corvida outlined by Sibley & Ahlquist (1990) (see also Ericson et al. 2002; Edwards & Boles 2002; Barker et al. 2004). Recently, Fain & Houde (2004) provided evidence for two major clades of Neoaves, Metaves and Coronaves, each of which constitute major parallel radiations. While there are many other suggestive monophyletic groups in the avian tree at this time, on the whole the assertion that ‘…Neoavian relationships… are decidedly uncertain’ (Cracraft et al. 2004, p. 483) represents a view with which most avian systematists would agree.
3. Current approaches to the tree
(a) Taxon and character sampling
In the first wave of large-scale analyses of avian relationships, ornithologists have tended to emphasize taxon sampling over character sampling, especially when compared with recent studies on mammals (see table 1 in Electronic Appendix). Part of the reason for this may be differences in phylogenetic structure of birds and mammals; whereas mammals possess a few morphologically well-delimited groups (18 orders and 48 families), birds comprise 27 orders and approximately 155 traditionally recognized families, the latter of which often have very vague boundaries. An additional characteristic of the bird datasets is that nuclear DNA studies tend to be taxon-rich, whereas mtDNA studies—at least those that sample the entire mitochondrial genome—tend to be character rich. Published nuclear DNA studies in birds have not yet achieved the balance of characters and taxa that mammal studies have achieved, although such character-rich studies will undoubtedly soon be appearing.
Has the steady increase in the size of DNA sequence datasets seen in figure 1b,c brought with it a concomitant increase in phylogenetic accuracy and resolution, sensu Hillis (1998)? Undoubtedly yes, but few efforts have specifically focused on testing this hypothesis using a consistent sampling of taxa. An unpublished simulation study indicated that, approximately 25 000 nucleotides would be required for resolution of an ‘average’ branch of the avian tree (E. L. Braun, R. T. Kimball and J. Harshman, personal communication). Current sequencing goals for the avian Tree of Life fall somewhere within this vicinity (J. Harshman, personal communication). The generally high degree of genetic similarity among avian species and genera compared with mammals implies that the number of nucleotides required to resolve intergeneric nodes could be in the tens of thousands, similar to the number estimated to be necessary to resolve the human-chimp-gorilla split approximately 6 Myr ago (Saitou & Nei 1986). The consensus among systematists is that large and complex phylogenies can be resolved most efficiently with increased taxon sampling (Pollock et al. 2002; Hillis et al. 2003). Still, avian systematists working today are adhering to a more conservative approach in which both dense taxon and character sampling are high priorities. The reigning zeitgeist is one in which gradual accumulation of sequence characters will result in a concomitant increases in statistical resolution of branches.
(b) Mitochondrial versus nuclear DNA trees
Which is better for achieving a robust phylogenetic tree for birds—mitochondrial or nuclear DNA? Initial trees based on complete mitochondrial genomes surprised the community by placing Paleognaths in a derived position as sister to the Galloanserae, and passerines near the root (Härlid et al. 1997; Härlid & Arnason 1999); these trees are now known to have been hampered by poor taxon sampling and consequent misplacement of the root, but had the positive effect of raising awareness of these issues. A taxonomically large study of cytochrome b sequences also yielded a basal position for passerines (Johnson 2001), suggesting in hindsight that sequence length also has an important role in resolving the avian tree. Initial suggestions that mtDNA per se was ill suited for resolving higher-level relationships within birds have been muted somewhat now that there are roughly 30 complete mitochondrial genomes for birds. Despite a few incongruent results, recent analyses of complete avian and non-avian reptile mitochondrial genomes (Braun & Kimball 2002; Harrison et al. 2004b; S. Edwards, J. Gasper, W. Nelson, J. Avise and D. Pollock, unpublished data) recover arrangements in which basal branches are composed of Paleognaths and Galloanseriformes, conforming to the consensus view. In addition, the view that mtDNA evolves too rapidly for the analysis of birds at higher levels is also not entirely consistent with recent theoretical work suggesting that the optimal rate of character evolution under parsimony—for a wide range of conditions as much as 0.6 substitutions per site—is much higher than previously thought (Yang 1998). Even so, the extreme bias in substitution patterns makes application to higher‐level questions problematic, and mtDNA is clearly very sensitive to the type of analysis performed (Braun & Kimball 2002). The utility of mtDNA will undoubtedly increase with informed methods of analysis, such as transversion parsimony (Harrison et al. 2004b), and with further taxon sampling—the sequencing of complete mtDNAs is now straightforward enough that the approach is being applied to phylogenetic analysis of individual avian families (R. Carson and G. Spicer, personal communication) and on large scales in both mammals and fishes (Miya et al. 2001; Shevchuk & Allard 2001).
With their more even base composition and slower rate of substitution, nuclear genes appear to hold exceptional promise for resolving higher‐level relationships within birds, and a number of recent studies have produced substantial datasets with encouraging results. A variety of nuclear genes have now been employed in avian studies; some of the most commonly used are c-mos, c-myc, RAG-1 and 2, 18SrRNA and β-fibrinogen introns (see table 2 in Electronic Appendix). Intronless genes such as RAG-1 and 2 are useful because alignment is very straightforward as is amplification using conserved primers, whereas18 SrRNA, and less so, introns such as β-fibrinogen intron 7, inevitably present some difficulties in alignment. On the other hand, introns have the added value that they present numerous insertions and deletions (indels), which can be of substantial use as cladistic characters. A comparison of information content of nuclear and mtDNA in those studies that have employed both character sets on the same taxa (see table 2 in Electronic Appendix) suggests that mtDNA generally contains greater information content than nuclear genes. However, such measures of information content—including bootstrap values—are widely appreciated to be susceptible to convergence on the wrong relationships in cases of lineage-specific changes in base composition, rapid sequence evolution or long branch attraction (Braun & Kimball 2002). Many mitochondrial genome trees published to date possess bootstrap values frequently approaching 95% or 100%, yet result in trees that are highly incongruent with other data. As emphasized in other recent reviews, congruence must be the final arbiter of avian relationships. Several studies (e.g. Birks & Edwards 2002; Pereira et al. 2002; Russello & Amato 2004) have shown complementarity of nuclear and mitochondrial datasets, with these partitions resolving the base and the tips of trees, respectively.
(c) Insertions/deletions
It was hoped that differences in mitochondrial gene order would provide strong corroborating information on avian relationships, but the most comprehensive study to date suggested the contrary—that gene order had undergone convergence multiple times within birds (Mindell et al. 1998), in contrast to the interpretation from phylogenetic studies in invertebrates (Boore 1999). However, indels in nuclear genes have begun to provide a number of compelling markers for specific clades of birds (Groth & Barrowclough 1999; Irestedt et al. 2004). The first significant example of such characters was Ericson et al.'s (2000) study of the c-myc gene. Two insertions of one and three amino acids, both of which preserved a functional reading frame of the protein, were found at various levels within the Passerida of Sibley and Ahlquist. This study was noteworthy for its phylogenetic delimitation of two groups above the family level in a section of the passerines (Passerida) that is otherwise very depauperate in synapomorphies. The single amino acid insertion was found in all Passerida tested, a result that has now been confirmed in 170 representatives for the clade, essentially confirming its status as a synapomorphy (Ericson & Johansson 2003). The three amino acid insertion was found in Motacillidae (pipits and wagtails), Fringillidae (New World seed eaters), Emberizidae (buntings), Parulidae (New World warblers) and Icteridae (New World blackbirds). New sequence data do not significantly conflict with clades defined by both c-myc insertions (Barker et al. 2002, 2004). Other indels that appear congruent with sequence include a 15 bp deletion in the RAG-1 gene for all Neoaves (all modern birds that are not ratites or Galloanseriforms; Groth & Barrowclough 1999; their Plethornithae). However, RAG-1 also exhibits a large number of other indels in various regions of the gene, only some of which are congruent with other data (Groth & Barrowclough 1999; Barker et al. 2002). β-fibrinogen intron 7 possesses numerous highly informative indels (Prychitko & Moore 2003), which appear to increase in consistency with increasing length (Fain & Houde 2004). Some indels provide resolution where sequence data do not, even at relatively low taxonomic levels (Kimball et al. 2001). Thus, the first wave of indel analyses in genes encoding proteins suggests that these markers will need to be evaluated on a case-by-case basis for congruence with other datasets. Johnson (2004) found that deletions were six times more common than insertions in introns of pigeons and doves (Columbidae); this and the fact that convergent deletions should be more common than convergent insertions may mean that deletions may be less reliable as cladistic markers, but further data are needed to test this hypothesis.
(d) Gene duplications in phylogenetic analysis
Gene duplications and complex gene trees of multigene families are being increasingly used to infer phylogenetic relationships in a variety of animal and plant clades (Mathews & Donoghue 1999; Cotton & Page 2002). Garcia-Moreno & Mindell (2000) used paralogous (‘gametologous’) relationships on the avian sex chromosomes to root trees of copies of the CHD gene found on both the Z and W chromosomes. They retrieved a gene tree implying paleognaths at the base of the avian tree, congruent with other data. Rooting with paralogs is a powerful approach that bypasses many of the traditional problems associated with rooting, such as long-branch attraction, particularly when the paralogs or gametologs have clearly duplicated prior to the diversification of the clade of interest and have accumulated substitutions without gene conversion.
(e) Sources of conflict among genes and the problem of concatenation
There is a growing appreciation that higher‐level molecular studies of birds, despite their focus on processes far above the species level, are still subject to normal processes of population genetics, which can sometimes confound the search for species trees (Edwards 1997). Incomplete lineage sorting (ILS) of gene fragments is a well-known process by which a gene tree can fail to ‘track’ the species tree if the time between speciation events is short (relative to the effective population size along diverging lineages; Avise 2000). What is less well appreciated is that (i) the phylogenetic effects of ILS, although often transient, can in principle become permanent fixtures in the genetic record, rather than eventually ‘sorting out’ as lineages continue to diverge (Avise 2000; Funk & Omland 2003; Poe & Chubb 2004; Shedlock et al. 2004); and (ii) that ILS depends primarily on the length of the internode, regardless of the depth of that internode in the tree. For higher-level avian trees, there is good reason to be sceptical that ILS will cause substantial problems, because the time between divergence events of the exemplars sampled for phylogenetic analysis is often sufficiently long to preclude problems. However, as taxon sampling becomes denser—a worthy goal for any phylogenetic analysis—the chance that gene trees will not track species trees become higher, because the time between divergence events of sampled exemplars becomes smaller. Although focusing on much lower taxonomic levels than those reviewed here, Funk & Omland (2003) found that over 16% of 331 avian species studied in recent years displayed species-level polyphyly or paraphyly of gene (usually mtDNA) lineages. Ongoing studies of nuclear genes in birds may be even more subject than mtDNA to incongruence of gene and species trees, especially when taxa are densely sampled at the level of genera. It is currently unknown how deep in the phylogenetic hierarchy for birds ILS becomes a problem; the suggestion that avian orders diverged in rapid succession from one another means that ILS could be a problem both at the base and at the tips of the avian tree (Poe & Chubb 2004). Patterns of para- and polyphyly deep within trees can also be caused by hybridization, but this process is rarely invoked.
It is common in avian studies analysing multiple nuclear genes to concatenate gene fragments sequenced for a given taxon into a single composite sequence. The motivation behind concatenation is often to better resolve what is assumed to be a single gene tree. With mtDNA, the practice of multigene concatenation is straightforward; even in the unlikely event that mitochondrial recombination is ongoing, such recombination should only take place among mitochondrial lineages within species, and therefore should not strongly compromise higher-level phylogenetic analyses (reviewed in Arbogast et al. 2002). On the other hand, concatenation of independent nuclear genes, or of nuclear and mitochondrial datasets, may under circumstances of ILS or divergent substitution processes have adverse effects on phylogenetic reconstruction. Irestedt et al. (2004) found extensive incongruence between data partitions in antbirds (Thamnophilus) using Bayes factors, but cautioned that limitations of substitution models, rather than incongruence per se, may explain these results (Nylander et al. 2004).
It is still unknown how necessary concatenation of sequences is for increasing resolution of species trees, because alternative methods of estimating species trees have not been explored. An alternative approach, in which the species tree is estimated via combining signal from multiple independent (but non-concatenated) gene trees, has received scant attention in systematics, despite its fundamental utility (Nielsen 1998; Slowinksi & Page 1999; Felsenstein 2003). It may be that the species tree for birds is best inferred without concatenation, by assembling datasets for multiple genes, each of which is long enough to provide some resolution of its own gene tree. Even if each individual gene tree is poorly resolved, recent methods permit the summing up of weak signals in multiple gene trees to provide more robust estimates of tree parameters (Rannala & Yang 2003), an approach that could eventually be extended to tree topologies themselves. In such approaches, conflicts between gene trees are useful data points for estimating branching orders and lineage lengths, rather than sources of error in phylogenetic analysis. The focus of the avian Tree of Life—the species tree, rather than the individual gene tree(s) comprising the avian genome—should always remain paramount.
4. The future: the tools of phylogenomics
(a) Contiguous versus dispersed sequence sampling strategies
Like the PCR in the mid- to late-1980s, the tools of modern genomics promise to revolutionize many fields in systematics and evolutionary biology. These tools offer systematic ornithology the promise of increased throughput, increased sequencing accuracy and a greater integration of phylogenetics and genome evolution. Many of these tools are focused on increasing the efficiency of obtaining long contiguous stretches of DNA sequence, rather than multiple, smaller DNA segments. As such, it is worth asking: what are the consequences of sampling sequence data across multiple loci dispersed across the genome, as is typically achieved in PCR-based studies, as opposed to sampling single long, contiguous sequences, as is typically achieved in shotgun sequencing studies?
Sampling multiple dispersed loci is usually considered a bonus, because contiguous regions will not represent the entire genome and many dispersed sites will incorporate multiple independent rates, base compositions and processes of substitution that when combined will ultimately aid phylogenetic analysis (Cummings et al. 1995; Otto et al. 1996). The question then becomes, is the heterogeneity available in, for example, approximately 40 kb of contiguous avian DNA sequence comparable to that found in approximately 40 kb of dispersed sequence? The draft chicken genome (International Chicken Genome Sequencing Consortium 2004) provides partial answers to these questions, showing that G+C content is higher on micro- than on macrochromosomes, but further analysis is required. For example, Nekrutenko & Li (2000) have posed this question for eukaryotic genomes, including the human genome, and found that compositional heterogeneity is positively correlated with regional GC content, and that regions of compositional homogeneity are typically on the order of 40 kb or more in length, although much smaller than the approximately 300 kb postulated in classical isochore models. Birds may depart from this pattern; indeed, their smaller genomes and high frequency of GC-rich microchromosomes may render them more base-compositionally heterogeneous at smaller scales than mammals. Recent shotgun sequencing studies in chickens, quail and several passerines suggest that substitution processes (as judged by base composition) on the scale of approximately 40 kb are indeed very heterogeneous: GC-content tends to vary dramatically over such regions, with peaks within coding regions and troughs between genes (Kaufman et al. 1999; Gasper et al. 2001; Shiina et al. 2004). However, it would be premature to generalize from these studies, which focused on genomically idiosyncratic and highly dynamic regions containing histocompatibility genes. Regardless, fitting models of sequence evolution is likely to be as challenging for contiguous data as it is for dispersed data; although dispersed data are better for defining data partitions than contiguous data (each gene could naively be considered a separate partition, for example), likelihood, Bayesian or pseudocount analyses of contiguous data may soon be able to define partitions in an iterative manner so as to optimize model fitting and phylogenetic performance.
Sequencing of multiple dispersed regions is likely to remain the tool of choice for phylogenetic questions for purely practical reasons, such as targeting of phylogenetically informative regions and maximizing ease of alignment. For example, Thomas et al. (2003) produced sequences spanning a 1.8 Mb region surrounding the CFTR gene for 12 vertebrates, including chicken and eight mammals. The region contained 10 genes, with the remainder being introns, regulatory regions and non-coding DNA. However, in comparisons of human versus rat or mouse, less than 40% of this region was alignable (and presumably amenable to phylogenetic analysis). Intriguingly, the vast majority of this alignable fraction was non-annotated and presumably outside of coding regions, suggesting that such regions for birds should also be amenable to phylogenetic analysis. With the extreme uniformity of genome size and structure within birds (Burt 2002; Waltari & Edwards 2002), the fraction of alignable sequence in such large-scale comparisons will almost certainly be greater than in mammals. We therefore suggest that both contiguous and dispersed datasets have their inherent advantages for avian phylogenetics, with the former having a greater ability to bridge phylogenetics and comparative genomics. Large-scale contiguous sequencing will also undoubtedly reveal qualitatively new types of character data for birds, such as the discovery and characterization of retroelements and genomic rearrangements (see below).
(b) BAC and other large-insert libraries for birds
BAC libraries—genomic libraries with inserts in the range of 100–200 kb—may have only limited use in avian phylogenetics owing to the considerable time, expertise and laboratory equipment required for construction, storage, manipulation and distribution. Nonetheless, they offer abundant opportunities for large-scale genome analysis, and recent initiatives sponsored by NSF and NIH promise an ever-growing list of libraries for phylogenetically informative taxa (Couzins 2002). BAC libraries for several taxa of birds are currently or soon to be available (white leghorn domestic chicken (Gallus gallus) and wild turkey (Meleagris gallopavo), http://bacpac.chori.org/; zebra finch (Poephila guttata), http://www.genome.gov/10001852; Red Jungle Fowl (Gallus gallus), http://hbz7.tamu.edu/homelinks/bac_est/bac.htm; California Condor (Gymnogyps californianus) and emu (Dromaius novaehollandiae), http://www.benaroyaresearch.org/bri_investigators/amemiya/libraries.htm, http://www.jgi.doe.gov/programmes/comparative/top_level/BAC.html) as well as outgroup taxa (painted turtle (Chrysemys picta), American alligator (Alligator mississipiensis) and Tuatara (Sphenodon punctatus), see JGI Web site above). Together these libraries provide an excellent start to broad taxonomic coverage of major branches of the tree for birds and relatives. Cosmid and fosmid libraries are technically straightforward to construct and should be used more widely within systematic ornithology and other areas of animal phylogenetics (Edwards et al. 2000).
(c) Shotgun sequencing
One potential use of large-insert libraries in avian systematics is for obtaining large-scale sequencing coverage of targeted regions of the genome (Thomas et al. 2003). Shotgun sequencing is the method by which large-insert clones of a specific genomic region can be sequenced in their entirety. It was the method of choice for the chicken and other vertebrate genome projects, but in fact has been used extensively for a variety of research questions for over two decades (Bodenteich et al. 1994). The method has been used in recent years by molecular ecologists interested in acquiring a ‘landscape’ view of their particular genomic regions such as the MHC, and is very useful for learning about the genomic neighbourhood surrounding a particular target gene that has relevance to systematics (Edwards et al. 2000). For example, the RAG-1 locus has recently been shown to deliver a strong phylogenetic signal at a variety of higher levels within birds (Groth & Barrowclough 1999; Paton et al. 2003). It is known that RAG-1 is closely linked to an evolutionarily related and phylogenetically useful locus, RAG-2, in model species in which their chromosomal locations have been investigated (Gellert 1996). Chromosomal proximity of genes of use in phylogenetic inference is of interest to systematists because this knowledge can help clarify how evolutionarily independent two loci are and whether they are contained in the same or different isochores (Barker et al. 2004). Birds appear to be conserved in their chromosomal synteny, even in comparisons with the human genome (Burt et al. 1999; Burt 2002; International Chicken Genome Sequencing Consortium 2004), yet gene order and presence/absence in specific regions will undoubtedly provide useful phylogenetic information.
Shotgun sequencing, particularly when combined with robotics approaches, is becoming faster, simpler and cheaper. Still, ornithologists will ask whether the investment of time and resources is worth the effort. Resolving the avian tree will almost certainly not require a shotgun sequencing approach—the piecemeal PCR approaches in use currently appear adequate for the task. Thus the major reason for considering shotgun sequencing is to integrate phylogenetic analysis and molecular evolution of target regions, as is beginning to be done for mammals (Thomas et al. 2003). Like clone-end sequencing (see below), shotgun sequencing can also provide a wealth of information about potential cladistic markers such as retroposons. Because only a fraction of the DNA sites generated in a typical multi-species large-scale sequencing study can actually be used for phylogenetic analysis, the efficiency with which the avian community can acquire shotgun sequences will undoubtedly influence their impact on the field. However, shotgun sequencing, with its high redundancy and analysis of subcloned DNA, is generally considered more accurate than direct sequencing of PCR products (see § 4(h) below and Fig. 3). An additional result of a shotgun approach is that heterozygous sites are eliminated, yielding fully resolved haplotypes (alleles); depending on how data are analysed, this may or may not be advantageous, since scored polymorphisms can be a help in cases of limited taxon sampling.
(d) Accessing large-scale multi-species sequences
A deceptively challenging hurdle in gaining access to large-scale multi-species sequences is gaining access to regions of the genome that are precisely homologous to one another across an array of species. Traditional library screening (Thomas et al. 2003) may prove cumbersome unless conducted on a scale possible only in genome centres. Recent advances in homologous recombination in yeast offer new hope for rapidly obtaining homologous regions of DNA from a novel genome (Raymond et al. 2002b). These methods take advantage of yeast's tendency to facilitate recombination between two linear templates of double-stranded DNA as part of its natural double-strand break repair system. This recombination is accomplished when three types of DNA fragment are transfected simultaneously into yeast: (i) linearized fragments of genomic DNA from a focal avian species of interest; (ii) 80 bp ‘linkers’ consisting of 40 bp homologous to a vector into which the genomic fragments will be cloned in the yeast and 40 bp homologous to single-copy regions flanking the region of interest in the focal avian species; and (iii) the appropriate subcloning vector (figure 2). The result is the generation of a recombinant DNA clone that contains precisely the region of interest from the focal species and that can then be easily manipulated for sequencing or other characterization. A recent study of genetic variation in the human pathogen Pseudomonas aeruginosa used this approach to sequence the homologous region of the O-antigen biosynthetic locus in 20 isolates (Raymond et al. 2002a). Although the locus varied in length from 5 to 25 kb between isolates and no phylogenies were built, the region was precisely delimited by homologous flanking sequences and contained a multigene family whose members were alignable to one another and amenable to phylogenetic analysis.
Figure 2.

Subcloning of large DNA fragments via yeast-mediated recombination (after Raymond et al. 2002b). The method is useful for subcloning a specific region of DNA from a larger BAC clone or in principle from genomic DNA. Linearized DNA from a focal species is depicted at the top; pairs of symbols on DNA strands indicate unique 40 bp motifs present throughout the genome; these need not occur at the ends of the linear DNA but can occur anywhere within the DNA segments. The open square and black diamond on the focal DNA segment are known to flank precisely the region of interest. These motifs are incorporated into the single-stranded linkers, the other half of which is homologous to ends of the vector into which the fragment will be cloned (bottom). The 80 bp linker mediates recombination between the linear fragments and the vector. The resulting product is a recombinant clone containing precisely the DNA between the flanking 40 bp motifs in the original DNA fragment, which can then be manipulated for DNA sequencing. (Modified from fig. 2 of Raymond et al. (2002b)).
The relevance of these methods for avian systematics is that they would allow the researcher to capture from any species of interest large segments of DNA—much larger than can be routinely amplified by long PCR techniques—that are also precisely homologous to one another. The only requirement is knowledge of approximately 40 bp of sequence flanking both sides of the target region of interest—information that is rapidly accumulating as new genomic regions and genome projects are advancing for birds. Such an approach is still a long way away from being implemented into comparative biology of birds or other vertebrates. Still, the discovery of conserved non-genic sequences in the mammalian genome that are even more conserved than exons (Dermitzakis et al. 2003), combined with suggestions for increased synteny in avian versus mammalian genomes, suggest that the recombinational cloning approach could work well for avian comparative genomics. Other sequencing methods, such as chip-based sequencing, may also find relevance in large-scale systematics projects, although such methods have generally been used for comparison of closely related genomes, and the ease with which distantly related genomes can be sequenced and compared in this way is still unclear (Shendure et al. 2004).
Aligning and analysing large genomic regions across multiple species is another area of comparative genomics that ornithologists will eventually need to grapple with. Fortunately, there are a number of new and powerful bioinformatics tools specifically designed for comparative analysis in a phylogenetic framework (Miller et al. 2004). For example, the Ensemble (http://www.ensembl.org/) and University of California Santa Cruz Genome Browser (http://genome.ucsc.edu/cgi-bin/hgGateway) are two gateways for a variety of tools for comparative genomics. Out of the many useful tools becoming available, we mention two that appear particularly promising in their ability to handle very long (multimegabase) sequences for multiple species: Phylo-Vista (Shah et al. 2004) and MAVID (Bray & Pachter 2004), two relatively new platforms for conducting large-scale comparative sequence analysis. Our review of available bioinformatics tools is far from complete, but we predict that, because of the higher gene density and greater genomic synteny within birds, large-scale alignment and comparative sequence analysis in birds will be relatively straightforward compared with mammals.
(e) Plasmid-, cDNA- and BAC-end sequencing
Sequences gleaned from the ends of cloned inserts can be an important source of phylogenetic information. When performed on a large scale, such an approach can yield literally hundreds, if not thousands, of stretches of DNA for which PCR primers can be immediately designed. The vast majority of loci produced in this way will be from the non-coding portion of the genome when a BAC library is used as a starting point, and will encompass a variety of non-coding and intergenic genomic features, including retroposons and regulatory regions. By contrast, when a cDNA library is used to generate so-called expressed sequence tags, the loci are all expressed, in most cases translated into functional proteins, and likely to be conserved and unambiguously alignable across birds, particularly given the slow rate of molecular evolution of avian‐coding regions (Mindell et al. 1996). A number of Tree of Life projects are pursuing this strategy, particularly for invertebrate clades (G. Giribet, personal communication). Large collections of clone-end sequences, or heterogeneous sequences from databases, can also be used to estimate phylogenies using motif-counting approaches, even when the sampled loci do not overlap between species (Stuart et al. 2002; Chu et al. 2004). Such methods have recovered at least one major branch within birds, that separating Paleognaths from Neognaths (Edwards et al. 2002), and their power may lie most clearly in exploration of patterns of genome evolution and motif usage. As strictly phylogenetic tools, their utility is unclear because there are no clear models of character change and identifying homologies and characters is problematic.
Because end-sequencing will provide loci for only one species in a given clade, it is not guaranteed that all loci will be amplifiable across the clade of interest (which for Neoaves probably encompasses more than 100 Myr). Presumably such loci would be more challenging to characterize across all birds than would exon-spanning intron crossing loci, which use conserved sequences in exons flanking introns to anchor PCR primers. Still, many non-coding anonymous regions are now known to be highly conserved between major lineages of mammals (Boffelli et al. 2003; Margulies et al. 2003; Thomas et al. 2003). The compact genomes of birds, with their much lower fraction of repetitive DNA, will lend themselves even better to this approach.
(f) Phylogenetic utility of gene order and synteny
Although data are still sparse, a few studies have suggested that the preservation of gene order along avian chromosomes may be highly conserved and substantially higher than in mammals. For example, a number of studies have documented strong correspondence between whole chromosomes of different bird species using fluorescent in situ hybridization techniques (Shetty et al. 1999; Raudsepp et al. 2002; Guttenbach et al. 2003). Despite the apparent conservatism of the avian karyotype, these studies have also uncovered examples of chromsome fusion and fission, and homologies between macro- and microchromosomes (Gru¨tzner et al. 2001; Nanda et al. 2002). Although the era of avian ‘cytophylogenetics’ has barely begun, the field can take confidence from new methodological and analytical advances that promise to speed up data acquisition and analysis considerably, as well as examples from mammals (Robinson et al. 2004) that clearly demonstrate the utility of karyological data for higher‐level systematics.
(g) Retroelements in avian systematics
Comparative genomics provides a platform for investigating the vast number of dispersed mobile repetitive DNA elements in eukaryotes that collectively may drive genome evolution and shape genome architecture to a much greater degree than previously appreciated (Weiner 2002; Kazazian 2004). Mobile elements are classified into two broad categories: (i) DNA transposons, so-called ‘jumping genes’, that autonomously relocate via a ‘cut-and-paste’ mechanism; and (ii) retroposons, which rely on a replicative ‘copy-and-paste’ process that requires an RNA intermediate to support movement of newly amplified copies of elements from a parent to target locus (Weiner et al. 1986; Kajikawa & Okada 2002). Retroposons are further divided into groups of long and short interspersed elements (SINEs and LINEs, respectively) that are of particular interest to evolutionary biologists because copies of these molecules shared at the same locus in two different taxa are derived from the same element originally inserted into the germline of a common ancestor, and thus can be used as clade markers (Shedlock & Okada 2000; Okada et al. 2003; Shedlock et al. 2004).
Because retroposons are not precisely removed from the genome, have a known ancestral condition (i.e. absence of insertion at a particular locus), do not use a specific sequence recognition site for insertion, and are identical by descent, they hold considerable promise for inferring systematic relationships with minimal noise owing to character reversal or parallel insertion events at the same locus in different lineages (Batzer & Deininger 2002). The utility of a given retroelement for inferring phylogeny depends critically on its taxonomic distribution and the profile of element diversification in the genome during the evolutionary time period in question (Shedlock & Okada 2000; Shedlock et al. 2004). Practical drawbacks to retroposon analysis include mutational decay of reliable PCR priming sites for loci in relatively old taxa that have diverged well beyond approximately 100 Myr. However, the traditional and somewhat cumbersome cloning methods for isolating phylogenetically informative elements for many taxa will probably be superceded by large-scale end-sequencing surveys, which can yield hundreds of elements with little effort, as well as bioinformatics analyses of model genomes (Shedlock et al. 2004).
Initial study of the molecular biology of retroposons in humans and in experimentally important mammal genomes is now being extended to non-model organisms (Malik et al. 1999; Smit 1999; Kajikawa & Okada 2002; Okada et al. 2003; Thomas et al. 2003). The case for studying retroelements in birds is being primed by completion of the chicken genome and, in particular, the characterization of CR1 retrotransposons, a large family of mobile chicken repeats that appear to be distributed widely among vertebrates (Chen et al. 1991; Burch et al. 1993; International Chicken Genome Sequencing Consortium 2004). CR1s were originally considered SINEs based on the extensive truncation of their 5′ ends; however, longer CR1s have now been identified that contain the characteristic open reading frames of LINEs. Although birds have substantially reduced genome size relative to mammals, it is estimated from the draft chicken genome that ∼200 000 CR1s exist in the chicken (Wicker et al. 2004; International Chicken Genome Sequencing Consortium 2004) and it is reasonable to expect that diagnostic subgroups and related CR1-like elements will be readily characterized in diverse bird species as well as in other reptilian taxa. Experimental access to a wide variety of avian retroelements is being facilitated by the influx of comprehensive chicken genome data and by expanded sequencing of large-insert BAC libraries constructed for investigating avian genomes. Cot clone sequences have recently been used to rapidly survey the diversity of CR1 LINEs in over 2.8 Mb of the high-copy, repetitive fraction of the chicken genome (Wicker et al. 2004). Sequencing of songbird cosmid clones have already yielded CR1 and L1 elements that may prove to be phylogenetically useful (Hess et al. 2000; Gasper et al. 2001). Likewise, subfamilies of retroposons such those in the Pol-III family of SINEs previously characterized in turtles may also provide useful systematic information for bird evolutionary studies (Kajikawa et al. 1997; Sasaki et al. 2004b). Moreover, novel families of SINEs not detected thus far in chickens have already been isolated in penguins and lizards by conventional genomic library screening and are providing useful phylogenetic information for each of these groups (N. Okada, unpublished data).
Making the best use of new data from mobile elements for solving problems in avian systematics will require a targeted strategy of integrating retroposon insertion patterns of diagnostic subfamilies of SINEs and LINEs with results from other molecular, morphological and palaeontological studies, as has been done for the cetacean-artiodactyl mammal radiations (Nikaido et al. 1999, 2001). Parallels in the evolutionary radiation of birds and mammals since the late Cretaceous have been noted previously (Hedges et al. 1996; Harrison et al. 2004a), and we are optimistic that access to new phylogenetically informative CR1-like elements will help resolve patterns of avian macroevolution over the past 100 Myr, especially in light of recent results for mammals and fishes (Shedlock & Okada 2000). Retroposons are just as susceptible to incomplete lineage sorting and the challenges of short internodes as single nucleotide changes; however, because of their negligible rate of convergence and researchers' ability to molecularly dissect putative cases of convergence, even a single retroposon integration arguably provides more certain evidence of synapomorphy than do a plethora of nucleotide sequences. Finally, in addition to contributing to a more accurate tree for birds, avian retroposon dynamics should provide a critical comparison to mammalian and non-avian reptiles as we test alternative hypotheses of vertebrate genome evolution. Characterizing the density and distribution of mobile DNA elements in different lineages of modern birds will provide insight into the molecular pathways underlying the nearly fivefold range in genome size apparent among living amniotes. As such, the study of retroelements offers an opportunity to enrich our understanding of not only the pattern, but also the process, of avian diversification.
(h) Chromatogram analysis and sequence accuracy
Sequence accuracy is an issue with obvious relevance to avian systematics. Data quality and sequencing errors are undeniably an issue in the molecular systematics of birds: a host of examples of chimeric DNA sequences, nuclear copies of putatively mtDNA (numts) and simple sequencing errors have surfaced in the 15 year application of PCR (Avise et al. 1995; Edwards & Arctander 1996; see comments in Sorenson et al. 2003). In addition, the analysis of nuclear gene sequence data in birds is complicated somewhat by the presence of heterozygous sites and the need to incorporate such sites into phylogenetic analysis (see below; such ‘heterozygous’ sites are also present in mtDNA sequences in the form of cryptic heteroplasmy (Rieder et al. 1998), but are generally less frequent and go unreported in most avian studies). Heterozygosity is an issue that is only inconsistently reported in avian phylogenetic studies of nuclear genes. Groth and Barrowclough (1999) deemed 42 out of 46 114 sites (approximately 0.1%) as heterozygous; they found up to nine differences between putative alleles within individuals and an average interallelic divergence of 2.6 differences (0.09%). Barker et al. (2002) and Sorenson et al. (2003) used ambiguity codes and specified their treatment as polymorphisms, an approach that would have particular power using transversion-parsimony and in ML analyses.
The tools of sequence capture and chromotogram analysis currently used in genome projects can help provide a standardized protocol for identifying heterozygous sites in avian studies, and can also aid in reducing sequencing errors (Nickerson et al. 1997; Ewing & Green 1998; Rieder et al. 1998). However, such tools have been used in only a handful of studies on birds (reviewed in Brumfield et al. 2003), and not yet for an avian systematic study. For example, Phred/Phrap is a standard chromatogram analysis tool that analyses a variety of commonly employed sequence trace types (such as ABI chromatograms; Ewing & Green 1998). Indeed, Phred/Phrap is known to call bases from raw chromatogram data more accurately than does the software frequently provided with DNA sequencers. Phred/phrap performs a variety of sequence trimming and aligning functions, and, crucially, provides an objective, chromatogram-based quality value (QV) for each sequenced nucleotide. The software can combine the QVs for a given nucleotide across multiple reads to provide a consensus QV for a site. In principle, QVs could be directly integrated into phylogenetic analysis itself, and, if present in the avian databases would provide a uniform standard by which sequence quality could be judged.
A display of QVs measured for a 2000 bp segment of mtDNA from a downy woodpecker is shown in figure 3. The sequence was obtained from shotgun sequencing of a cloned fragment of downy woodpecker DNA (S. Edwards, J. Gasper, W. Nelson, J. Avise and D. Pollock, unpublished data); thus most nucleotides were sequenced in multiple chromotogram traces. The display indicates that overall the QVs of each nucleotide are very high, indicating a high overall confidence in the sequence. Many of the QVs are higher than 50, indicating that these bases have a 1 in 100 000 chance of being incorrect. Evaluation of an avian DNA sequence in this way could help to identify numts, whose presence is frequently detected in heterogeneous or low‐quality sequencing reads (Sorenson & Quinn 1998), and could help to reduce other routine sequencing errors that are undoubtedly present in the avian database, as they are in the databases for humans and other species (Clark & Whittam 1992). Phred/phrap QV analysis can be useful for more traditional small‐scale analyses as well; it does not require multiple overlapping reads and can be profitably applied to traditional single-read direct sequences.
Figure 3.
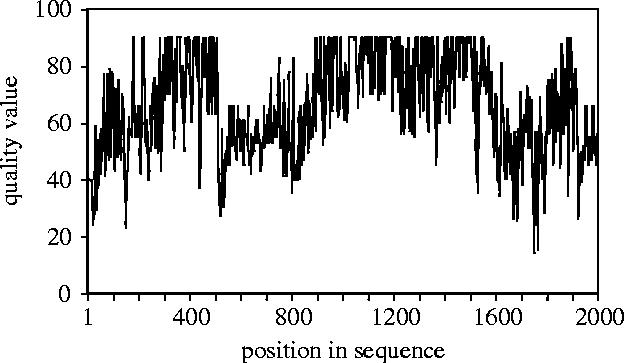
Plot of quality values (QVs) for approximately 2000 bp of mitochondrial DNA from a downy woodpecker (S. Edwards, unpublished data). The sequence was determined by the shotgun approach starting with a long PCR product (S. Edwards, J. Gasper, W. Nelson, J. Avise and D. Pollock, unpublished data). QVs are based on details of peak morphology, height and consistency across similar nucleotides both in the trace under scrutiny as well as across traces spanning homologous bases, and are equivalent to QV=−10 log(Pe), where Pe is the probability that the base is an error. Thus a QV of 30 indicates that there is a 1 in 1000 chance that the base in question is incorrect, or, alternatively that the probability of a correct call is 99.9%. Typically, NIH genome projects require a QV of 40, although some genome publications have used minimum QVs of 20. The flatness of the threshold of QV at approximately 90 is a function of the base‐calling method.
(i) Community genomics
Comparative genomics approaches lend themselves well to community efforts and pooling of resources, although the sheer complexity of some of the protocols would necessarily entail some centralization in the early stages of such projects. For example, BAC and other large-insert libraries would need to be produced by a centralized facility, as they are now, as would the preparation of shotgun subclone libraries. However, the sequencing of individual BAC subclones could easily be parcelled out to different laboratories, since this involves standard sequencing methodology (figure 4). For example, individual laboratories might focus on BACs prepared from focal taxa of particular interest to that laboratory. The approach outlined in figure 4 is undoubtedly slower and less cost-effective than sequencing of targeted regions of focal taxa by large genome centres, but acknowledges that many of the questions that may interest avian systematists may never achieve the priority required for investment by genome centres. Although such community-wide collaborative arrangements are a long way off—the Green Plant Phylogeny Research Coordination Group still sets the standard for inclusiveness and networking among systematists—such an arrangement would generate a greater atmosphere of community effort than do current practices.
Figure 4.

A community genomics approach to producing large-scale multispecies sequences from targeted genomic regions. The approach is based on the assumption that the focal species are from non-model clades of interest primarily to systematists and evolutionary biologists, and is tailored to questions that interest systematists, but could not achieve the priority required for tackling by a genome centre. The programme emphasizes utilization of genomics resources from non-model species in such a way as to maximize involvement by the systematic community while minimizing reliance on large genome centres to produce the actual data. The programme envisions preparation of source libraries and templates for DNA sequences by genome centres, which then transfer subclones widely throughout the community of systematists. Individual laboratories would be capable of sequencing the subclones required to cover one or a few BAC clones. The figure of 2500 subclones assumes approximately 7–8-fold coverage of a ∼150 kb BAC clone. The approach is undoubtedly slower and less cost-effective than sequencing of the same regions by a large genome centre, but is able to achieve greater inclusiveness and taxonomic coverage than current practice.
5. Conclusion
Regardless of the technologies chosen for the Tree of Life for birds, a major and well-known obstacle is the apparently rapid diversification of most modern avian orders in the late Cretaceous, perhaps facilitated by isolation of multiple lineages following continental break-up (Poe & Chubb 2004; Cracraft 2004). Indeed, some authors have proposed the existence of hard polytomies among major clades of the Neoaves in the absence of more convincing evidence (Poe & Chubb 2004), although others remain optimistic that the short internodes now frustrating avian systematists will be slowly and steadily resolved with confidence given more data. This review has evaluated various conceptual and genomics approaches of relevance to future avian systematic studies. In addition to abetting the avalanche of primary sequence data expected in the next few years, we encourage exploration of new types of characters and large-scale approaches in the interest of both phylogenetics and genome evolution.
Acknowledgments
We thank Charles Godfrey for inviting this review and John Langdon for editorial assistance. K. Barker, J. Harshman, S. Hackett, K. Omland, U. Johansson, I. Lovette, G. Barrowclough, G. Voelker, J. Cracraft, M. Sorenson, G. Giribet, J. Kulski, T. Shiina and H. Ellegren provided helpful discussion, and J. Cracraft and G. Spicer gave us advance access to unpublished information. The manuscript benefited greatly from comments by J. Avise, E. Braun, M. Sorenson, J. Harshman, P. Ericson and two very insightful anonymous reviewers. This work was supported in part by NSF grants DEB-0108249 and IBN-0207870 to S.V.E.
Supplementary Material
References
- Arbogast B.S, Edwards S.V, Wakeley J, Beerli P, Slowinski J.B. Estimating divergence times from molecular data on population genetic and phylogenetic time scales. Annu. Rev. Ecol. Syst. 2002;33:707–740. [Google Scholar]
- Avise J.C. Harvard University Press; Cambridge, MA: 2000. Phylogeography: the history and formation of species. [Google Scholar]
- Avise J, Nelson W, Sibley C. Correction: DNA sequence support for a close phylogenetic relationship between some storks and New World vultures. Proc. Natl Acad. Sci. USA. 1995;92:3076. doi: 10.1073/pnas.91.11.5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker F.K, Barrowclough G.F, Groth J.G. A phylogenetic hypothesis for passerine birds: taxonomic and biogeographic implications of an analysis of nuclear DNA sequence data. Proc. R. Soc. B. 2002;269:295–308. doi: 10.1098/rspb.2001.1883. doi:10.1098/rspb.2001.1883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker F.K, Cibois A, Schikler P, Feinstein J, Cracraft J. Phylogeny and diversification of the largest avian radiation. Proc. Natl Acad. Sci. USA. 2004;101:11 040–11 045. doi: 10.1073/pnas.0401892101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzer M, Deininger P.L. Alu repeats and human genomic diversity. Nature Rev. Genet. 2002;3:370–379. doi: 10.1038/nrg798. [DOI] [PubMed] [Google Scholar]
- Birks S.M, Edwards S.V. A phylogeny of the megapodes (Aves: Megapodidae) based on nuclear and mitochondrial DNA sequences. Mol. Phylogenet. Evol. 2002;23:408–421. doi: 10.1016/s1055-7903(02)00002-7. [DOI] [PubMed] [Google Scholar]
- Bodenteich A, Chissoe S, Wang Y.-F, Roe B.A. Shotgun cloning as the strategy of choice to generate templates for high-throughput dideoxynucleotide sequencing. In: Adams M.D, Fields C, Ventner J.C, editors. In Automated DNA sequencing and analysis. Academic Press; San Diego, CA: 1994. pp. 42–50. [Google Scholar]
- Boffelli D, McAuliffe J, Ovcharenko D, Lewis K.D, Ovcharenko I, Pachter L, Rubin E.M. Phylogenetic shadowing of primate sequences to find functional regions of the human genome. Science. 2003;299:1391–1394. doi: 10.1126/science.1081331. [DOI] [PubMed] [Google Scholar]
- Boore J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999;27:1767–1780. doi: 10.1093/nar/27.8.1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun E.L, Kimball R.T. Examining basal avian divergences with mitochondrial DNA sequences: model complexity, taxon sampling and sequence length. Syst. Biol. 2002;51:614–625. doi: 10.1080/10635150290102294. [DOI] [PubMed] [Google Scholar]
- Bray N, Pachter L. MAVID: constrained ancestral alignment of multiple sequences. Genome Res. 2004;14:693–699. doi: 10.1101/gr.1960404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brumfield R, Nickerson D, Beerli P, Edwards S.V. The utility of single nucleotide polymorphisms in inferences of population history. Trends Ecol. Evol. 2003;18:249–256. [Google Scholar]
- Burch J.B.E, Davis D.L, Haas N.B. Chicken repeat 1 elements contain a pol-like open reading frame and belong to the non-long terminal repeat class of retrotransposons. Proc. Natl Acad. Sci. USA. 1993;90:8199–8203. doi: 10.1073/pnas.90.17.8199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burt D.W. Origin and evolution of avian microchromosomes. Cytogenet. Genome Res. 2002;96:97–112. doi: 10.1159/000063018. [DOI] [PubMed] [Google Scholar]
- Burt D.W, et al. The dynamics of chromosome evolution in birds and mammals. Nature. 1999;402:411–413. doi: 10.1038/46555. [DOI] [PubMed] [Google Scholar]
- Chen Z.-Q, Ritzel R.G, Lin C.C, Hodgetts R.B. Sequence conservation in avian CR1: an interspersed repetitive DNA family evolving under functional constraints. Proc. Natl Acad. Sci. USA. 1991;88:5814–5818. doi: 10.1073/pnas.88.13.5814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu K.H, Qi J, Yu Z.-G, Anh V. Origin and phylogeny of chloroplasts revealed by a simple correlation analysis of complete genomes. Mol. Biol. Evol. 2004;21:200–206. doi: 10.1093/molbev/msh002. [DOI] [PubMed] [Google Scholar]
- Clark A, Whittam T. Sequencing errors and molecular evolutionary analysis. Mol. Biol. Evol. 1992;9:744–752. doi: 10.1093/oxfordjournals.molbev.a040756. [DOI] [PubMed] [Google Scholar]
- Cooper A, Penny D. Mass survival of birds across the Cretaceous-tertiary boundary: molecular evidence. Science. 1997;275:1109–1113. doi: 10.1126/science.275.5303.1109. [DOI] [PubMed] [Google Scholar]
- Cotton J.A, Page R.D. Going nuclear: gene family evolution and vertebrate phylogeny reconciled. Proc. R. Soc. B. 2002;269:1555–1561. doi: 10.1098/rspb.2002.2074. doi:10.1098/rspb.2002.2074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couzins J. NSF's ark draws alligators, algae and wasps. Science. 2002;297:1638–1639. doi: 10.1126/science.297.5587.1638. [DOI] [PubMed] [Google Scholar]
- Cracraft J. Avian evolution, Gondwana biogeography and the Cretaceous–Tertiary mass extinction event. Proc. R. Soc. B. 2001;268:459–469. doi: 10.1098/rspb.2000.1368. doi:10.1098/rspb.2000.1368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cracraft J, et al. Phylogenetic relationships among modern birds (Neornithes) In: Cracraft J, Donoghue M.J, editors. In Assembling the tree of life. Oxford University Press; New York: 2004. pp. 468–489. [Google Scholar]
- Cummings M.P, Otto S.P, Wakeley J. Sampling properties of DNA sequence data in phylogenetic analysis. Mol. Biol. Evol. 1995;12:814–822. doi: 10.1093/oxfordjournals.molbev.a040258. [DOI] [PubMed] [Google Scholar]
- Dermitzakis E.T, Reymond A, Scamuffa N, Ucla C, Kirkness E, Rossier C, Antonarakis S.E. Evolutionary discrimination of mammalian conserved non-genic sequences (CNGs) Science. 2003;302:1033–1035. doi: 10.1126/science.1087047. [DOI] [PubMed] [Google Scholar]
- Desjardins P, Morais R. Sequence and gene organization of the chicken mitochondrial genome, a novel gene order in higher vertebrates. J. Biol. Chem. 1990;121:599–634. doi: 10.1016/0022-2836(90)90225-B. [DOI] [PubMed] [Google Scholar]
- Edwards S.V. Relevance of microevolutionary processes for higher level molecular systematics. In: Mindell D.P, editor. In Avian molecular systematics and evolution. Academic Press; New York: 1997. pp. 251–278. [Google Scholar]
- Edwards S.V, Arctander P. Retraction of a thrush (Catharus guttatus) cytochrome b sequence. Proc. R. Soc. B. 1996;263:323. doi: 10.1098/rspb.1996.0049. [DOI] [PubMed] [Google Scholar]
- Edwards S.V, Boles W.E. Out of Gondwana: the origin of passerine birds. Trends Ecol. Evol. 2002;17:347–349. [Google Scholar]
- Edwards S.V, Wilson A.C. Phylogenetically informative length polymorphism and sequence variability in mitochondrial DNA of Australian songbirds (Pomatostomus) Genetics. 1990;126:695–711. doi: 10.1093/genetics/126.3.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S.V, Arctander P, Wilson A.C. Mitochondrial resolution of a deep branch in the genealogical tree for perching birds. Proc. R. Soc. B. 1991;247:99–107. doi: 10.1098/rspb.1991.0017. [DOI] [PubMed] [Google Scholar]
- Edwards S.V, Nusser J, Gasper J. Characterization and evolution of Mhc genes from non-model organisms, with examples from birds. In: Baker A.J, editor. In Molecular methods in ecology. Blackwell Scientific; Cambridge: 2000. pp. 168–207. [Google Scholar]
- Edwards S.V, Fertil B, Giron A, Deschavanne P.J. A genomic schism in birds revealed by phylogenetic analysis of DNA strings. Syst. Biol. 2002;51:599–613. doi: 10.1080/10635150290102285. [DOI] [PubMed] [Google Scholar]
- Ericson P.G.P, Johansson U.S. Phylogeny of Passerida (Aves: Passeriformes) based on nuclear and mitochondrial sequence data. Mol. Phylogenet. Evol. 2003;29:126–138. doi: 10.1016/s1055-7903(03)00067-8. [DOI] [PubMed] [Google Scholar]
- Ericson P.G.P, Johansson U.S, Parsons T.J. Major divisions in oscines revealed by insertions in the nuclear gene c-myc: a novel gene in avian phylogenetics. Auk. 2000;117:1069–1078. [Google Scholar]
- Ericson P.G, Christidis L, Cooper A, Irestedt M, Jackson J, Johansson U.S, Norman J.A. A Gondwanan origin of passerine birds supported by DNA sequences of the endemic New Zealand wrens. Proc. R. Soc. B. 2002;269:235–241. doi: 10.1098/rspb.2001.1877. doi:10.1098/rspb.2001.1877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B, Green P. Base-calling of automated sequencer traces using PHRED. II. Error probabilities. Genome Res. 1998;8:186–194. [PubMed] [Google Scholar]
- Fain M.G, Houde P. Parallel radiations in the primary clades of birds. Evolution. 2004;58:2558–2573. doi: 10.1111/j.0014-3820.2004.tb00884.x. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. Sinauer Associates, Inc.; Sunderland, MA: 2003. Inferring phylogenies. [Google Scholar]
- Funk D.J, Omland K.E. Species-level paraphyly and polyphyly: frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2003;34:397–423. [Google Scholar]
- Garcia-Moreno J, Mindell D.P. Rooting a phylogeny with homologous genes on opposite sex chromosomes (gametologs): a case study using avian CHD. Mol. Biol. Evol. 2000;17:1826–1832. doi: 10.1093/oxfordjournals.molbev.a026283. [DOI] [PubMed] [Google Scholar]
- Gasper J, Shiina T, Inoko H, Edwards S.V. Songbird genomics: analysis of 45-kb upstream of a polymorphic Mhc class II gene in Red-winged Blackbird (Agelaius phoeniceus) Genomics. 2001;75:26–34. doi: 10.1006/geno.2001.6596. [DOI] [PubMed] [Google Scholar]
- Gellert M. Recent advances in understanding V(D)J recombination. Adv. Immunol. 1996;64:39–64. doi: 10.1016/s0065-2776(08)60886-x. [DOI] [PubMed] [Google Scholar]
- Gru¨tzner F, Zend-Ajusch E, Stout K, Munsche S, Niveleau A, Nanda I, Schmid M, Haaf T. Chicken microchromosomes are hypermethylated and can be identified by specific painting probes. Cytogenet. Cell Genet. 2001;93:265–269. doi: 10.1159/000056996. [DOI] [PubMed] [Google Scholar]
- Groth J.G, Barrowclough G.F. Basal divergences in birds and the phylogenetic utility of the nuclear RAG-1 gene. Mol. Phylogenet. Evol. 1999;12:115–123. doi: 10.1006/mpev.1998.0603. [DOI] [PubMed] [Google Scholar]
- Guttenbach M, Nanda I, Feichtinger W, Masabanda J.S, Griffin D.K, Schmid M. Comparative chromosome painting of chicken autosomal paints 1–9 in nine different bird species. Cytogenet. Genome Res. 2003;103:173–184. doi: 10.1159/000076309. [DOI] [PubMed] [Google Scholar]
- Härlid A, Arnason U. Analyses of mitochondrial DNA nest ratite birds within the Neognathae: supporting a neotenous origin of ratite morphological characters. Proc. R. Soc. B. 1999;266:305–309. doi:10.1098/rspb.1999.0638 [Google Scholar]
- Härlid A, Janke A, Arnason U. The mtDNA sequence of the ostrich and the divergence between paleognathous and neognathous birds. Mol. Biol. Evol. 1997;14:754–761. doi: 10.1093/oxfordjournals.molbev.a025815. [DOI] [PubMed] [Google Scholar]
- Härlid A, Janke A, Arnason U. The complete mitochondrial genome of Rhea americana and early avian divergences. J. Mol. Evol. 1998;46:669–679. doi: 10.1007/pl00006347. [DOI] [PubMed] [Google Scholar]
- Harrison G.L, McLenachan P.A, Phillips M.J, Slack K.E, Cooper A, Penny D. Four new mitochondrial genomes help get to basic evolutionary questions in the late Cretaceous. Mol. Biol. Evol. 2004a;21:974–983. doi: 10.1093/molbev/msh065. [DOI] [PubMed] [Google Scholar]
- Harrison G.L, McLenachan P.A, Phillips M.J, Slack K.E, Cooper A, Penny D. Four new avian mitochondrial genomes help get to basic evolutionary questions in the late Cretaceous. Mol. Biol. Evol. 2004b;21:974–983. doi: 10.1093/molbev/msh065. [DOI] [PubMed] [Google Scholar]
- Hedges S.B. Molecular evidence for the origin of birds. Proc. Natl Acad. Sci. USA. 1994;91:2621–2624. doi: 10.1073/pnas.91.7.2621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedges S.B, Parker P.H, Sibley C.G, Kumar S. Continental breakup and the ordinal diversification of birds and mammals. Nature. 1996;381:226–229. doi: 10.1038/381226a0. [DOI] [PubMed] [Google Scholar]
- Hess C.M, Gasper J, Hoekstra H, Hill C, Edwards S.V. MHC class II pseudogene and genomic signature of a 32-kb cosmid in the house finch (Carpodacus mexicanus) Genome Res. 2000;10:613–623. doi: 10.1101/gr.10.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillis D.M. Taxonomic sampling, phylogenetic accuracy, and investigator bias. Syst. Biol. 1998;47:3–8. doi: 10.1080/106351598260987. [DOI] [PubMed] [Google Scholar]
- Hillis D.M, Pollock D.D, McGuire J.A, Zwickl D.J. Is sparse taxon sampling a problem for phylogenetic inference? Syst. Biol. 2003;52:124–126. doi: 10.1080/10635150390132911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International Chicken Genome Sequencing Consortium. Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature. 2004;432:695–716. doi: 10.1038/nature03154. [DOI] [PubMed] [Google Scholar]
- Irestedt M, Johansson U.S, Parsons T.J, Ericson P.G.P. Phylogeny of major lineages of suboscines (Passeriformes) analysed by nuclear DNA sequence data. J. Avian Biol. 2001;32:15–25. [Google Scholar]
- Irestedt M, Fjeldså J, Nylander J.A.A, Ericson P.G.P. Phylogenetic relationships of typical antbirds (Thamnophilidae) and test of incongruence based on Bayes factors. BMC Evol. Biol. 2004;4:23. doi: 10.1186/1471-2148-4-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson K.P. Taxon sampling and the phylogenetic position of passeriformes: evidence from 916 avian cytochrome b sequences. Syst. Biol. 2001;50:128–136. [PubMed] [Google Scholar]
- Johnson K.P. Deletion bias in avian introns over evolutionary timescales. Mol. Biol. Evol. 2004;21:599–602. doi: 10.1093/molbev/msh051. [DOI] [PubMed] [Google Scholar]
- Kajikawa M, Okada N. LINEs mobilize SINEs in the Eel through a shared 3′ sequence. Cell. 2002;111:433–444. doi: 10.1016/s0092-8674(02)01041-3. [DOI] [PubMed] [Google Scholar]
- Kajikawa M, Ohshima K, Okada N. Determination of the entire sequence of turtle CR1: the first open reading frame of the turtle CR1 element encodes a protein with a novel zinc finger motif. Mol. Biol. Evol. 1997;14:1206–1217. doi: 10.1093/oxfordjournals.molbev.a025730. [DOI] [PubMed] [Google Scholar]
- Kaufman J, Milne S, Göbel, Walker B.A, Jacob J.P, Auffrey C, Zoorob R, Beck S. The chicken B locus is a minimal-essential major histocompatibility complex. Nature. 1999;401:923–925. doi: 10.1038/44856. [DOI] [PubMed] [Google Scholar]
- Kazazian H.H.J. Mobile elements: drivers of genome evolution. Science. 2004;303:1626–1632. doi: 10.1126/science.1089670. [DOI] [PubMed] [Google Scholar]
- Kimball R.T, Braun E.L, Ligon J.D, Lucchini V, Randi E. A molecular phylogeny of the peacock-pheasants (Galliformes: Polyplectron spp.) indicates loss and reduction of ornamental traits and display behaviours. Biol. J. Linn. Soc. 2001;73:187–198. [Google Scholar]
- Kocher T.D, Thomas W.K, Meyer A, Edwards S.V, Pääbo S, Villablanca F.X, Wilson A.C. Dynamics of mitochondrial DNA evolution in animals: amplification and sequencing with conserved primers. Proc. Natl Acad. Sci. USA. 1989;86:6196–6200. doi: 10.1073/pnas.86.16.6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik H.S, Burke W.D, Eickbush T. The age and evolution of non-LTR retrotransposable elements. Mol. Biol. Evol. 1999;16:793–805. doi: 10.1093/oxfordjournals.molbev.a026164. [DOI] [PubMed] [Google Scholar]
- Margulies E.H, Blanchette M, Haussler D, Green E.D. Identification and characterization of multi-species conserved sequences. Genome Res. 2003;13:2507–2518. doi: 10.1101/gr.1602203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews S, Donoghue M.J. The root of angiosperm phylogeny inferred from duplicate phytochrome genes. Science. 1999;286:947–950. doi: 10.1126/science.286.5441.947. [DOI] [PubMed] [Google Scholar]
- Matthee C.A, Burzlaff J.D, Taylor J.F, Davis S.K. Mining the mammalian genome for artiodactyl systematics. Syst. Biol. 2001;50:367–390. [PubMed] [Google Scholar]
- Miller W, Makova K.D, Nekrutenko A, Hardison R.C. Comparative genomics. Annu. Rev. Genomics Hum. Genet. 2004;5:15–56. doi: 10.1146/annurev.genom.5.061903.180057. [DOI] [PubMed] [Google Scholar]
- Mindell D.P, Knight A, Baer C, Huddleston C.J. Slow rates of molecular evolution in birds and the metabolic rate and body temperature hypotheses. Mol. Biol. Evol. 1996;13:422–426. [Google Scholar]
- Mindell D.P, Sorenson M.D, Dimcheff D.E. Multiple independent origins of mitochondrial gene order in birds. Proc. Natl Acad. Sci. USA. 1998;95:10 693–10 697. doi: 10.1073/pnas.95.18.10693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell D.P, Sorenson M.D, Dimcheff D.E, Hasegawa M, Ast J.C, Yuri T. Interordinal relationships of birds and other reptiles based on whole mitochondrial genomes. Syst. Biol. 1999;48:138–152. doi: 10.1080/106351599260490. [DOI] [PubMed] [Google Scholar]
- Miya M, Kawaguchi A, Nishida M. Mitogenomic exploration of higher teleostean phylogenies: a case study for moderate-scale evolutionary genomics with 38 newly determined complete mitochondrial DNA sequences. Mol. Biol. Evol. 2001;18:1993–2009. doi: 10.1093/oxfordjournals.molbev.a003741. [DOI] [PubMed] [Google Scholar]
- Nanda I, Schrama D, Feichtinger W, Haaf T, Schartl M, Schmid M. Distribution of telomeric (TTAGGG)(n) sequences in avian chromosomes. Chromosoma. 2002;111:215–227. doi: 10.1007/s00412-002-0206-4. [DOI] [PubMed] [Google Scholar]
- Nekrutenko A, Li W.H. Assessment of compositional heterogeneity within and between eukaryotic genomes. Genome Res. 2000;10:1986–1995. doi: 10.1101/gr.10.12.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickerson D.A, Tobe V.O, Taylor S.L. PolyPhred: automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 1997;25:2745–2751. doi: 10.1093/nar/25.14.2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R. Maximum likelihood estimation of population divergence times and population phylogenies under the infinite sites model. Theor. Popul. Biol. 1998;53:143–151. doi: 10.1006/tpbi.1997.1348. [DOI] [PubMed] [Google Scholar]
- Nikaido M, Rooney A.P, Okada N. Phylogenetic relationships among cetartiodactyls based on evidence from insertions of SINEs and LINEs: hippopotamuses are the closest extant relatives of whales. Proc. Natl Acad. Sci. USA. 1999;96:10 261–10 266. doi: 10.1073/pnas.96.18.10261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikaido M, et al. Retroposon analysis of major cetacean lineages: the monophyly of toothed whales and the paraphyly of river dolphins. Proc. Natl Acad. Sci. USA. 2001;98:7384–7389. doi: 10.1073/pnas.121139198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylander J.A, Ronquist F, Huelsenbeck J.P, Nieves-Aldrey J.L. Bayesian phylogenetic analysis of combined data. Syst. Biol. 2004;53:47–67. doi: 10.1080/10635150490264699. [DOI] [PubMed] [Google Scholar]
- Okada N, Shedlock A.M, Nikaido M. Retroposon mapping in molecular systematics. In: Miller W.J, Capy P, editors. Methods in molecular biology. Mobile genetic elements. vol. 260. Humana Press Inc; Tatowa, NJ: 2003. pp. 189–226. [DOI] [PubMed] [Google Scholar]
- Otto S.P, Cummings M.P, Wakeley J. Inferring phylogenies from DNA sequence data: the effects of sampling. In: Harvey P.H, Brown A.J.L, Maynard Smith J, Nee S, editors. In New uses for new phylogenies. Oxford University Press; New York: 1996. pp. 103–115. [Google Scholar]
- Paton T, Haddrath O, Baker A.J. Complete mitochondrial DNA genome sequences show that modern birds are not descended from transitional shorebirds. Proc. R. Soc. B. 2002;269:839–846. doi: 10.1098/rspb.2002.1961. doi:10.1098/rspb.2002.1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paton T.A, Baker A.J, Groth G, Barrowclough G.F. RAG-1 sequences resolve phylogenetic relationships within Charadriiform birds. Mol. Phylogenet. Evol. 2003;29:268–278. doi: 10.1016/s1055-7903(03)00098-8. [DOI] [PubMed] [Google Scholar]
- Pereira S.L, Baker A.J, Wajntal A. Combined nuclear and mitochondrial DNA sequences resolve generic relationships within the Cracidae (Galliformes, Aves) Syst. Biol. 2002;51:946–958. doi: 10.1080/10635150290102519. [DOI] [PubMed] [Google Scholar]
- Poe S, Chubb A.L. Birds in a bush: five genes indicate explosive evolution of avian orders. Evolution. 2004;58:404–415. [PubMed] [Google Scholar]
- Pollock D.D, Eisen J.A, Doggett N.A, Cummings M.P. A case for evolutionary genomics and the comprehensive examination of sequence biodiversity. Mol. Biol. Evol. 2000;17:1776–1788. doi: 10.1093/oxfordjournals.molbev.a026278. [DOI] [PubMed] [Google Scholar]
- Pollock D.D, Zwickl D.J, McGuire J.A, Hillis D.M. Increased taxon sampling is advantageous for phylogenetic inference. Syst. Biol. 2002;51:664–671. doi: 10.1080/10635150290102357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prychitko T.M, Moore W.S. The utility of DNA sequences of an intron from the B-fibrinogen gene in phylogenetic analysis of woodpeckers (Aves: Picidae) Mol. Phylogenet. Evol. 1997;8:193–204. doi: 10.1006/mpev.1997.0420. [DOI] [PubMed] [Google Scholar]
- Prychitko T.M, Moore W.S. Alignment and phylogenetic analysis of b-fibrinogen intron 7 sequences among avian orders reveal conserved regions within the intron. Mol. Biol. Evol. 2003;20:762–771. doi: 10.1093/molbev/msg080. [DOI] [PubMed] [Google Scholar]
- Rannala B, Yang Z. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics. 2003;164:1645–1656. doi: 10.1093/genetics/164.4.1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raudsepp T, Houck M.L, O'Brien P.C, Ferguson-Smith M.A, Ryder O.A, Chowdhary B.P. Cytogenetic analysis of California condor (Gymnogyps californianus) chromosomes: comparison with chicken (Gallus gallus) macrochromosomes. Cytogenet. Genome Res. 2002;98:54–60. doi: 10.1159/000068532. [DOI] [PubMed] [Google Scholar]
- Raymond C.K, Sims E.H, Kas A, Spencer D.H, Kutyavin T.V, Ivey R.G, Zhou Y, Kaul R, Clendenning J.B, Olson M.V. Genetic variation at the O-antigen biosynthetic locus in Pseudomonas aeruginosa. J. Bacteriol. 2002a;184:3614–3622. doi: 10.1128/JB.184.13.3614-3622.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raymond C.K, Sims E.H, Olson M.V. Linker-mediated recombinational subcloning of large DNA fragments using yeast. Genome Res. 2002b;12:190–197. doi: 10.1101/gr.205201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieder M.J, Taylor S.L, Tobe V.O, Nickerson D.A. Automating the identification of DNA variations using quality-based fluorescence re-sequencing: analysis of the human mitochondrial genome. Nucleic Acids Res. 1998;26:967–973. doi: 10.1093/nar/26.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson T.J, Fu B, Ferguson-Smith M.A, Yang F. Cross-species chromosome painting in the golden mole and elephant-shrew: support for the mammalian clades Afrotheria and Afroinsectiphillia but not Afroinsectivora. Proc. R. Soc. B. 2004;271:1477–1484. doi: 10.1098/rspb.2004.2754. doi:10.1098/rspb.2004.2754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russello M.A, Amato G. A molecular phylogeny of Amazona: implications for neotropical parrot biogeography, taxonomy, and conservation. Mol. Phylogenet. Evol. 2004;30:421–437. doi: 10.1016/s1055-7903(03)00192-1. [DOI] [PubMed] [Google Scholar]
- Saitou N, Nei M. The number of nucleotides required to determine the branching order of three species, with special reference to the human-chimpanzee-gorilla divergence. J. Mol. Evol. 1986;24:189–204. doi: 10.1007/BF02099966. [DOI] [PubMed] [Google Scholar]
- Sasaki T, Takahashi K, Nikaido M, Miura S, Yasukawa Y, Okada N. First application of the SINE (short interspersed repetitive element) method to infer phylogenetic relationships in reptiles: an example from the turtle superfamily Testudinoidea. Mol. Biol. Evol. 2004;21:705–715. doi: 10.1093/molbev/msh069. [DOI] [PubMed] [Google Scholar]
- Shah N, Couronne O, Pennacchio L.A, Brudno M, Batzoglou S, Bethel E.W, Rubin E.M, et al. Phylo-VISTA: interactive visualization of multiple DNA sequence alignments. Bioinformatics. 2004;20:636–643. doi: 10.1093/bioinformatics/btg459. [DOI] [PubMed] [Google Scholar]
- Shedlock A.M, Okada N. SINE insertions: powerful tools for molecular systematics. Bioessays. 2000;22:148–160. doi: 10.1002/(SICI)1521-1878(200002)22:2<148::AID-BIES6>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Shedlock A.M, Takahashi K, Okada N. SINEs of speciation: tracking lineages with retroposons. Trends Ecol. Evol. 2004;19:545–553. doi: 10.1016/j.tree.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Shendure J, Mitra R.D, Varma C, Church G.M. Advanced sequencing technologies: methods and goals. Nature Rev. Genet. 2004;5:335–344. doi: 10.1038/nrg1325. [DOI] [PubMed] [Google Scholar]
- Shetty S, Griffin D.K, Marshall-Graves J.A. Comparative painting reveals strong chromosome homology over 80 million years of bird evolution. Chromosome Res. 1999;7:289–295. doi: 10.1023/a:1009278914829. [DOI] [PubMed] [Google Scholar]
- Shevchuk N.A, Allard M.W. Sources of incongruence among mammalian mitochondrial sequences: COII, COIII, and ND6 genes are main contributors. Mol. Phylogenet. Evol. 2001;21:43–54. doi: 10.1006/mpev.2001.0989. [DOI] [PubMed] [Google Scholar]
- Shiina T, Shimizu S, Hosomichi K, Kohara S, Watanabe S, Hanzawa K, Beck S, Kulski J.K, Inoko H. Comparative genomic analysis of two avian (quail and chicken) MHC regions. J. Immunol. 2004;172:6751–6763. doi: 10.4049/jimmunol.172.11.6751. [DOI] [PubMed] [Google Scholar]
- Sibley C.G, Ahlquist J.E. Yale University Press; New Haven: 1990. The phylogeny and classification of birds: a study in molecular evolution. [Google Scholar]
- Slowinksi J, Page R.D.M. How should species phylogenies be inferred from sequence data? Syst. Biol. 1999;48:814–825. doi: 10.1080/106351599260030. [DOI] [PubMed] [Google Scholar]
- Smit A.F.A. Interspersed repeats and other mementos of transposable elements in mammalian genomes. Curr. Opin. Genet. Dev. 1999;9:657–663. doi: 10.1016/s0959-437x(99)00031-3. [DOI] [PubMed] [Google Scholar]
- Sorenson M.D, Quinn T.W. Numts: a challenge for avian systematics and population biology. Auk. 1998;115:214–221. [Google Scholar]
- Sorenson M.D, Oneal E, García-Moreno J. More taxa, more characters: the hoatzin problem is still unresolved. Mol. Biol. Evol. 2003;20:1484–1499. doi: 10.1093/molbev/msg157. [DOI] [PubMed] [Google Scholar]
- Stapel S.O, Leunissen J.A.M, Versteeg M, Wattel J, de Jong W.W. Ratites as oldest offshoot of avian stem-evidence from a crystallin sequences. Nature. 1984;311:257–259. doi: 10.1038/311257a0. [DOI] [PubMed] [Google Scholar]
- Stuart G.W, Moffett K, Leader J.J. A comprehensive vertebrate phylogeny using vector representations of protein sequences from whole genomes. Mol. Biol. Evol. 2002;19:554–562. doi: 10.1093/oxfordjournals.molbev.a004111. [DOI] [PubMed] [Google Scholar]
- Thomas J.W, et al. Comparative analyses of multi-species sequences from targeted genomic regions. Nature. 2003;424:788–793. doi: 10.1038/nature01858. [DOI] [PubMed] [Google Scholar]
- van Tuinen M, Sibley C.G, Hedges S.B. The early history of modern birds inferred from DNA sequences of nuclear and mitochondrial ribosomal genes. Mol. Biol. Evol. 2000;17:451–457. doi: 10.1093/oxfordjournals.molbev.a026324. [DOI] [PubMed] [Google Scholar]
- Waltari E, Edwards S.V. Evolutionary dynamics of intron size, genome size, and physiological correlates in archosaurs. Am. Nat. 2002;160:539–552. doi: 10.1086/342079. [DOI] [PubMed] [Google Scholar]
- Weiner A.M. SINEs and LINEs: the art of biting the hand that feeds you. Curr. Opin. Gen. Dev. 2002;14:343–350. doi: 10.1016/s0955-0674(02)00338-1. [DOI] [PubMed] [Google Scholar]
- Weiner A.M, Deininger P.L, Efstratiadis A. Nonviral retroposons: genes, pseudogenes, and transposable elements generated by the reverse flow of genetic information. Annu. Rev. Biochem. 1986;55:631–661. doi: 10.1146/annurev.bi.55.070186.003215. [DOI] [PubMed] [Google Scholar]
- Wicker T, et al. The repetitive landscape of the chicken genome. Genome Res. 2004;14:000–000. doi: 10.1101/gr.2438005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. On the best evolutionary rate for phylogenetic analysis. Syst. Biol. 1998;47:125–133. doi: 10.1080/106351598261067. [DOI] [PubMed] [Google Scholar]
- Zimmer R, Verrinder Gibbins A.M. Construction and characterization of a large-fragment chicken bacterial artificial chromosome library. Genomics. 1997;42:217–226. doi: 10.1006/geno.1997.4738. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.