

Abstract
The thioredoxin system plays an important role in maintaining a reducing environment in the cell. Recently, several thioredoxin binding partners have been identified and proposed to mediate aspects of redox signaling, but the significance of these interactions is unclear in part due to incomplete understanding of the mechanism for thioredoxin binding. Txnip (thioredoxin-interacting protein) is a protein critical for regulation of glucose metabolism whose only currently known function is to bind and inhibit thioredoxin. We explored the mechanism of the Txnip-thioredoxin interaction and present evidence that Txnip and thioredoxin form a stable disulfide-linked complex. We identified two Txnip cysteines that are important for thioredoxin binding and showed that this interaction is consistent with a disulfide exchange reaction between oxidized Txnip and reduced thioredoxin. These cysteines are not conserved in the broader family of arrestin-domain-containing proteins, and we demonstrated that the thioredoxin-binding property of Txnip is unique. These data suggest that Txnip is a target of reduced thioredoxin and provide insight into Txnip’s potential role as a redox-sensitive signaling protein.
INTRODUCTION
Thioredoxin is a ubiquitous disulfide oxidoreductase that, along with the glutathione system, plays a major role in maintaining the cytoplasm in a reducing environment. Thioredoxin activity is mediated by a pair of cysteine thiols at its active site (human thioredoxin C32 and C35) that are oxidized during reduction of the substrate. By maintaining this reducing environment, thioredoxin is a critical defense against excess concentrations of reactive oxygen species, which are deleterious to cells and implicated in the pathophysiology of diseases such as atherosclerosis (1,2), diabetes (3), and arthritis (4,5). The reducing environment of the cell is also important for keeping protein thiols reduced, so that under normal conditions proteins contain many free sulfhydryl groups and relatively rare accessible disulfides (6).
In addition to the classical reducing activity of thioredoxin, recent evidence suggests that the redox state of thioredoxin may itself be an important component of redox signaling pathways. Thioredoxin reportedly binds a number of transcription factors and signaling molecules including NF-κB p50 subunit (7), Ref-1 (8), Jab-1 (9), Oct-4 (10), and PTEN (11). One of the better characterized interactions is that of reduced thioredoxin with apoptosis signal-regulating kinase 1 (ASK1)1, which plays a key role in promoting stress-induced apoptosis (12). Since only reduced thioredoxin is thought to bind ASK1, the interaction can be controlled by the redox state of the cell: during conditions of oxidative stress, ASK1 is released from thioredoxin and promotes apoptosis. The identification of thioredoxin and ASK1 cysteines required for the interaction (13) supports the hypothesis that thioredoxin forms a mixed disulfide complex with ASK1. However, the implications of this interaction are not clear in part due to i) the lack of direct evidence that thioredoxin can form a stable mixed disulfide and ii) the lack of a plausible mechanism for formation of this disulfide in the normal reducing environment of the cell.
Txnip (thioredoxin-interacting protein, also called “vitamin-D3-upregulated protein-1” (14) and “thioredoxin-binding protein 2”), is a 50-kDa protein with structural homology to the arrestins. Despite the arrestin homology, currently the only known function of Txnip is to bind thioredoxin and inhibit thioredoxin reducing activity (15-17). Txnip, like ASK1, appears to form a disulfide bond with thioredoxin since the interaction requires the presence of the thioredoxin active-site cysteines (17). Consistent with this role, overexpression of Txnip decreases thioredoxin reducing activity, increases redox stress, inhibits growth and hypertrophy, and causes increased apoptosis (16,18,19), while loss of Txnip expression is associated with tumor growth and metastasis (20-22). In addition, increasing evidence suggests major physiological roles for Txnip in glucose metabolism and cell differentiation. Both a naturally occuring mouse strain “hyplip1” with a truncation in Txnip (23-25) and mice with targeted deletion of Txnip (26) have low blood glucose, hyperlipidemia, and a severely dysregulated response to fasting. It is possible that these roles for Txnip are not mediated by inhibition of thioredoxin function alone. Understanding Txnip function will require that the mechanism for the interaction of Txnip and thioredoxin be better defined.
In the present work we explored the mechanism of the Txnip-thioredoxin interaction and present evidence that Txnip and thioredoxin form a stable disulfide-liked complex. We identified two Txnip cysteines that are important for thioredoxin binding and showed that this interaction is consistent with a disulfide exchange reaction between oxidized Txnip and reduced thioredoxin. These two critical cysteines are not conserved in the broader family of arrestin-domain-containing proteins, suggesting that Txnip is a unique redox-sensitive signaling protein.
METHODS
293 Cell culture and transfection
HEK 293 cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with high glucose, no sodium pyruvate, 10% fetal bovine serum, and antibiotics (100 U/ml penicillin and streptomycin). Plasmid-encoded proteins were expressed by transfection with Fugene 6 (Roche) using 3% Fugene and a 1:3 ratio of μg DNA to μl Fugene. Three days after transfection, cells were washed in ice-cold phosphate-buffered saline (PBS), harvested in lysis buffer (Tris-buffered saline with 0.5% Triton X-100, 1 mM phenylmethyl-sulphonylfluoride, and protease inhibitor cocktail [Sigma]), and clarified at 16,000 g for 10 minutes.
Adipocyte culture
3T3-L1 mouse fibroblasts (ATCC, Manassas, VA) were cultured in DMEM with high glucose (25 mM), penicillin, streptomyocin and 10% newborn calf serum (Invitrogen). Differentiation was induced by adding 5 μg/ml insulin, 0.25 μM dexamethasone, and 0.5 mM 3-isobutyl-1-methylxanthine to the media according to a standard protocol (27). By 8 days after induction, greater than 95% of cells were mature adipocytes based on morphology and Oil Red O uptake.
Adipocyte transduction
Txnip constructs were overexpressed in mature adipocytes by infection with a vector based on replication-deficient human immunodeficiency virus. Human Txnip was subcloned into the pCDH1-MCS1-EF1-Puro vector (System Biosciences, Mountain View, CA) and the Txnip C247S mutant was made by site-directed mutagenesis. Pseudoviral particles were made by transfection of Txnip, C247S, or an empty vector into 293TN cells (ATCC). A vector expressing green fluorescent protein (GFP) (pSIH1-H1-copGFP, System Biosciences) was co-transfected with each vector in a 1:100 ratio. Multiplicity of infection (MOI) of the resulting particles was determined by the number of cells expressing GFP on fluorescence microscopy 96 h after transduction. For experimental studies, 8 days after induction cells were cultured in low glucose (5.6 mM), transduced with an MOI of 1-2, and allowed to express viral proteins for 96 h. Realtime PCR and Western analysis verified about a two-fold overexpression of Txnip and Txnip C247S in transduced cells (data not shown).
Cloning of mouse arrestin-domain-containing genes
The mouse Arrdc2 gene, with a GC content of 65%, was amplified from gut cDNA by polymerase chain reaction (PCR) with PfuTurbo polymerase (Stratagene, La Jolla, CA) in the presence of 1.1 M betaine and 4 mM Mg+2 and subcloned into pcDNA3.1/V5/His (Invitrogen, Carlsbad, CA). Primers were: 5’-caccgcaatgctgttcgataaggt-3’ (forward) and 5’-taccgaagttgtcagcccatggat-3’ (reverse) and the product matched GenBank sequence NM_027560. Mouse Arrdc4 was amplified from gut cDNA and cloned into pcDNA4/HisMax (Invitrogen). Primers were: 5’-atgggaggcgaggcgggagcgaat-3’ (forward) and 5’-tcagagaatgaaggatacaggctgggtctc-3’ (reverse) and matched GenBank sequence AK037198. Mouse Txnip was cloned from embryonic fibroblast cDNA into pcDNA3.1/V5/His using primers: 5’-accatggtgatgttcaagaagatcaag-3’ (forward) and 5’-ctgcacgttgttgttgttgttgttgtt-3’ (reverse) and matched GenBank sequence AK089403.
Site-directed mutagenesis
Cysteine-to-serine mutants were introduced into human Txnip (in pcDNA3.1+) and thioredoxin (in pGEX) by whole-plasmid PCR with PfuTurbo polymerase, followed by digestion of template DNA with DpnI (New England Biolabs, Ipswich, MA). Constructs were verified by bidirectional sequencing.
GST binding assays
Human thioredoxin was cloned from HeLa cell cDNA and subcloned between the EcoRI and XhoI sites of pGEX-4T3 (Amersham Biosciences), carboxy-terminal to the glutathione-S-transferase (GST). GST fusion proteins were induced in E. coli BL21 cells by 0.5 mM isopropyl-ß-D-thiogalactopyranoside for 3 hours. Cell pellets were lysed in B-PER (Pierce Biotechnology, Rockford, IL) and 40 U/ml DNAse and the cleared lysates were incubated with glutathione-sepharose 4B beads (BioWorld, Dublin, OH) for 30 minutes at 4 °C followed by three washes with 0.5% Triton X-100 in Tris-buffered saline. For GST binding, 293 cell lysates were incubated with beads containing equal amounts of GST protein. Binding proceeded overnight with rotation at 4 °C followed by 3 washes with lysis buffer. Bound proteins were released by boiling in gel-loading sample bufffer. All experiments were replicated at least once and essentially identical results were obtained to those shown.
Western analysis
Protein samples were electrophoresed through 10% SDS-polyacrylamide Bis-Tris gels (Invitrogen). Immunodetection of Txnip was performed with a custom-made monoclonal antibody (JY2, available from MBL Intl., Woburn, MA). Monoclonal antibodies to the V5 and Xpress epitopes were from Invitrogen.
Thioredoxin insulin-reduction activity assay
Thioredoxin activity in cell lysates was measured by the insulin-reduction assay of Holmgren et al. (28) with modifications. Briefly, cell lysates were incubated in excess insulin, thioredoxin reductase, and NADPH at 37 °C for 20 min. The reaction was terminated by the addition of guanidine and 5,5′-dithiobis-(2-nitrobenzoic acid) [DTNB]. Reduction of DTNB to 5-thio-2-nitrobenzoic acid was detected by optical density at 412 nm. Recombinant human thioredoxin-C73S-GST was used as a standard.
Statistical analysis
Differences between groups were tested by t-tests with unequal variance assumed. All tests were corrected for multiple comparisons using α = 1-(1-α0)1/n, where α0 = 0.05 and n = total number of comparisons.
RESULTS AND DISCUSSION
Wildtype Txnip and thioredoxin form a dithiothreitol-sensitive complex
Previous studies have shown that Txnip does not interact with the thioredoxin active-site double-cysteine mutant (C32,35S) (15,17), suggesting that Txnip and thioredoxin interact by formation of a disulfide bond with one of the thioredoxin active-site cysteines. However, there is little direct evidence that wildtype thioredoxin forms stable mixed disulfides with its putative binding partners under physiological conditions. Indeed, thioredoxin is thought to be an active disulfide reductase in large part due to the instability of the intermediate mixed disulfide (29,30). Nuclear magnetic resonsance studies have identified interactions of NFκB and Ref-1 peptides with thioredoxin, but since the peptides were cross-linked to a thioredoxin C35A mutant, they do not provide evidence regarding the physiological relevance of the mixed disulfide complex (7,8). Indirect evidence supports the generation of a mixed disulfide between thioredoxin and ASK1: ASK1 binds selectively to reduced thioredoxin; the interaction requires one thioredoxin active-site cysteine and an ASK1 cysteine; and the interaction is stabilized by single mutation of either thioredoxin C35 or C32 to serine (31). However, despite the structural similarity of the reduced and oxidized thioredoxin forms (32,33), there is a precedent for selective binding to reduced thioredoxin solely by noncovalent interactions in the E. coli T7 polymerase complex (34).
We therefore tested whether we could identify a stable disulfide complex of Txnip and thioredoxin by a thioredoxin-GST binding assay. Human Txnip was over-expressed in 293 cells, which have nearly undetectable levels of endogenous Txnip, and the cell lysates were incubated overnight with thioredoxin-GST bound to glutathione-Sepharose beads. Bound proteins were released by boiling in non-reducing sample buffer, incubated in a range of dithiothreitiol (DTT) concentrations, and subjected to Western analysis with a monoclonal antibody against Txnip (JY2) (Fig. 1). On short exposure, primarily monomeric Txnip at ∼45 kDa was observed (left panel). The intensity of this band increased with increasing concentrations of DTT, consistent with release of the monomeric Txnip from disulfide-linked complexes by DTT. Fainter bands were also visible at the expected size of the Txnip-thioredoxin-GST complex (∼83 kDa). On longer exposure (middle panel), this 83-kDa band was clearly visible with 0 to 0.1 mM DTT but disappeared in a dose-dependent manner from 0.1 to 10 mM DTT. The 83-kDa band was not visible in the input lysates (right panel). The identification of an 83-kDa JY2-reactive band that is sensitive to DTT is thus consistent with the identification of a stable Txnip-thioredoxin disulfide-linked complex.
Figure 1.

Detection of the thioredoxin-Txnip complex by nonreducing gel electrophoresis. Txnip was overexpressed in 293 cells and the lysates were incubated overnight with thioredoxin-C73S-GST beads. After three washes in lysis buffer, the complex was released by incubation in nonreducing sample buffer at 80 ° for 10 min. The supernatant was divided and dithiothreitiol (DTT) was added to 0, 0.1, 1, or 10 mM. The original lysates and the bound protein samples were then subject to SDS-PAGE and Western analysis for Txnip with the monoclonal antibody JY2. The experiment was replicated with essentially identical results as those shown.
We also observed that the Txnip monomer showed a mobility shift with increasing levels of DTT (Fig. 1), suggesting that Txnip contains one or more intramolecular disulfides. This observation is similar to the mobility shift seen for the redox-active disulfide of PTEN (35). However, this evidence was limited by the inability of the Western analysis to resolve the Txnip bands that would be expected for separate dithiol and disulfide forms of Txnip.
Txnip cysteine 267 is conserved in four human/mouse homologs
We then hypothesized that if Txnip formed a disulfide bond with thioredoxin, at least one Txnip cysteine would be required for the interaction. Furthermore, if thioredoxin interaction were conserved among Txnip homologs, this cysteine should be conserved. A search of the PubMed mouse and human genome database revealed five proteins homologous to Txnip of unknown function (Fig. 2A). Txnip and these homologs all contain structural homology to the arrestins, which are intracellular scaffolding proteins that have important roles modulating receptor endocytosis, ubiquitination, and signaling (36). The homologs have been assigned official gene names arrestin domain containing (Arrdc) 1 through 5. Three have so far been cloned and briefly characterized: human Arrdc4 as “down-regulated in hepatocellular carcinoma 1(DRH1)” (37), rat Arrdc2 as “induced by lysergic acid diethylamide-1 (ILAD-1)” (38), and most recently, human Arrdc3 as “thioredoxin-binding-protein-2-like inducible membrane protein (TLIMP)” (39). The most distantly related protein, Arrdc5, is notable for its sperm-specific expression. The functions of these proteins and the significance of the arrestin domains remain unknown.
Figure 2.

The arrestin-domain-containing (Arrdc) family of proteins. (A) In addition to Txnip, at least five arrestin-domain-containing proteins of unknown function are present in the mouse and human genomes. All have predicted structural homology to the arrestins and contain both N-terminal and C-terminal arrestin domains. Percentage identity to Txnip is shown. (B) Among the three proteins with highest homology to Txnip (Arrdc2, 3, and 4), alignment of the predicted protein sequences revealed that only one cysteine (Txnip C267) is conserved.
Alignment of the three proteins (Arrdc2-4) with the most identity to Txnip demonstrated a single conserved cysteine (Txnip C267) (Fig. 2B; full alignments in Fig. S1A). This cysteine is also largely conserved in arrestin-domain-containing homologs through evolution to C. elegans and D. melanogaster (Fig. S1B). This suggested that the Txnip cysteine 267 would be required for interaction with thioredoxin and that the arrestin-domain-containing proteins describe a family of thioredoxin-interacting proteins.
Txnip cysteine 247, not cysteine 267, is critical for the Txnip-thioredoxin interaction
Using yeast two-hybrid screening, Yamanaka et al. identified that deletion of the amino-terminal arrestin domain (residues 1-154) did not affect binding to thioredoxin (15). We therefore generated a Txnip cysteine-to-serine mutant for each of the 7 cysteine residues in the remainder of the protein (residues 155-391) and overexpressed them in 293 cells. Thioredoxin-GST bound wildtype Txnip and six of the mutant Txnips, including the C267S mutant. Surprisingly, thioredoxin-GST instead failed to bind a mutation of Txnip cysteine 247 (C247). Thus Txnip C247, not the conserved C267, is critical for the interaction with thioredoxin.
While Txnip C267 is highly conserved through evolution, Txnip C247 is not present in any of the arrestin-domain-containing proteins other than Txnip orthologs (Fig. 3B, bottom, and Fig. S1). Even within Txnip orthologs it is not present in the sequenced avian (G. gallus) and amphibian (X. laevis) species (Fig. 3B). This suggests that the family of Arrdc proteins is not defined by interaction with thioredoxin.
Figure 3.

Binding of thioredoxin-GST beads to Txnip Cys-to-Ser mutants. (A) Wildtype and mutant Txnip proteins were overexpressed in 293 cells and the cell lysates were incubated with thioredoxin-GST beads. Wildtype Txnip was incubated with GST alone as a negative control. Bound Txnip was detected by Western analysis with JY2. Only Txnip C247S did not bind to thioredoxin-GST. The experiment was replicated with essentially identical results as those shown. (B) Alignment of Txnip homologs demonstrated that C247 is relatively conserved in vertebrate Txnip orthologs (shown are sequences from Gallus gallus, Xenopus laevis, Oncorhynchus mykiss, Danio rerio, Tetraodon nigroviridus, and Fugu rubripes) but not present in other Arrdc proteins and invertebrate arrestin-domain-containing proteins (shown are sequences from Caenorhabditis elegans and Drosophila melanogaster) despite conservation of C267.
Txnip-like proteins do not bind to thioredoxin
To test the hypothesis that the Txnip-like proteins do not interact with thioredoxin, we cloned the three mouse homologs with closest homology to Txnip (Arrdc2, 3, 4), as well as mouse Txnip, into tagged expression vectors. The vectors were then overexpressed in 293 cells and detection of the tagged proteins was confirmed in cell lysates (Fig. 4, left panels). Thioredoxin-GST failed to detect an interaction with either Arrdc2 or Arrdc4 under conditions that bound Txnip (Fig. 4, right panels). Consistent with these observations, Oka et al. have recently reported that human Arrdc3 also does not bind thioredoxin (39).
Figure 4.
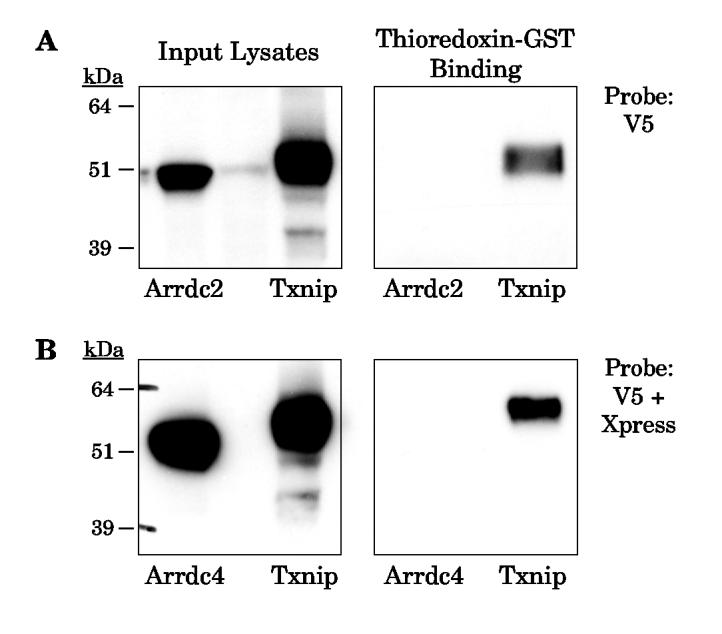
Binding of thioredoxin-GST beads to mouse Arrdc2 and Arrdc4. Epitope-tagged mouse Arrdc2, Arrdc4, and Txnip over-expressed in 293 cells. Lysates were incubated overnight with thioredoxin-C73S-GST. After washing in lysis buffer, the bound proteins were subject to Western analysis. (A) Western analysis shows that Arrdc2 was detected in the input lysate with anti-V5, but no binding to thioredoxin-GST was detected. As a positive control, under these conditions binding of mouse Txnip was successfully detected. (B) Mouse Arrdc4 was expressed with an N-terminal Xpress epitope. Input lysates and bound proteins were therefore subjected to Western analysis with anti-Xpress and anti-V5. Arrdc4 was detected in the lysate but no binding to thioredoxin was observed under conditions of successful Txnip binding. The experiments were replicated with essentially identical results as those shown.
These results confirm that the arrestin-domain-containing Txnip-like protein family is not defined by thioredoxin binding. The structural homology with the arrestins suggests a role as scaffolding proteins for protein-protein interaction and signaling, as described for the beta-arrestins (36). It may be that each homolog has evolved for interaction with different substrates and that studies of their binding partners will be important for defining their functions. In addition, since Txnip’s ability to bind thioredoxin is unique, Txnip may retain other conserved scaffolding and/or signaling functions.
The effect of redox conditions on the Txnip-thioredoxin interaction suggests Txnip is redox active
Our evidence that Txnip forms a DTT-sensitive complex with thioredoxin, together with the identification of a Txnip cysteine critical for the interaction, strongly suggested that the interaction is mediated by formation of a disulfide bond. However, in the reducing environment of the normal eukaryotic cytosol, oxidation by de novo formation of protein disulfides is uncommon in the absence of an electron acceptor to complete the redox reaction (40). Most protein disulfides are generated in the endoplasmic reticulum (ER) in a process that requires both protein disulfide isomerase and ER oxidoreductase-1 (Ero1) to catalyze electron transfer to molecular oxygen(41,42).
We therefore hypothesized that if Txnip formed a disulfide bond with thioredoxin, a disulfide exchange mechanism would be most likely. This suggested two possibilities: either Txnip interacts with oxidized thioredoxin, or Txnip contains a disulfide bond and interacts with reduced thioredoxin. Nishiyama et al. have previously reported that the interaction of Txnip with thioredoxin is abolished by oxidation of thioredoxin (17), suggesting that Txnip interacts with reduced thioredoxin. However, oxidation of human thioredoxin in vitro can cause inactivation of thioredoxin by dimerization at cysteine 73, thus preventing binding to Txnip (43,44). To eliminate this possibility, we tested the effect of redox state on the interaction of thioredoxin-C73S with Txnip.
Thioredoxin-C73S-GST beads were pre-incubated in lysis buffer and either reduced by addition of 100 μM DTT, left untreated, or oxidized by addition of 100 μM H2O2. The DTT or H2O2 was then removed by extensive washing prior to incubation with cell lysates containing overexpressed Txnip. As a negative control, thioredoxin-C73S did not bind Txnip C247S (Fig. 5A, left). Binding of wildtype Txnip was not affected by reduction of thioredoxin-C73S with DTT (Fig. 5A, right). In contrast, oxidation of thioredoxin-C73S strongly blocked binding to Txnip, confirming that thioredoxin must be reduced for efficient interaction with Txnip.
Figure 5.
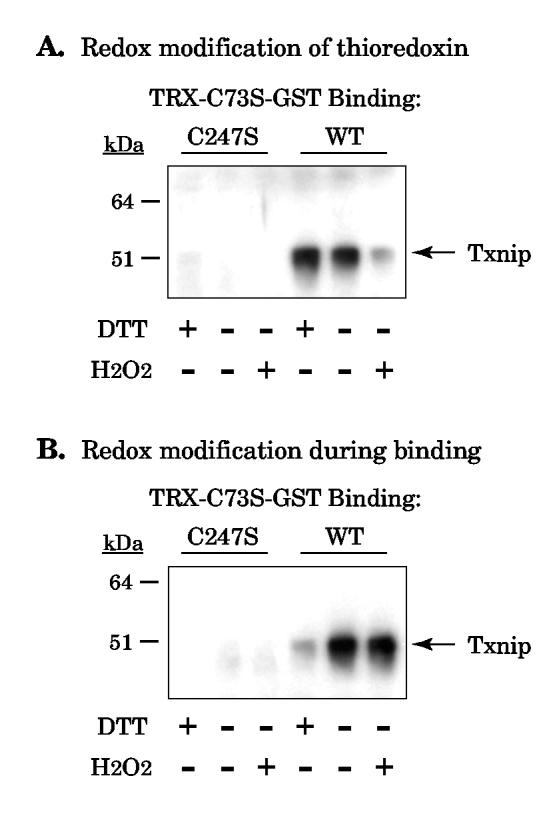
Effect of redox state on the thioredoxin-Txnip interaction. (A) Thioredoxin-C73S-GST beads were incubated in 100 μM of dithiothreitol (DTT) or hydrogen peroxide (H2O2) for two hours and washed 3 times in at least 20 bed volumes of lysis buffer. The beads were then incubated with cell lysates containing wildtype Txnip or C247S Txnip as a negative control. Bound proteins were probed for Txnip with JY2. (B) Lysates containing wildtype or C247S Txnip were added to unmodified thioredoxin-C73S-GST beads in the presence or absence of 100 μM DTT and H2O2. Bound proteins were probed for Txnip. The experiments were replicated with essentially identical results as those shown.
We also tested whether manipulating the redox conditions during binding affected the Txnip-thioredoxin interaction. Thioredoxin-GST beads were incubated with 293 cell lysate in the presence of DTT, no additive, or H2O2 (Fig. 5B). Again, Txnip C247S served as a negative control and no Txnip C247S binding was detected (Fig. 5B, left). However, redox manipulation during binding had the opposite effect from redox manipulation of thioredoxin alone: addition of 100 μM H2O2 had no effect while addition of 100 μM DTT inhibited binding (Fig. 5B, right).
The results of these two experiments are consistent with a disulfide exchange mechanism and suggest that Txnip is present as either an intramolecular or intermolecular disulfide. Since Txnip interacts with reduced thioredoxin, the disulfide does not come from thioredoxin. Furthermore, addition of DTT during binding inhibits the reaction, suggesting that DTT reduces the putative Txnip disulfide and prevents disulfide exchange.
We therefore considered evidence that Txnip might have an intramolecular or intermolecular disulfide. Western analysis for Txnip in fresh lysate from 293 cells overexpressing Txnip (Fig. 1, input lysates) or endogenous Txnip from NIH 3T3 and Thp1 monocytic cell lines (not shown) showed little evidence for JY2 reactivity at sizes larger than the monomer under nonreducing conditions. The absence of evidence for an intermolecular disulfide, take together with the redox-sensitive mobility shift of Txnip (Fig. 1), was thus most consistent with the hypothesis that Txnip contains one or more redox-active intramolecular disulfides.
A second Txnip cysteine is critical for efficient thioredoxin binding
If an intramolecular Txnip disulfide were required for interaction with thioredoxin, the simplest hypothesis would be that the disulfide involves cysteine 247 and that its partner Txnip cysteine would also be required for interaction with thioredoxin. We therefore generated cysteine-to-serine mutants of the four cysteines in the Txnip N-terminal arrestin domain and tested them for interaction with thioredoxin-GST (Fig. 6). The Txnip C63S mutant nearly abolished binding by thioredoxin-GST, identifying a second Txnip cysteine that is important for thioredoxin interaction. This result therefore supports the hypothesis that Txnip contains an intramolecular disulfide between C63 and C247 that allows interaction with reduced thiredoxin by disulfide exchange.
Figure 6.
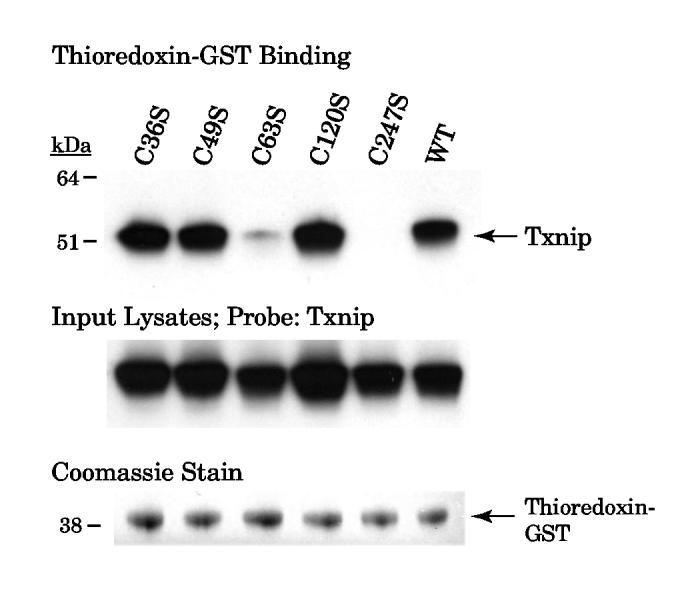
Binding of thioredoxin-GST beads to Txnip N-terminal Cys-to-Ser mutants. Wildtype and mutant Txnip proteins were overexpressed in 293 cells and the cell lysates were incubated with thioredoxin-C73S-GST. After washing, beads were incubated in sample buffer and the supernatant was probed for Txnip with JY2. Input cell lysates were probed for Txnip and demonstrated similar immunoreactivity. Similar amounts of thioredoxin-GST were observed in the supernatant by Coomassie staining. The experiment was replicated with essentially identical results as those shown.
Furthermore, the Western analysis suggested that while some binding of the Txnip C63S to thioredoxin was still possible, no binding at all of the Txnip C247S was detected. We therefore hypothesized that Txnip C247 is more critical for thioredoxin binding because it forms the disulfide with thioredoxin, while Txnip C63 is required for efficient reaction by disulfide exchange but mutation of this cysteine does not prevent the interaction completely. This hypothesis is also consistent with the yeast two-hybrid analysis that found a Txnip truncation construct with deletion of the amino-terminal 155 residues still bound thioredoxin (15).
Txnip C247 is critical for inhibition of thioredoxin activity in adipocytes
To test whether the Txnip mixed disulfide is important for the inhibition of thioredoxin activity by Txnip, we overexpressed wildtype and Txnip C247S in mature 3T3-L1 adipocytes by lentiviral transduction. Since these cells are post-mitotic, any confounding effect of proliferation rate on thioredoxin activity is avoided. Four days after transduction, thioredoxin-mediated insulin-reducing activity in the cell lysates was measured, normalized to total protein content, and then normalized to the mean activity of the control cells (transduced with virus expressing an empty vector) (Fig. 7). In cells transduced with wildtype Txnip, thioredoxin activity was reduced to 48 ± 13% (mean ± standard deviation; N = 5) of control levels (p < 0.001). In contrast, in cells transduced with Txnip C247S, thioredoxin activity was 120 ± 2% of control levels (p < 0.001 for comparison with wildtype Txnip; not significantly different for comparison with controls).
Figure 7.
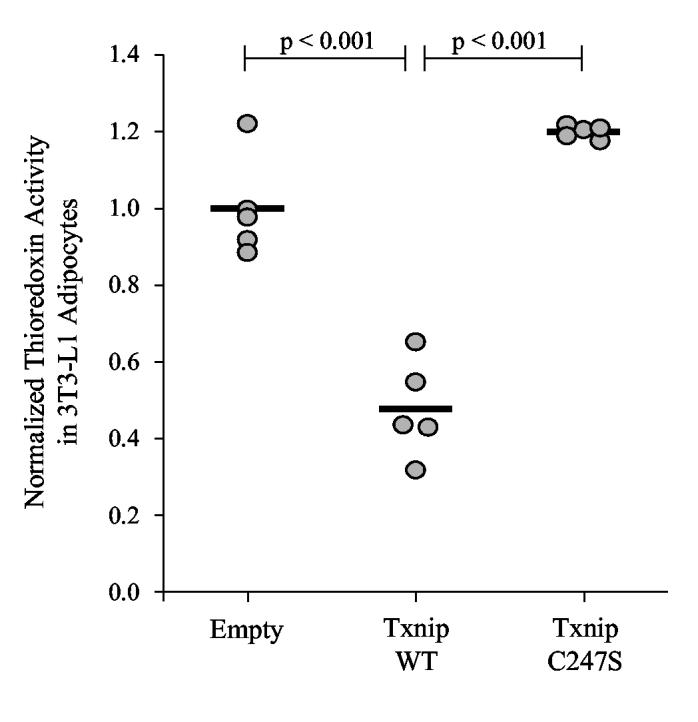
Effect of Txnip on thioredoxin activity in mature adipocytes. Differentiated 3T3-L1 adipocytes were transduced with lentiviral pseudoparticles expressing human wildtype Txnip (“Txnip WT”), Txnip C247S, or an empty vector (“Empty”). After 4 days the cells were lysed and the insulin-reducing activity of thioredoxin was measured. Data were expressed as activity per total protein content and then normalized to the mean level of the empty-vector control lysates. Bars represent the mean for each group. Significant differences between groups are indicated with p-values.
These data provide evidence that Txnip-thioredoxin mixed disulfide formation is functionally important for the inhibition of thioredoxin’s reducing activity by Txnip. However, since these experiments may involve more than physiological levels of Txnip expression, the effects of the Txnip-thioredoxin interaction in vivo may be different (though the over-expression obtained here was only about two-fold). In addition, other functions of Txnip and thioredoxin may not be dependent on or affected by mixed disulfide formation.
The evidence shown here for a stable mixed disulfide between Txnip and thioredoxin also strengthens the more general hypothesis that thioredoxin’s role in redox signaling is not limited to involvement in reduction reactions but may also include protein-protein interactions that are regulated by the state of its redox-active cysteines. In particular, several aspects of the Txnip-thioredoxin interaction are similar to the interaction of ASK1 with thioredoxin. Like Txnip, ASK1 does not bind the thioredoxin active-site mutant (C32S, C35S) and binds preferentially to reduced thioredoxin, suggesting the thioredoxin active-site cysteines are involved. Furthermore, mutation of a single thioredoxin active-site cysteine appears to stabilize the complex, consistent with the technique of “trapping” the mixed disulfide complex by C35A mutation (7). Finally, mutagenesis of ASK1 has identified a single cysteine-to-serine mutation that is critical for the interaction with thioredoxin. Thus based on the results here we speculate that ASK1 may also bind thioredoxin by a disulfide exchange reaction. However, support for this hypothesis would require evidence to demonstrate either an intramolecular or intermolecular ASK1 disulfide.
In summary, the results shown here provide evidence that Txnip and thioredoxin form a stable mixed disulfide by disulfide exchange. We propose that oxidized Txnip contains a disulfide linkage between cysteines 63 and 247, and the oxidized Txnip reacts with thioredoxin to form a disulfide between Txnip cysteine 247 and thioredoxin cysteine 32 (Fig. 8). In this model, either reduction of Txnip or oxidation of thioredoxin would prevent the interaction. We speculate that identification of the mechanism and generation of mutants that do not bind thioredoxin may allow identification of thioredoxin-independent functions of Txnip, based on its arrestin domain structure. These results also support the hypothesis that thioredoxin, in addition to its reducing activity, can participate in redox signaling through the formation of mixed disulfide complexes with other signaling proteins in a redox-dependent manner.
Figure 8.
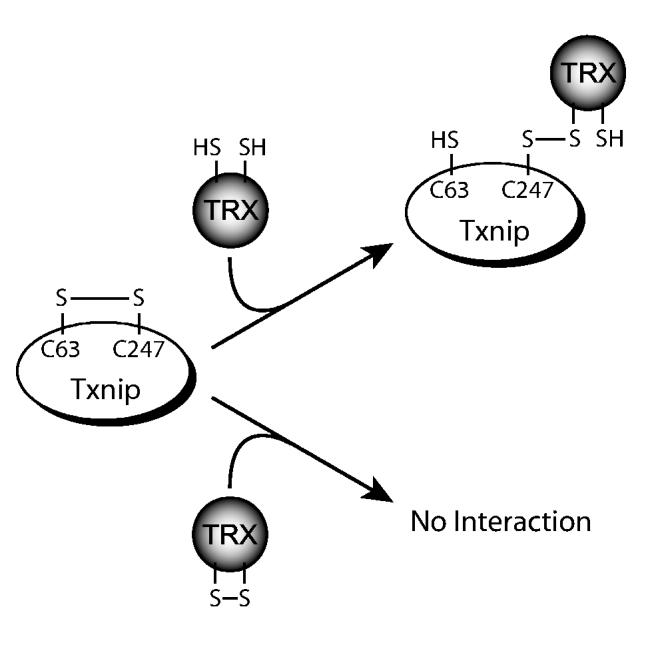
Proposed mechanism for Txnip-thioredoxin interaction. Txnip contains an intramolecular disulfide bond between C63 and C247 that allows efficient interaction with thioredoxin. Txnip forms a disulfide bond with reduced thioredoxin at C247 by disulfide exchange, yielding a stable thioredoxin mixed disulfide.
Supplementary Material
Footnotes
Acknowledgments:
This work was supported by grants from the National Institutes of Health (NIH) to R. Lee and an NIH National Research Service Award to P. Patwari.
- ASK1
- apoptosis signal-regulating kinase 1
- ER
- endoplasmic reticulum
- GST
- glutathione-S-transferase
- DTT
- dithiothreitiol
- H2O2
- hydrogen peroxide
- PCR
- polymerase chain reaction
- PBS
- phosphate-buffered saline
REFERENCES
- 1.Griendling KK, FitzGerald GA. Circulation. 2003;108:1912–1916. doi: 10.1161/01.CIR.0000093660.86242.BB. [DOI] [PubMed] [Google Scholar]
- 2.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, Tuncman G, Gorgun C, Glimcher LH, Hotamisligil GS. Science. 2004;306:457–461. doi: 10.1126/science.1103160. [DOI] [PubMed] [Google Scholar]
- 3.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hitchon CA, El-Gabalawy HS. Arthritis Res Ther. 2004;6:265–278. doi: 10.1186/ar1447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Henrotin Y, Kurz B, Aigner T. Osteoarthritis Cartilage. 2005;13:643–654. doi: 10.1016/j.joca.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 6.Gilbert HF. Adv Enzymol Relat Areas Mol Biol. 1990;63:69–172. doi: 10.1002/9780470123096.ch2. [DOI] [PubMed] [Google Scholar]
- 7.Qin J, Clore GM, Kennedy WM, Huth JR, Gronenborn AM. Structure. 1995;3:289–297. doi: 10.1016/s0969-2126(01)00159-9. [DOI] [PubMed] [Google Scholar]
- 8.Qin J, Clore GM, Kennedy WP, Kuszewski J, Gronenborn AM. Structure. 1996;4:613–620. doi: 10.1016/s0969-2126(96)00065-2. [DOI] [PubMed] [Google Scholar]
- 9.Hwang CY, Ryu YS, Chung MS, Kim KD, Park SS, Chae SK, Chae HZ, Kwon KS. Oncogene. 2004;23:8868–8875. doi: 10.1038/sj.onc.1208116. [DOI] [PubMed] [Google Scholar]
- 10.Guo Y, Einhorn L, Kelley M, Hirota K, Yodoi J, Reinbold R, Scholer H, Ramsey H, Hromas R. Stem Cells. 2004;22:259–264. doi: 10.1634/stemcells.22-3-259. [DOI] [PubMed] [Google Scholar]
- 11.Meuillet EJ, Mahadevan D, Berggren M, Coon A, Powis G. Arch Biochem Biophys. 2004;429:123–133. doi: 10.1016/j.abb.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 12.Saitoh M, Nishitoh H, Fujii M, Takeda K, Tobiume K, Sawada Y, Kawabata M, Miyazono K, Ichijo H. EMBO J. 1998;17:2596–2606. doi: 10.1093/emboj/17.9.2596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang R, Al-Lamki R, Bai L, Streb JW, Miano JM, Bradley J, Min W. Circ Res. 2004;94:1483–1491. doi: 10.1161/01.RES.0000130525.37646.a7. [DOI] [PubMed] [Google Scholar]
- 14.Chen KS, DeLuca HF. Biochim Biophys Acta. 1994;1219:26–32. doi: 10.1016/0167-4781(94)90242-9. [DOI] [PubMed] [Google Scholar]
- 15.Yamanaka H, Maehira F, Oshiro M, Asato T, Yanagawa Y, Takei H, Nakashima Y. Biochem Biophys Res Commun. 2000;271:796–800. doi: 10.1006/bbrc.2000.2699. [DOI] [PubMed] [Google Scholar]
- 16.Junn E, Han SH, Im JY, Yang Y, Cho EW, Um HD, Kim DK, Lee KW, Han PL, Rhee SG, Choi I. J Immunol. 2000;164:6287–6295. doi: 10.4049/jimmunol.164.12.6287. [DOI] [PubMed] [Google Scholar]
- 17.Nishiyama A, Matsui M, Iwata S, Hirota K, Masutani H, Nakamura H, Takagi Y, Sono H, Gon Y, Yodoi J. J Biol Chem. 1999;274:21645–21650. doi: 10.1074/jbc.274.31.21645. [DOI] [PubMed] [Google Scholar]
- 18.Schulze PC, De Keulenaer GW, Yoshioka J, Kassik KA, Lee RT. Circ Res. 2002;91:689–695. doi: 10.1161/01.res.0000037982.55074.f6. [DOI] [PubMed] [Google Scholar]
- 19.Yoshioka J, Schulze PC, Cupesi M, Sylvan JD, MacGillivray C, Gannon J, Huang H, Lee RT. Circulation. 2004;109:2581–2586. doi: 10.1161/01.CIR.0000129771.32215.44. [DOI] [PubMed] [Google Scholar]
- 20.Goldberg SF, Miele ME, Hatta N, Takata M, Paquette-Straub C, Freedman LP, Welch DR. Cancer Res. 2003;63:432–440. [PubMed] [Google Scholar]
- 21.Han SH, Jeon JH, Ju HR, Jung U, Kim KY, Yoo HS, Lee YH, Song KS, Hwang HM, Na YS, Yang Y, Lee KN, Choi I. Oncogene. 2003;22:4035–4046. doi: 10.1038/sj.onc.1206610. [DOI] [PubMed] [Google Scholar]
- 22.Nishinaka Y, Nishiyama A, Masutani H, Oka S, Ahsan KM, Nakayama Y, Ishii Y, Nakamura H, Maeda M, Yodoi J. Cancer Res. 2004;64:1287–1292. doi: 10.1158/0008-5472.can-03-0908. [DOI] [PubMed] [Google Scholar]
- 23.Bodnar JS, Chatterjee A, Castellani LW, Ross DA, Ohmen J, Cavalcoli J, Wu C, Dains KM, Catanese J, Chu M, Sheth SS, Charugundla K, Demant P, West DB, de Jong P, Lusis AJ. Nat Genet. 2002;30:110–116. doi: 10.1038/ng811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hui TY, Sheth SS, Diffley JM, Potter DW, Lusis AJ, Attie AD, Davis RA. J Biol Chem. 2004;279:24387–24393. doi: 10.1074/jbc.M401280200. [DOI] [PubMed] [Google Scholar]
- 25.Sheth SS, Castellani LW, Chari S, Wagg C, Thipphavong CK, Bodnar JS, Tontonoz P, Attie AD, Lopaschuk GD, Lusis AJ. J Lipid Res. 2005;46:123–134. doi: 10.1194/jlr.M400341-JLR200. [DOI] [PubMed] [Google Scholar]
- 26.Oka SI, Liu W, Masutani H, Hirata H, Shinkai Y, Yamada SI, Yoshida T, Nakamura H, Yodoi J. FASEB J. 2005 doi: 10.1096/fj.05-4439fje. [DOI] [PubMed] [Google Scholar]
- 27.Rubin CS, Lai E, Rosen OM. J Biol Chem. 1977;252:3554–3557. [PubMed] [Google Scholar]
- 28.Holmgren A, Bjornstedt M. Methods Enzymol. 1995;252:199–208. doi: 10.1016/0076-6879(95)52023-6. [DOI] [PubMed] [Google Scholar]
- 29.Wynn R, Cocco MJ, Richards FM. Biochemistry. 1995;34:11807–11813. doi: 10.1021/bi00037a019. [DOI] [PubMed] [Google Scholar]
- 30.LeMaster DM, Springer PA, Unkefer CJ. J Biol Chem. 1997;272:29998–30001. doi: 10.1074/jbc.272.48.29998. [DOI] [PubMed] [Google Scholar]
- 31.Liu Y, Min W. Circ Res. 2002;90:1259–1266. doi: 10.1161/01.res.0000022160.64355.62. [DOI] [PubMed] [Google Scholar]
- 32.Jeng MF, Campbell AP, Begley T, Holmgren A, Case DA, Wright PE, Dyson HJ. Structure. 1994;2:853–868. doi: 10.1016/s0969-2126(94)00086-7. [DOI] [PubMed] [Google Scholar]
- 33.in J, Clore GM, Gronenborn AM. Structure. 1994;2:503–522. doi: 10.1016/s0969-2126(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 34.Doublie S, Tabor S, Long AM, Richardson CC, Ellenberger T. Nature. 1998;391:251–258. doi: 10.1038/34593. [DOI] [PubMed] [Google Scholar]
- 35.Kwon J, Lee SR, Yang KS, Ahn Y, Kim YJ, Stadtman ER, Rhee SG. Proc Natl Acad Sci U S A. 2004;101:16419–16424. doi: 10.1073/pnas.0407396101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lefkowitz RJ, Shenoy SK. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 37.Yamamoto Y, Sakamoto M, Fujii G, Kanetaka K, Asaka M, Hirohashi S. Clin Cancer Res. 2001;7:297–303. [PubMed] [Google Scholar]
- 38.Nichols CD, Sanders-Bush E. J Neurochem. 2004;90:576–584. doi: 10.1111/j.1471-4159.2004.02515.x. [DOI] [PubMed] [Google Scholar]
- 39.Oka SI, Masutani H, Liu W, Horita H, Wang D, Kizaka-Kondoh S, Yodoi J. Endocrinology. 2005 doi: 10.1210/en.2005-0679. [DOI] [PubMed] [Google Scholar]
- 40.Sevier CS, Kaiser CA. Nat Rev Mol Cell Biol. 2002;3:836–847. doi: 10.1038/nrm954. [DOI] [PubMed] [Google Scholar]
- 41.Tu BP, Weissman JS. J Cell Biol. 2004;164:341–346. doi: 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dias-Gunasekara S, Gubbens J, van Lith M, Dunne C, Williams JA, Kataky R, Scoones D, Lapthorn A, Bulleid NJ, Benham AM. J Biol Chem. 2005;280:33066–33075. doi: 10.1074/jbc.M505023200. [DOI] [PubMed] [Google Scholar]
- 43.Ren X, Bjornstedt M, Shen B, Ericson ML, Holmgren A. Biochemistry. 1993;32:9701–9708. doi: 10.1021/bi00088a023. [DOI] [PubMed] [Google Scholar]
- 44.Andersen JF, Sanders DA, Gasdaska JR, Weichsel A, Powis G, Montfort WR. Biochemistry. 1997;36:13979–13988. doi: 10.1021/bi971004s. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.