

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) lytic switch protein, Rta, is a ligand-independent inducer of the Notch signal transduction pathway, and KSHV cannot reactivate from latency in cells null for the Notch target protein RBP-Jk. Here we show that Rta promotes DNA binding of RBP-Jk, a mechanism that is fundamentally different from that established for the RBP-Jk-activating proteins, Notch intracellular domain (NICD) and Epstein-Barr virus EBNA2. Although constitutively active RBP-Jk and NICD do not transactivate KSHV promoters independently, cotransfection of an Rta mutant lacking its transactivation domain robustly restores transcriptional activation. Cooperation requires intact DNA binding sites for Rta and RBP-Jk and trimeric complex formation between the three molecules in vitro. In infected cells, RBP-Jk is virtually undetectable on a series of viral and cellular promoters during KSHV latency but is significantly enriched following Rta expression during viral reactivation. Accordingly, Rta, but not EBNA2 and NICD, reactivates the complete viral lytic cycle.
The phylogenetically conserved Notch signal transduction pathway restricts the differentiated state of cells during fate decisions in many tissues in both developing and adult organisms (48, 66). Ligand binding to the Notch receptor leads to proteolytic release of the Notch intracellular domain (NICD; also known as [aka] ICN and RAMIC), which translocates to the cell nucleus to activate transcription of target genes (33, 43, 51, 78, 80). NICD has no intrinsic DNA binding activity, so is tethered to DNA by physical interaction with the sequence-specific DNA binding protein recombination signal binding protein Jk (RBP-Jk) (aka Epstein-Barr virus [EBV] C promoter binding factor 1 [CBF1]/Suppressor of Hairless [Su (H)]/Lag-1 [CSL]) (35).
RBP-Jk is a monomeric 60-kDa protein that binds to the consensus DNA sequence (C/T)GTGGGAA with nanomolar affinity (11, 70). The DNA binding domain of RBP-Jk is located in its highly conserved, 425-amino-acid (aa) central core (2, 10, 18). This core folds into two c-Rel-like IPT/TIG domains, separated by a modified beta-trefoil domain (BTD), and includes four invariant residues that contact the DNA element (45, 70). In the absence of a Notch signal, DNA-bound RBP-Jk represses transcription by binding to histone deacetylase-associated transcription corepressors, resulting in transcriptionally silent chromatin (34, 39, 104, 105). Following Notch activation, NICD binds to the transcriptional repression domain of RBP-Jk to release the transcriptional corepressor complexes and nucleates the “Notch enhancer complex.” This multiprotein complex recruits histone acetyltransferase proteins and elongation factors to transactivate transcription (24, 25, 36, 44, 47, 88, 95, 103).
Uncontrolled Notch signaling is associated with malignancies in both natural and experimental systems (1, 3). Transformation of primary B cells by the Epstein-Barr virus requires the EBV nuclear antigen 2 (EBNA2) protein (14, 30, 75). EBNA2 orchestrates the viral and cellular gene expression program required for successful establishment and maintenance of latent EBV infection (37, 77, 82, 86, 89, 106). Similar to NICD, EBNA2 tethers to DNA through interactions with RBP-Jk, masks the RBP-Jk repression domain, and activates promoters containing RBP-Jk binding sites through interactions with the cellular protein SKIP (22, 32, 38, 58, 103). Nontransforming mutations of EBNA2 map to its transcriptional activation domain and a central 7-amino-acid motif that is highly conserved among all EBNA2 and NICD homologs (GPPWWPP) (12, 13, 57). This conserved motif is required for functional interactions of both EBNA2 and NICD with RBP-Jk. The signature is also found in the murine protein KyoT2, which counteracts Notch signals (85). As NICD only partially replaces inactivating mutations of EBNA2 in models of EBV transformation (27, 31), key aspects of RBP-Jk-dependent function remain a mystery.
Kaposi's sarcoma-associated herpesvirus (KSHV; aka human herpesvirus 8) is the etiologic agent responsible for the malignancies primary effusion lymphoma (PEL) and Kaposi's sarcoma (KS) (69). We and others have demonstrated that the KSHV protein Rta (named for replication and transcriptional activator, expressed from open reading frame 50 [ORF50]) is both necessary and sufficient for reactivation of KSHV in tissue culture models of latency (28, 62, 64, 81, 96). Rta is a 120-kDa protein that directly transactivates viral and cellular promoters (9, 16, 17, 62, 64, 87), and truncated mutants of Rta lacking the transactivation domain cannot reactivate the virus (62). Rta transactivates viral promoters by binding DNA either independently or in combination with cellular transcription factors, including RBP-Jk, Octamer-1, and C/EBPα (7, 8, 53, 55, 61, 76, 79, 91, 92).
The KSHV genome contains at least 44 RBP-Jk elements, and Rta-mediated transactivation of four viral promoters containing RBP-Jk sites is abrogated in RBP-Jk null cells (53-55). Similar to NICD and EBNA2, KSHV Rta directly binds to RBP-Jk, and Rta supershifts DNA-bound RBP-Jk (48, 53, 66). However, unlike NICD and EBNA2, Rta binds both to the N terminus of RBP-Jk and its central repression domain (53), and Rta lacks the GPPWWPP signature peptide used by NICD and EBNA2 to bind RBP-Jk. Indeed, NICD induces only a subset of the viral genes activated by Rta in cells latently infected by KSHV (6). In contrast to the requirement for RBP-Jk during EBV latency, KSHV establishes latent infection in RBP-Jk null cells, suggesting that transcriptional repression by RBP-Jk is not significant for turning off viral lytic promoters (54). Nevertheless, KSHV requires RBP-Jk to successfully reactivate from latency (54). Therefore, the mechanism by which Rta and RBP-Jk interact with the KSHV genome to activate transcription and regulate viral reactivation is unclear.
In this paper, we demonstrate that Rta regulation of RBP-Jk activity is fundamentally different from that established for the cellular Notch and EBV EBNA2 proteins. Our data suggest that a major component of RBP-Jk-mediated transactivation by Rta is that Rta promotes DNA binding by RBP-Jk, a novel mechanism for activation of the Notch pathway.
MATERIALS AND METHODS
Plasmids.
All plasmids were propagated and purified as described previously; those not described below were previously published (53, 61, 64). All inserts cloned by PCR were confirmed by DNA sequencing.
The reporter plasmids pTK-WT-hsp-luc (KSHV TK RBP-JK) and 2xRBP-hsp-luc (EBV Cp RBP-Jk) were constructed by annealing complementary oligonucleotides corresponding to dimers of the sequences shown in Fig. 1A, flanked by restriction sites for cloning into the hsp-luc plasmid.
FIG. 1.

Transcriptionally deficient Rta restores the ability of constitutively active RBP-Jk to transactivate KSHV genes in a promoter-specific manner. (A) Sequence alignment of the RBP-Jk-containing elements of the indicated promoters and the cloning strategy for each element in the reporter vector. Numbers represent the positions of each sequence relative to the transcriptional start site (Mta and Cp) or ORF (TK only) of each gene. (B and C) Expression vectors pCMX-VP16-RBP-Jk (expressing RBP-Jk/VP16) and pcDNA3.1-Myc-HisC-NICD (expressing NICD) were cotransfected into BL-41 cells with reporter plasmids p57Δ5-5Dwt-hsp-luc (B) and 2xRBP-hsp-luc (C) as indicated. Calculations are described in Materials and Methods. Error bars represent standard deviations of the means of at least three experiments. (D to G) Expression vectors V5-50ΔSTAD and those from panels B and C were cotransfected into BL41 cells (D, F, and G) or BJAB cells (E) with reporter plasmids p57Δ5-5Dwt-hsp-luc (D and E), p57WT-GL3 (F), and pTK-WT-hsp-luc (G). Extracts were analyzed as described above. For BL41 cells, + or ++ represents 3 or 9 μg of V5-50ΔSTAD, respectively. For BJAB cells, +, ++, or +++ represents 1, 3, or 9 μg of V5-50ΔSTAD, respectively. −, none.
The pV5-50ΔAatII plasmid contains Rta cDNA nucleotides (nt) 1 to 1242 cloned into pcDNA3.1-V5-HisA (Invitrogen) between EcoRI and XhoI, using PCR (forward [F]primer, GCGAGGCCTAATACGACTCACTATAGGG; reverse [R] primer, GCGCTCGAGCGTCAGGAAAGAGCT).
The pMalc2X-FL50 plasmid (expressing maltose binding protein [MBP]-Rta FL) contains the full-length Rta cDNA cloned into pMalc2X (New England Biolabs) between EcoRI and PstI. pMalc2X-50ΔAatII (expressing MBP-50DAatII) contains Rta cDNA nt 1 to 1242 with a stop codon in pMalc2x.
pRSET-A-RBP-Jk (expressing His-RBP-Jk) contains RBP-Jk (53) cloned into pRSET-A between BamHI and PstI.
pSG5-EBNA2, pCMX-VP16-RBP-Jk, pGST-RBP-Jk, and pcDNA3.1-Myc-HisC-NICD were gifts from B. Sugden (University of Wisconsin), T. Honjo (Kyoto University), and T. Kadesch (University of Pennsylvania).
Cell lines, transfections, and luciferase assays.
BC-3 cells were a gift of P. Moore (University of Pittsburgh) and were propagated, electroporated, and analyzed exactly as described for BCBL-1 cells in reference 64. BL-41 cells were a gift of Y. Yuan (University of Pennsylvania) and were electroporated at 224 V. BJAB cells were electroporated at 150 V. For all transfections, total DNA was normalized with pcDNA3, and transfection and lysis control were by β-galactosidase assay from cotransfected plasmid pcDNA3.1-His-lacZ. Each transfection was performed in triplicate at least twice.
For stable transfections, 1 × 106 293 cells were seeded in 10-cm tissue culture dishes. After overnight growth at 37°C, 10 μg of either BAC36wt or BAC36Δ50 (gifts of S. J. Gao and Greg Pari, respectively [96, 102]) were transfected using Transit-LT 1 reagent (Mirus) according to the manufacturer's directions. After 2 days, transfectants were selected with hygromycin (100 μg/ml) until a uniform, green cell population remained (about 3 weeks).
Protein expression and purification.
Rta (1-272) (Rta aa 1 to 272) (from plasmid pET28b-N50), and His-RBP-Jk (from plasmid pRSET-RBP-Jk) were expressed and purified as described previously (61).
Glutathione S-transferase (GST)-RBP-Jk was expressed and purified in NETN+ as described previously (63) and dialyzed versus 1× DNA binding buffer (61).
Maltose binding protein fusions (FL50-MBP and 50ΔAatII-MBP) were expressed and purified from Escherichia coli BL21 codon plus RIL/DE3 (Stratagene), following the manufacturer's directions (New England Biolabs), with the following modifications. Column binding buffer was supplemented with protease inhibitor cocktail (1:500 [Sigma]). Following freeze-thawing, resuspended pellets were sonicated with 10 pulses, 8 seconds each (with a 5-second rest in between) at 13% amplitude. Fractions were analyzed, dialyzed, and quantitated as described previously (61).
Nuclear extracts were prepared as described previously (52) from parallel cultures of BC-3 cells that were grown for 18 h in the presence or absence of tetradecanoyl phorbol acetate (TPA) as described previously (62).
EMSA.
32P-labeled DNA probes were prepared and tested by an electrophoretic mobility shift assay (EMSA) as described previously (61). Nuclear extracts were normalized for total protein, as determined by Bradford assay, prior to analysis. Gels were visualized by autoradiography or phosphorimager (Typhoon 9410 variable mode imager).
35S-labeled RBP-Jk was tested by an EMSA following programming of TnT T7 Quick-Coupled transcription/translation system (Promega) with the pcDNA3.1-RBP-Jk plasmid according to the manufacturer's directions. Unincorporated l-[35S]methionine was removed using Sephadex G-50 chromatography (Roche Biomedicals). All components were assembled in a manner similar to that described above for EMSA with 32P-labeled DNA probes.
ChIP
Chromatin immunoprecipitation (ChIP) was performed as described in reference 91 with minor modifications. Briefly, 9.5 × 105 BC-3, BCBL-1, or 293 cells were stimulated with TPA (20 ng/μl) for 16 to 18 h or left untreated. After cell extracts were precleared, protein/DNA complexes were immunoprecipitated with an RBP-Jκ-specific antibody (Chemicon) or rabbit immunoglobulin G (IgG) (Sigma) at 4°C overnight with nutation. For BC-3 cells, protein-DNA complexes were washed three times with 1 ml dilution buffer at 4°C for 30 min each time and one time with 1 ml dilution buffer at 24°C. Chromatin was isolated and resuspended in 100 μl of water.
For BCBL-1, 293, BL-41, or BJAB cells, protein-DNA complexes were immunoprecipitated as described above or with anti-VP16 antibody (Sigma). Immunoprecipitated complexes were collected with 60 μl of 50% (vol/vol) protein A agarose for 2 h at 4°C. Washes and DNA isolation were performed exactly as described previously (90). All analysis was by real-time PCR with quantitation by the ΔΔCT method (60).
All of the ChIPs were performed using IgG and phosphate-buffered saline (PBS) as negative controls for the anti-RBP-Jk antibody and PCR, respectively. In all cases, the increase in DNA binding measured using anti-RBP-Jk exceeded that for control IgG. In many cases, the actual increase in DNA binding for control IgG was between 0 and 1 (i.e., when the Δ cycle threshold [ΔCT] value without TPA exceeded the ΔCT value with TPA; more background chromatin was immunoprecipitated from uninduced cells than from TPA-induced cells). Since the agarose gel represents an end point analysis of the PCR, rather than a kinetic analysis (i.e., real-time PCR), the IgG control lanes were not shown for all ChIPs.
Reactivation of KSHV.
A total of 107 BCBL-1 cells were stimulated with TPA and/or transfected with the indicated plasmids at 150 V, 975 μF. After growth in 10 ml complete RPMI for 6 days, virions were harvested from the supernatant as described in reference 42, and DNA was resuspended in 100 μl water. Cellular DNA was harvested from cell pellets using the Puregene DNA isolation kit according to the manufacturer's directions and resuspended in 100 μl water. Virions and cellular DNA were assayed by real-time PCR using primers specific for ORF57 (KSHV) or endogenous retrovirus-3 (ERV-3) (cellular; as described previously (99). Quantitation was performed by the ΔΔCT method (60).
Real-time PCR.
Real-time PCR was performed using AmpliTaq Gold polymerase (Applied Biosystems) and a Corbett RotorGene 3000 instrument or Bio-Rad iCycler per the manufacturer's suggestions. The cycling parameters follow: 10 min at 95°C and then 40 to 60 cycles, with 1 cycle consisting of 20 s at 95°C, 30 s at 55°C, and 40 s at 72°C.
Primer sequences (5′ to 3′) were as follows: for Mta, F, GCGGCTAGCAACCTGGCAGCCA; R, GCGCAGATCTCCTCTGCATCAACCAT); for K-bZIP, F, GGTGGAGAGTATACGCAACTGCAAC; R, GGTTATAGTCGACAACGGAGGAAATAC; for K14, F, GCAGTATATTCACATTATGCAATACC; R, CGTAAGGCACCCTTATCTTTGAAAT; for human interleukin 6 (IL-6), F, GTAAAACTTCGTGCATGACTTCAGC; R, GACATCTCCAGTCCTATATTTATTG; for HES-1 (Hairy/enhancer of split 1), F, CAAGACCAAAGCGGAAAGAA; R, GGATCCTGTGTGATCCCTAGGC; for CD23, F, CCCGGATAACATTACACGCATGGCCT; R, GCCTATTTGCTCAATCATCAAGAG; and for ERV-3, F, CATGGGAAGCAAGGGAACTAATG; R, CCCAGCGAGCAATACAGAATTT.
The ΔΔCT method (60) was used for quantitation with the following modifications. For chromatin immunoprecipitations, ΔΔCT was calculated individually for each primer pair for cells treated with TPA or cells left untreated using the formula ΔCT = CT (IP) − CT (input chromatin), in which CT (IP) is the cycle threshold of immunoprecipitated chromatin. Next, ΔΔCT was calculated using the formula ΔΔCT = ΔCT (for TPA-treated cells) − ΔCT (for untreated cells). The change in immunoprecipitated chromatin was thus calculated using the formula 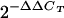 . This value was calculated using either the anti-RBP-Jk antibody or control IgG for each chromatin preparation. Where stated in the text, increase in enrichment equals the increase (2−ΔΔCT) determined from anti-RBP-Jk divided by the increase (2−ΔΔCT) determined from the IgG control.
. This value was calculated using either the anti-RBP-Jk antibody or control IgG for each chromatin preparation. Where stated in the text, increase in enrichment equals the increase (2−ΔΔCT) determined from anti-RBP-Jk divided by the increase (2−ΔΔCT) determined from the IgG control.
Similarly, for viral reactivation, the following formulas were used: ΔCT = CT (virion DNA) − CT (cellular DNA), which was calculated for each transfection, and ΔΔCT = ΔCT (for cells transfected with expression vectors) − ΔCT (for cells transfected with empty vector). Increase in reactivation was calculated as described above.
Design and synthesis of molecular beacons.
All molecular beacons were designed as described previously (67) on the basis of the KSHV genomic DNA sequence (GenBank accession no. U75698) or human genomic DNA sequence, except for the ERV-3 probe sequence, which was described previously (99). Molecular beacons were prepared by solid-phase DNA synthesis on a PE Biosystems 394 DNA synthesizer. A controlled-pore glass column (Biosearch Technologies) was used to incorporate a quencher moiety, 4-dimethylaminoazobenzene-4′-sulfonyl (dabcyl), at the 3′ end of the oligodeoxyribonucleotides, and a fluorescein phosphoramidite (Glen Research) was used to incorporate a fluorescein moiety at the 5′ end of the oligonucleotide. The oligonucleotides were purified by high-pressure liquid chromatography (Beckman) on a C18 reverse-phase column (Waters). Details of the purification steps are available at www.molecular-beacons.org. Molecular beacon sequences were as follows (lowercase letters denote nucleotides of stems, uppercase letters denote nucleotides of a region complementary to genomic sequence of the indicated genes): for KSHV Mta, cccacccGTTCCCACGGCCCATTTTTCGTTTGTGgggtggg; for KSHV K14, cgctgcCCATGGCCACAGGATGTAGAgcagcg; for CD23, ccgtcgTGCCTCTGCCTAGAGCCTTCCcgacgg; and for ERV-3, ccccccaTCTTCCCTCGAACCTGCACCATCAAGTCAtgggggg.
Western blotting.
Western blotting was performed exactly as described previously (62), using PBS with 0.1% Tween 20 for washes.
RESULTS
Constitutively active RBP-Jk does not activate transcription of a consensus RBP-Jk DNA element from a KSHV promoter.
We previously demonstrated that Rta, the KSHV lytic switch protein, requires RBP-Jk to activate transcription of the viral Mta (ORF57) promoter (53). We identified an essential 7-bp RBP-Jk site as part of the 26-bp Rta-responsive element (RRE) in this promoter (53, 61). However, mutations flanking the consensus RBP-Jk binding site in the RRE (Fig. 1A) of the Mta promoter have dramatic effects on Rta-mediated transactivation (61; also data not shown) but have little effect on DNA binding affinity of RBP-Jk in vitro (not shown). To begin to analyze DNA binding of RBP-Jk to this element in vivo, we tested the ability of constitutively active RBP-Jk (fused to the VP16 activation domain [RBP-JK/VP16] [41]) to activate the wild-type (WT) and mutant RREs (containing the RBP-Jk element) to different levels in BL-41 cells. Surprisingly, RBP-JK/VP16 only weakly activated the WT Mta RRE to a maximum of approximately fourfold (Fig. 1B); the orientation of the RRE relative to the TATA box did not affect this result (data not shown). Furthermore, we obtained similar results in RBP-Jk null cells (OT11; data not shown), suggesting that endogenous RBP-Jk was not occluding the ability of ectopically expressed RBP-Jk/VP16 to access the RBP-Jk site. Expression of constitutively active NICD showed similar weak effects on Mta RRE-dependent reporter gene expression (Fig. 1B).
For a control, we replaced the Mta RRE in the reporter plasmid with the RBP-Jk-containing element from the EBV latency C promoter (Cp) (Fig. 1A, bottom line). The number of 5′ and 3′ flanking base pairs and the spacing to the TATA box were conserved. As expected, we found that both RBP-Jk/VP16 and NICD strongly activated the Cp reporter independently (Fig. 1C). This confirmed that the RBP-Jk element cloned at that position was competent to respond to activation. It also confirmed that RBP-Jk/VP16 was expressed in all transfectants. These data suggested that, although RBP-Jk alone binds avidly to the Mta RBP-Jk site in vitro, it may be unable to bind in transfected cells.
Transcriptionally deficient Rta restores the ability of constitutively active RBP-Jk to transactivate KSHV genes in a promoter-specific manner.
Since we had previously shown that KSHV Rta binds independently to the Mta RRE (61), we reasoned that Rta might bind DNA cooperatively with RBP-Jk. ORF50ΔSTAD is a C-terminal truncation mutant of Rta that lacks the transcriptional activation domain (62) but maintains the ability to bind to both DNA and RBP-Jk (53, 61). ORF50ΔSTAD is thus an ideal protein to test Rta's effects on RBP-Jk independently of Rta-mediated transactivation. Figure 1D shows that neither RBP-Jk/VP16 nor ORF50ΔSTAD activates the Mta RRE alone in transfected BL-41 cells. However, if we cotransfect a constant amount of RBP-Jk/VP16 expression vector with increasing amounts of the ORF50ΔSTAD expression vector, we achieve a maximum of about 30- to 40-fold activation. The effects of RBP-Jk/VP16 and ORF50ΔSTAD, alone or together, were similar in BJAB cells; however, the magnitude of transactivation when the plasmids were cotransfected was about half that in BL-41 cells (Fig. 1E). These data suggest that Rta restores or enhances the ability of RBP-Jk/VP16 to bind to the Mta promoter. Definitive proof of this DNA binding mechanism came from chromatin immunoprecipitations to detect the amount of Mta RRE of the reporter plasmid bound to RBP-Jk/VP16. When the transfections represented in Fig. 1D and E were repeated using the full-length Mta promoter, but followed by ChIPs using an anti-VP16 antibody, the amount of Mta promoter recovered in the VP16 immunoprecipitation was enriched in the presence of ORF50ΔSTAD (data not shown). Moreover, the increase in enrichment of DNA-bound RBP-Jk/VP16 when coexpressed with ORF50ΔSTAD (divided by that from RBP-Jk/VP16 expressed alone) was virtually identical to the increase in transactivation in the presence or absence of ORF50ΔSTAD as measured by luciferase activity (Fig. 1D and E; approximately sixfold in BL-41 cells and threefold in BJAB cells).
Confirming that endogenous RBP-Jk is regulated similarly, Fig. 1F shows that constitutively active NICD also weakly activates the Mta promoter when expressed alone. However, NICD cooperates with coexpressed ORF50ΔSTAD to a maximum of nearly 40-fold. This result proves that the stimulation of RBP-Jk/VP-16 DNA binding by ORF50ΔSTAD is not simply a peculiarity of the RBP-Jk/VP16 fusion molecule.
Among promoters previously identified as targets of Rta (62, 64), the KSHV thymidine kinase (TK) promoter contains an RBP-Jk element located in a position similar to that of the Mta RRE (Fig. 1A). Figure 1G shows that neither RBP-JK/VP16 nor ORF50ΔSTAD transactivates the TK element when expressed individually or together. Therefore, the ability of Rta to restore DNA binding of RBP-Jk is promoter specific.
Rta and cellular RBP-Jk bind with alternative specificity to the RRE of the Mta promoter.
Figure 2 shows a comparison of the relative magnitudes of Rta-mediated transactivation of the WT Mta RRE and a series of linker scanning mutants (previously published in reference 61). The RBP-Jk element from the TK promoter is shown at the bottom of Fig. 2 and was maximally transactivated by Rta to only about 18% of the Mta RRE in BL-41 cells. The relative magnitudes of activation of each reporter shown are quantitatively similar in CV-1 and BL41 cells (61; this study). To compare these transactivation data with DNA binding by Rta and RBP-Jk, the proteins were expressed in E. coli and purified to homogeneity and then tested by electrophoretic mobility shift assays. As summarized in Fig. 2, RBP-Jk binds to all of the DNAs containing its consensus sequence, including the TK element, but not to any DNAs that have mutations in the element. Data for the WT element and m1 to m3 are shown in Fig. 3, top panel, and demonstrate dose-dependent binding of GST-RBP-Jk to the WT and m1 probes.
FIG. 2.

RBP-Jk binding is essential for Rta-mediated activation of the Mta promoter, but trimeric complex formation is necessary for optimal activation. (Left) Alignment of the sequences of the top strands of the WT and mutant RREs from the TK and Mta promoters. Mutated base pairs are indicated by bold underlined type. (Middle) Summary of results of EMSA experiments depicted in Fig. 3 and 4 and described in the text. (Right) Percent activation summarizes the ability of WT Rta to transactivate reporter vectors for each of the indicated sequences (adapted from reference 61). The relative transactivation values are similar in CV-1 cells (61) or BL-41 cells (this study).
FIG. 3.

RBP-Jk and Rta bind independently to overlapping sites in the Mta promoter. Purified recombinant GST-RBP-Jk (top), MBP-Rta FL (middle), or His6-Rta (aa 1 to 272) (bottom) were mixed in increasing amounts with the indicated 32P-labeled oligonucleotides (indicated by the black triangles above the lanes) and analyzed by EMSAs. Lanes 0, buffer alone plus probe.
The middle panel of Fig. 3 shows that MBP-FL Rta also binds avidly to the WT and m3 DNAs and binds weakly but detectably to the m1 and m2 DNAs, each in a dose-dependent fashion. Furthermore, MBP-FL Rta forms two or three complexes with each of the DNAs, which may represent different oligomeric forms of the DNA-bound Rta.
The bottom panel of Fig. 3 shows that the Rta DNA binding domain, Rta (1-272), binds to the Mta RRE with a specificity that resembles MBP-Rta FL, but with much more pronounced preference for the WT and m3 DNAs. In general, both Rta FL and truncated Rta share a preference for the AT-rich sequence located just upstream of the RBP-Jk consensus site, agreeing with other reports (56, 101). Conversely, both Rta proteins show reduced or no binding to probes in which the AT-rich sequence has been mutated by GC-rich linkers. This preference for AT-rich sequence is reflected in the inability of Rta (1-272) to bind the TK probe (Fig. 2). m2 is a better competitor than m1 for Rta (1-272) bound to labeled WT probe (data not shown), in agreement with the binding preference of MBP-Rta FL.
In total, the data summarized in Fig. 2 demonstrate that Rta and RBP-Jk bind with alternative but overlapping specificity to the Mta RRE. As expected, elements that do not bind to RBP-Jk in vitro are those that are not transactivated by Rta (m2, m3, and m8). Elements that bind to RBP-Jk, but not Rta (1-272) in vitro, are activated by Rta at less than WT Mta RRE levels (m1 and TK). Promoters that bind both proteins in vitro are activated by Rta at WT levels or greater (except for m9). Furthermore, the results for m3 and m8 affirm that DNA binding by Rta independently of RBP-Jk is not sufficient to activate the Mta promoter.
Maximal transactivation of RBP-Jk elements in the KSHV genome corresponds with trimeric complex formation among KSHV Rta, RBP-Jk, and DNA.
Having established the DNA binding preferences for the individual proteins to the Mta RRE, we reasoned that mixing the two proteins might help us understand how ORF50ΔSTAD restores RBP-Jk/VP16-mediated transactivation. Figure 4A, lanes 2 and 3, confirms that MBP-FL Rta and RBP-Jk bind independently to the WT Mta RRE. Figure 4A, lane 4, shows the complexes resulting from mixing the two proteins; comparison of lane 4 to lanes 2 and 3 reveals three effects: (i) a decrease in the RBP-Jk-specific complex, (ii) complete disappearance of the Rta-specific complexes, and (iii) formation of a new complex migrating at a position intermediate between the two slow-moving Rta-specific complexes seen in lane 2. These results suggest that Rta and RBP-Jk can bind simultaneously to the WT Mta RRE but that only specific oligomeric forms of Rta form the new supershifted complex. Addition of an MBP-specific antibody to the Rta/DNA complexes results in loss of the two slowly migrating complexes and appearance of a new supershifted complex, confirming the presence of Rta (Fig. 4A, compare lanes 2 and 6). Similarly, addition of the same antibody to the putative Rta/RBP-Jk/DNA complex of lane 4 results in the loss of that complex and the appearance of a complex that is retained in the well and does not enter the gel (lane 8; not visible in this figure), confirming the presence of Rta. Appearance of the Rta/antibody/DNA complexes in lane 8 (similar to lane 6) suggests that the antiserum can release a fraction of Rta from the RBP-Jk/DNA complex.
FIG. 4.

RBP-Jk and Rta form a trimeric complex when binding sites for both proteins are intact. (A) Mixtures of purified RBP-Jk, MBP-Rta FL, and anti-MBP antibody (as indicated) were analyzed by EMSAs as described in the legend to Fig. 3. Lanes 0, buffer alone plus probe. (B and C) Rabbit reticulocyte lysate was programmed with RBP-Jk cDNA and l-[35S]methionine to label RBP-Jk protein and then mixed with buffer alone (lanes 0) or with the indicated proteins. That mixture was incubated with the indicated unlabeled oligonucleotides and tested by EMSAs. A weak background band is seen in all lanes, regardless of DNA addition.
In the approach used in Fig. 4A, the Rta/DNA complexes (lane 2) migrate with a mobility very similar to that of the putative trimeric Rta/RBP-Jk/DNA complex (lane 4), so distinguishing the two types of complexes was difficult. Therefore, we sought a more conclusive method to distinguish the trimeric complex (Rta/RBP-Jk/DNA) from the Rta/DNA complex. We generated RBP-Jk as an 35S-labeled protein in rabbit reticulocyte lysates (RRL) and performed EMSAs using unlabeled DNA probes. In this way, the only labeled molecule was RBP-Jk. In Fig. 4B, lanes 1 and 2, the RBP-Jk-programmed RRL was preincubated with nonspecific DNA only or with nonspecific DNA and the WT Mta RRE oligonucleotide, respectively. Only lane 2 shows a robust RBP-Jk/DNA complex, migrating to a position similar to that of RBP-Jk bound to labeled DNA (i.e., Fig. 4A, lane 3). In the absence of the Mta RRE oligonucleotide, only weak background bands were seen (Fig. 4B, lane 1), but a robust 35S-labeled RBP-Jk is seen in lane 2, which suggests that RBP-Jk does not enter the gel in the absence of the WT Mta RRE probe.
Figure 4B, lanes 3 and 4, shows the results of further addition of MBP-FL-Rta or the MBP protein alone, respectively. Only MBP-Rta FL generates a new, supershifted complex (lane 3, indicated by arrows); since RBP-Jk is the only labeled molecule, RBP-Jk is clearly bound directly or indirectly to DNA. Addition of the anti-MBP serum results in a further retardation of the supershifted complex (compare lanes 3 and 5), indicating that MBP-Rta FL is associated with the RBP-Jk/DNA complex. The antiserum has no effect on complex formation in the presence of MBP alone (lane 6). These data confirm assembly of a trimeric Rta/RBP-Jk/DNA complex on the WT RRE from the Mta promoter.
We extended the analysis to the series of oligonucleotides shown in Fig. 2. RBP-Jk binds the unlabeled m1 DNA quantitatively similarly to the WT sequence (Fig. 4B, lane 7), but the addition of two different amounts of Rta does not result in formation of the trimeric complex (Fig. 4B, lanes 8 and 9). As a control for specificity, RBP-Jk does not form a complex with the m3 DNA, which is mutated in the RBP-Jk consensus sequence (Fig. 4B, lanes 12 to 16). The TK element, which is very weakly activated by Rta, binds to RBP-Jk, but the addition of Rta does not produce the trimeric complex (Fig. 4B, lanes 17 to 20). Taken together, these data suggest that formation of trimeric Rta/RBP-Jk/DNA complexes requires intact, individual binding sites for both Rta and RBP-Jk. By comparing Rta's ability to transactivate each sequence (Fig. 2), it is clear that trimeric complex formation corresponds with 100% activation of the RRE by Rta (as in the WT Mta RRE). However, formation of only an RBP-Jk/DNA complex, with no corresponding trimeric complex, results in reduced Rta transactivation (Mta m1 and TK).
These conclusions are further supported by the data of Fig. 4C. The trimeric complex assembles on the m4 DNA, but not on the m9 DNA (compare lanes 5 to 7 with lanes 8 and 9). Strong Rta transactivation corresponds with trimeric complex formation, and thus, independent DNA binding by Rta or RBP-Jk is not sufficient for robust Rta-mediated transactivation of the Mta RRE (summarized in Fig. 2). Furthermore, the protein/DNA interactions with m9 suggest that formation of the trimeric complex requires a more extended DNA sequence recognition relative to the DNA binding requirements of either protein alone.
Trimeric complex formation is required for ORF50ΔSTAD to restore the ability of RBP-Jk/VP-16 to transactivate the Mta RRE.
Since trimeric complex formation of Rta, RBP-Jk and the Mta or TK DNA (Fig. 4) corresponded with the ability of ORF50ΔSTAD to restore transactivation by RBP-Jk/VP16 on those promoters (Fig. 1D, E, and G), we extended the in vivo analyses using the Mta RRE mutants. As shown in Fig. 5, only the m4 element is transactivated when ORF50ΔSTAD is coexpressed with RBP-Jk/VP16; this effect is not seen for the m1, m2, m3, or m9 promoter. This is consistent with formation of the trimeric complex exclusively on the m4 element. Furthermore, since Rta binds to m3 but not m2, independent Rta binding to the promoter is not sufficient to recruit RBP-Jk to DNA in the absence of its binding site.
FIG. 5.

Trimeric complex formation is required for transcriptionally deficient Rta to restore the ability of constitutively active RBP-Jk to transactivate the Mta RRE. Transient cotransfections were performed with the indicated Mta RRE reporter plasmids and expression vectors and analyzed as described in the legend to Fig. 1D. The sequence of each of the RREs is given above each graph. The RBP-Jk consensus site is shaded. Mutated base pairs are indicated by boldface, underlined type.
The results presented above establish the cis-acting DNA sequence requirements for restoring RBP-Jk/VP16 binding to the Mta RRE by Rta. To test the trans-acting protein-protein requirements for this mechanism, we designed a series of C-terminal truncations of Rta that preserved the N-terminal DNA binding domain of the protein. Two of the mutants, ORF50ΔAatII and ORF50ΔSTAD (diagrammed in Fig. 6A) are stably expressed in E. coli and in BL-41 cells in the nuclei (Fig. 6B and data not shown). ORF50ΔAatII (aa 1 to 414) binds independently to the WT Mta RRE in vitro (data not shown). However, Fig. 6C shows that the ORF50ΔAatII truncation mutant fails to form the trimeric complex with 35S-labeled RBP-Jk and DNA.
FIG. 6.

DNA binding by Rta is not sufficient to form the trimeric complex and restore DNA binding by RBP-Jk/VP16. (A) Primary amino acid structure of indicated WT and truncated Rta proteins. The basic-amino-acid-rich domain (++), leucine repeat (LR), serine-threonine-rich domain (ST), and activation domain (AD) are indicated. (B) Western blot of BL-41 cells transfected with the indicated plasmids. The migration position of each protein is indicated by a small black square in the blot. (C) EMSA was performed as described in the legend to Fig. 4B, using the indicated proteins and unlabeled Mta WT RRE oligonucleotide. (D) BL-41 cells were cotransfected and analyzed as described in the legend to Fig. 1D, using the WT RRE reporter plasmid and indicated expression plasmids. Fold activation is shown on the y axis.
Furthermore, ORF50ΔAatII is unable to activate transcription of the Mta RRE in either the absence or presence of RBP-Jk/VP16 (Fig. 6D), suggesting that the mutant cannot restore RBP-Jk binding to DNA. These experiments confirm that trimeric complex formation of Rta with RBP-Jk and DNA in vitro corresponds with the ability of Rta to restore DNA binding of RBP-Jk in vivo. Furthermore, amino acids 414 to 530 demarcate the ends of the Rta domains required for both of these effects.
RBP-Jk is enriched on promoters in an Rta-dependent fashion during reactivation of KSHV without a change in its steady-state levels.
To determine whether the mechanism we defined above operates in virus-infected cells, we used ChIPs to evaluate the DNA binding status of RBP-Jk during viral latency and reactivation. We chose as our model PEL cells (BC-3 and BCBL-1), which harbor latent, episomal KSHV that reactivates when TPA is added to the cell growth medium in an Rta-dependent fashion. Figure 7A, top panel, shows that equal amounts of the Mta promoter are amplified from “input” chromatin in the absence or presence of TPA, but RBP-Jk is enriched on the Mta promoter only in the presence of Rta (i.e., following TPA addition to the cells). As shown in Table 1, the increase in enrichment over control IgG is approximately 73.2-fold in BCBL-1 cells, as quantitated by real-time PCR with molecular beacons. Amplification of chromatin captured by the control IgG or without template addition (NTC) shows no product for the Mta promoter. The same chromatin was used to detect the RBP-Jk-containing segments of the KSHV K-bZIP and K14 (viral G-protein-coupled receptor) promoters (Fig. 7A, middle and bottom panels, respectively). In both cases, input chromatin showed equivalent amounts of PCR amplification, while RBP-Jk was dramatically enriched only in the presence of Rta expression after TPA addition (49.3-fold enrichment over control IgG for K14 [Table 1]). For all three promoters, RBP-Jk was barely detectable during viral latency in the absence of Rta. Therefore, exclusion of RBP-Jk binding from at least three KSHV promoters appears to be a general phenomenon of KSHV latency, while induction of Rta expression during reactivation results in recruitment of RBP-Jk to these promoters.
FIG. 7.

RBP-Jk is selectively enriched on viral and cellular promoters during reactivation of KSHV. (A and B) Immunoprecipitations were performed using the indicated antibodies with chromatin from BC-3 cells treated with TPA (+) or not treated with TPA (−). DNA was detected by PCR using primers specific for each of the indicated viral (A) or cellular (B) promoters. All of the ChIPs were performed using IgG and PBS as negative controls for the anti-RBP-Jk antibody and PCR, respectively. All promoters except CD23 showed enrichment of RBP-Jk DNA binding only after TPA addition. For promoters in which the change for control IgG was between 0 and 1 (i.e., when the CT value in the absence of TPA exceeded the CT value in the presence of TPA), only the PBS control is shown. Lanes M, molecular size markers. hIL-6, human interleukin-6; HES-1, human hairy/enhancer-of-split; NTC, nontemplate control; PBS, phosphate-buffered saline substituted for an antibody. (C) The WT Cp oligonucleotide (depicted in Fig. 1A) was labeled with 32P and analyzed by EMSAs by mixing with RRL, histidine-tagged RBP-Jk, or extracts from BC-3 cells treated with TPA (+TPA) and not treated with TPA (−TPA) as indicated. In the right panel, the indicated protein samples were preincubated with WT or mutant (Mut) Cp oligonucleotide prior to addition of labeled WT Cp probe. The arrow points to the protein/DNA complex that is supershifted by addition of anti-RBP-Jk serum (not shown).
TABLE 1.
Increases in enrichment of RBP-Jk on viral promotersa
| Promoter | Fold enrichment in cells:
|
||
|---|---|---|---|
| BCBL-1 (PEL) | 293/BAC36wtb | 293/BAC36Δ50b | |
| ORF57/Mta | 73.2 | 191.3 | 0.6 |
| K14/vGPCR | 49.3 | 14.9 | 1.0 |
ChIPs were performed as described in Materials and Methods. Fold enrichment equals RBP-Jk DNA binding in the presence of TPA divided by control IgG (i.e., fold enrichment = 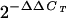 of RBP-Jk antibody/
of RBP-Jk antibody/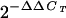 of IgG control).
of IgG control).
293 cells were stably transfected with the indicated bacmid constructs as described in Materials and Methods.
A critical question for these experiments is whether Rta is required for stimulation of RBP-Jk DNA binding to the viral genome. To test this question, we generated stable transfectants of 293 cells with either BAC36wt (a wild-type KSHV bacmid clone) or BAC36Δ50, a bacmid clone with most of Rta's open reading frame deleted. BAC36Δ50 is defective in KSHV reactivation in 293 cells (96). Stably transfected cell populations were induced by TPA treatment or left untreated (similar to BCBL-1 cells) and then analyzed by ChIP analyses using anti-RBP-Jk antibody or control IgG. Table 1 shows that RBP-Jk is enriched on both the ORF57/Mta and K14 promoters following TPA treatment but only in cells harboring the WT but not Rta-deleted (BAC36Δ50) virus. In the BAC36Δ50 transfectants, the amount of ORF57 or K14 promoter immunoprecipitated was equal regardless of TPA addition or antibody. This proves that enrichment of RBP-Jk on the ORF57 and K14 promoters requires expression of Rta.
To extend this analysis to cellular promoters, we analyzed RBP-Jk binding to the human IL-6, HES-1, and CD23 promoters. The top and middle panels of Fig. 7B, show that, similar to the viral promoters, RBP-Jk is virtually undetectable on the human IL-6 and HES-1 promoters during latency but becomes dramatically enriched on the promoters in the presence of Rta following TPA addition. Conversely, the amount of RBP-Jk bound to the CD23 promoter (Fig. 7B, bottom panel) remains unchanged regardless of TPA addition to the cells (note that the lanes corresponding to the IgG and anti-RBP-Jk ChIPs appear equivalent by this end point quantitation, but real-time quantitation using PCR with molecular beacons revealed that the anti-RBP-Jk antibody chromatin immunoprecipitates about ninefold more of the CD23 RBP-Jk site than the control IgG does). For all three cellular promoters, equal amounts of input chromatin were immunoprecipitated (input lanes); for a control, substitution of the RBP-Jk antibody with PBS resulted in no DNA amplification (for promoters in which the change for control IgG was between 0 and 1 [i.e., when the ΔCT in the absence of TPA exceeded the ΔCT in the presence of TPA]; only the PBS control is shown). Therefore, for at least the cellular IL-6 and HES-1 promoters, DNA binding of RBP-Jk is also regulated by Rta during reactivation.
To determine whether the steady-state level of RBP-Jk is altered by TPA addition to PEL cells, we prepared whole nuclear extracts from BC-3 cells with or without TPA treatment and analyzed the quantity of proteins that bind to the EBV Cp probe. The position of migration of recombinant RBP-Jk bound to the Cp probe is shown as a reference (Fig. 7C, lanes 1 to 7). BC-3 nuclear extracts form six protein/DNA complexes on the probe; however, the abundance of all of the complexes, including the RBP-Jk-specific complex, was similar in untreated or TPA-treated extracts (lanes 8 and 9). In data not shown, the anti-RBP-Jk serum supershifts only the protein/DNA complex indicated by the arrow in Fig. 7C.
A comparison of lanes 17 and 24 of Fig. 7C confirms that RBP-Jk protein levels are unchanged in PEL cells when treated with TPA. Furthermore, lanes 11 to 25 demonstrate that preincubation of the extract with an unlabeled Cp probe containing the WT RBP-Jk site, but not mutant site, results in loss of five of the six complexes. Since only one of those complexes has a migration position that corresponds to that of recombinant RBP-Jk (lanes 1 to 7), the identities of the four additional complexes that are specific for the WT RBP-Jk site are currently unknown.
EBNA2 and NICD do not induce KSHV lytic reactivation.
Our data suggest that Rta activates the Notch pathway in part by stimulating DNA binding of RBP-Jk, a mechanism fundamentally different from that established for NICD and EBNA2. One powerful means to compare the functional consequences of these mechanistic differences is to measure the magnitude of viral reactivation in response to Notch activation. As shown in Fig. 8, TPA treatment, the positive control, induces an approximately 17-fold increase in the release of mature virions 6 days after the addition of TPA. As measured by real-time PCR with molecular beacons, this agrees well with reactivation quantitated by Southern blotting (42). As previously shown (64), transfection of the expression vector for the Mta transactivator, a negative control, was unable to reactivate KSHV. As expected, transfection of the Rta expression vector resulted in ca. 7.5-fold induction of viral release from latently infected cells. In contrast, NICD and EBNA2 were both unable to reactivate the virus.
FIG. 8.

EBNA2 and NICD do not induce complete KSHV lytic reactivation. BCBL-1 cells were transfected with the indicated expression vectors, or treated with TPA, and encapsidated viral DNA was purified from the culture medium 6 days posttransfection. Viral DNA in each sample was quantitated using real-time PCR with molecular beacons for quantitation.
Although the overall amount of virus released is lower for transfection of Rta plasmid than for TPA treatment, the difference represents the lower percentage of cells that were transfected (5% Rta positive at 2 days posttransfection, 2.8% K8.1 positive at 2 days) compared to the percentage of cells that were induced by TPA (12.5% Rta positive 6 days post-TPA addition, 7.3% K8.1 positive at 6 days).
DISCUSSION
In this report, we show that Rta activates the Notch pathway in part by stimulating DNA binding of RBP-Jk, a novel mechanism not conserved with EBNA2 and NICD. RBP-Jk does not bind constitutively to the Mta promoter (Fig. 1B and F) in vivo, and only Rta, but not EBNA2 or NICD, activates transcription of the RRE in transient transfections (Fig. 1 and 2) (61). Constitutively active RBP-Jk and a truncation mutant of Rta lacking its transcriptional activation domain demonstrate that full activation of the Mta promoter is dependent on both the intact Rta and RBP-Jk binding sites in the RRE. Moreover, transactivation corresponds with trimeric complex formation of Rta, RBP-Jk, and the RRE, as measured by EMSAs (Fig. 2 and 4).
DNA binding of RBP-Jk is regulated similarly in KSHV-infected cells (Fig. 7 and Table 1), and only Rta induces complete KSHV lytic reactivation (Fig. 8), further distinguishing Rta's mechanism from that established for EBNA2 and NICD. In agreement, others have demonstrated that NICD induces only a subset of the viral genes activated by Rta in cells latently infected by KSHV (6). Together with our observation that ORF50ΔSTAD cooperates with NICD to stimulate transcription from the Mta promoter (Fig. 1F), we conclude that Rta enhances DNA binding of endogenous RBP-Jk. As RBP-Jk is a transcriptional repressor, we would expect that ORF50ΔSTAD expression in the absence of a Notch signal would enhance the repressing effect of RBP-Jk. Indeed, we have previously shown that ectopic overexpression of ORF50ΔSTAD completely suppressed the spontaneous KSHV lytic reactivation characteristically observed in latently infected PEL cells (62).
Transcriptional cooperation of the Rta mutant with NICD might reflect simultaneous binding of Rta and NICD to the RBP-Jk N terminus and BTD, respectively. Such a mechanism would further distinguish Rta, since NICD and EBNA2 suppress each other by competing for binding to the BTD (40). However, our data do not exclude the alternative hypothesis that only a transient interaction between the Rta mutant and RBP-Jk is required to target it to DNA. Nonetheless, we consider it likely that Rta also removes transcriptional corepressors from DNA-bound RBP-Jk, similar to the transcriptional activation mechanisms employed by EBNA2 and NICD. Indeed, other studies have shown that Rta also binds independently to the RBP-Jk central repression domain, supershifts DNA-bound RBP-Jk, and recruits chromatin remodeling proteins to activate transcription (29, 53). Stimulation of RBP-Jk DNA binding, as demonstrated in this report, describes an additional level of regulation of the Notch pathway by KSHV Rta that we propose is a pre- or corequisite for Rta-mediated transactivation on some promoters.
Although aa 1 to 272 of Rta bind to the Mta RRE in vitro (Fig. 3) and aa 170 to 400 of Rta bind RBP-Jk in GST pull-down experiments (53), neither region of Rta alone is sufficient to stimulate RBP-Jk DNA binding. Instead, aa 415 to 530 of Rta are required for trimeric complex formation with RBP-Jk/DNA in vitro and for restoring binding of RBP-Jk/VP16 to the Mta promoter in vivo (Fig. 6). This region shares little homology with EBNA2 or NICD and contains no motifs characteristic of DNA binding domains, so there are no bioinformatic clues to explain its function. Rta forms multimers in vitro, and oligomerization of Rta is critical for transactivation (56, 62). The G/C bp that is mutated in m9 is required for trimeric complex formation, but not independent DNA binding of Rta or RBP-Jk (Fig. 2). Therefore, we propose that homomultimers of Rta bind and straddle RBP-Jk to enable Rta to make critical contacts with the promoter on both sides of RBP-Jk (i.e., m1 and m9) and form the stable trimeric complex. It is likely that aa 415 to 530 are crucial for at least one of these interactions.
The literature suggests that the interaction of Rta with the Mta promoter is complex. A DNA binding mutant of Rta appears to transactivate the Mta promoter (with delayed kinetics) in PEL cells (8); assuming that this effect is independent of other KSHV transactivators, this suggests that a cellular protein is sufficient to mediate interaction of mutant Rta with the promoter in vivo. Rta has been shown to interact with the cellular protein C/EBPα as a DNA binding partner at the Mta promoter (92). In this model, Rta must first induce C/EBPα expression (92). In our experiments, C/EBPα is unlikely to function in regulation of Mta, since we show that a mutant of Rta lacking its transactivation domain stimulates DNA binding by RBP-Jk/VP16 (Fig. 1D and E). The significance of RBP-Jk-independent activation of the Mta promoter in full viral reactivation is unclear. However, it is possible that the inability of NICD and EBNA2 to participate in similar complex interactions with the Mta promoter might also contribute to their inability to fully reactivate KSHV from latency.
RBP-Jk bound avidly to the consensus elements of the Mta and TK promoters in vitro (Fig. 3), yet constitutively active RBP-Jk/VP16 did not activate transcription of these promoters in vivo (Fig. 1). Similar discrepancies have been reported in other systems. In mice, the HES-1 promoter contains two sites that bind RBP-Jk with high affinity in vitro, yet only one of them responds to activated Notch (35). Similarly, 11 of the 12 transcription units in the Enhancer of Split complex [E(spl)-c] in Drosophila contain at least one RBP-Jk element in their proximal promoters, yet each promoter responds differently to Notch activation (20, 71). In fact, in mouse RBP-Jk knockout studies, there has been no evidence of loss of promoter repression (83, 84). Our data do not reveal the mechanism by which RBP-Jk is excluded from promoters in the absence of Rta coexpression. As Rta is the first protein shown to promote DNA binding by RBP-Jk, it remains to be determined whether Rta counteracts any of the mammalian proteins that inhibit DNA binding of RBP-Jk (and if so, how). For example, murine protein KyoT2 inhibits RBP-Jk DNA binding and dislocates it from DNA (85), and NF-κB (p50/p65) and Ikaros bind DNA competitively with RBP-Jk when their recognition elements overlap (4, 72). We hypothesize that Rta either competes with an RBP-Jk-inhibiting protein for binding to RBP-Jk or DNA or catalyzes proteolysis of one of the inhibitory factors through its E3 ubiquitin ligase activity (98).
KSHV infection is associated primarily with two human malignancies, KS and PEL (69). Genes in the Notch pathway display both loss-of-function and gain-of-function phenotypes that may contribute to the pathophysiology of both cancers. KS tumors display a marked neoangiogenesis (69), and multiple genetic models implicate the Notch signaling pathway in various aspects of vascular biology, including angiogenesis, proliferation, migration, network formation, and quiescence (5). In most cases, targeted mutation of Notch pathway genes leads to vascular defects (46, 97). Notch-transformed cells further promote tumor maintenance and growth through Notch-dependent induction of angiogenesis in a juxtacrine fashion (65, 100). In the lymphoid compartment, uncontrolled Notch signaling is often associated with T-cell leukemias (21, 23, 26, 73, 74, 93). Ligand-independent activation of the Notch pathway by EBV EBNA2 is crucial for B-cell transformation through transactivation of viral and cellular gene expression (107). Therefore, there is substantial evidence to support a role for activated Notch signaling in KSHV-associated cancers. Although there is an epidemiologic connection between KSHV reactivation and KS risk (68, 94), Rta's RBP-Jk-dependent function as the lytic switch protein would have to be relatively inefficient to allow a reactivating cell to contribute to malignancy. It is likely that KSHV reactivation also promotes viral dissemination, horizontal spread, and increase in viral load.
The inability of RBP-Jk/VP16 and NICD to activate the Mta promoter in uninfected cells (Fig. 1) suggests that no KSHV proteins are required for this effect. However, KSHV LANA-1 (latency-associated nuclear antigen 1) can also compete with Rta for functional interactions with RBP-Jk (49). Why has KSHV evolved multiple mechanisms to disrupt normal RBP-Jk activity? A KSHV-infected cell likely has frequent exposure to activated Notch signaling. Vascular endothelial growth factor and fibroblast growth factor, two of the cytokines elevated in KS tumors, lie upstream of the Notch pathway and activate expression of Notch receptors and ligands (50, 59). Indeed, primary and immortalized KS cells overexpress activated Notch, which is essential to prevent apoptosis of these cells (15). Furthermore, 70% of PELs are coinfected with KSHV and EBV (19), suggesting that B cells are capable of supporting coincident latent infection by both viruses. EBNA2 is the first protein expressed following EBV infection, leading to ligand-independent activation of Notch signaling. Since RBP-Jk is excluded from key KSHV promoters, we propose that the virus has evolved this mechanism to maintain latency even in environments of Notch activation. The ability of Rta to recruit RBP-Jk to KSHV promoters of essential genes thus ensures that Rta is the only protein with the ability to efficiently reactivate the virus.
Acknowledgments
We thank Yan Yuan, Gregory Pari, S. J. Gao, Tom Kadesch, Bill Sugden, and Tasuku Honjo for reagents and cells and Vivian Bellofatto for critical reading of the manuscript.
This work was supported by the American Cancer Society (RSG MBC-103737) and the NJ Commission on Cancer Research.
REFERENCES
- 1.Allenspach, E. J., I. Maillard, J. C. Aster, and W. S. Pear. 2002. Notch signaling in cancer. Cancer Biol. Ther. 1:466-476. [DOI] [PubMed] [Google Scholar]
- 2.Amakawa, R., W. Jing, K. Ozawa, N. Matsunami, Y. Hamaguchi, F. Matsuda, M. Kawaichi, and T. Honjo. 1993. Human Jk recombination signal binding protein gene (IGKJRB): comparison with its mouse homologue. Genomics 17:306-315. [DOI] [PubMed] [Google Scholar]
- 3.Axelson, H. 2004. Notch signaling and cancer: emerging complexity. Semin. Cancer Biol. 14:317-319. [DOI] [PubMed] [Google Scholar]
- 4.Beverly, L. J., and A. J. Capobianco. 2003. Perturbation of Ikaros isoform selection by MLV integration is a cooperative event in Notch(IC)-induced T cell leukemogenesis. Cancer Cell 3:551-564. [DOI] [PubMed] [Google Scholar]
- 5.Bicknell, R., and A. L. Harris. 2004. Novel angiogenic signaling pathways and vascular targets. Annu. Rev. Pharmacol. Toxicol. 44:219-238. [DOI] [PubMed] [Google Scholar]
- 6.Chang, H., D. P. Dittmer, S. Y. Chul, Y. Hong, and J. U. Jung. 2005. Role of Notch signal transduction in Kaposi's sarcoma-associated herpesvirus gene expression. J. Virol. 79:14371-14382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chang, P. J., D. Shedd, L. Gradoville, M. S. Cho, L. W. Chen, J. Chang, and G. Miller. 2002. Open reading frame 50 protein of Kaposi's sarcoma-associated herpesvirus directly activates the viral PAN and K12 genes by binding to related response elements. J. Virol. 76:3168-3178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chang, P. J., D. Shedd, and G. Miller. 2005. Two subclasses of Kaposi's sarcoma-associated herpesvirus lytic cycle promoters distinguished by open reading frame 50 mutant proteins that are deficient in binding to DNA. J. Virol. 79:8750-8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen, J., K. Ueka, S. Sakakibara, T. Okuno, and K. Yamanishi. 2000. Transcriptional regulation of the Kaposi's sarcoma-associated herpesvirus viral interferon regulatory factor gene. J. Virol. 74:8623-8634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Christensen, S., V. Kodoyianni, M. Bosenberg, L. Friedman, and J. Kimble. 1996. lag-1, a gene required for lin-12 and glp-1 signaling in Caenorhabditis elegans, is homologous to human CBF1 and Drosophila Su(H). Development 122:1373-1383. [DOI] [PubMed] [Google Scholar]
- 11.Chung, C. N., Y. Hamaguchi, T. Honjo, and M. Kawaichi. 1994. Site-directed mutagenesis study on DNA binding regions of the mouse homologue of Suppressor of Hairless, RBP-J kappa. Nucleic Acids Res. 22:2938-2944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cohen, J. I., and E. Kieff. 1991. An Epstein-Barr virus nuclear protein 2 domain essential for transformation is a direct transcriptional activator. J. Virol. 65:5880-5885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen, J. I., F. Wang, and E. Kieff. 1991. Epstein-Barr virus nuclear protein 2 mutations define essential domains for transformation and transactivation. J. Virol. 65:2545-2554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cohen, J. I., F. Wang, J. Mannick, and E. Kieff. 1989. Epstein-Barr virus nuclear protein 2 is a key determinant of lymphocyte transformation. Proc. Natl. Acad. Sci. USA 86:9558-9562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Curry, C. L., L. L. Reed, T. E. Golde, L. Miele, B. J. Nickoloff, and K. E. Foreman. 2005. Gamma secretase inhibitor blocks Notch activation and induces apoptosis in Kaposi's sarcoma tumor cells. Oncogene 24:6333-6344. [DOI] [PubMed] [Google Scholar]
- 16.Deng, H., J. T. Chu, M. B. Rettig, O. Martinez-Maza, and R. Sun. 2002. Rta of the human herpesvirus 8/Kaposi sarcoma-associated herpesvirus up-regulates human interleukin-6 gene expression. Blood 100:1919-1921. [DOI] [PubMed] [Google Scholar]
- 17.Deng, H., M. J. Song, J. T. Chu, and R. Sun. 2002. Transcriptional regulation of the interleukin-6 gene of human herpesvirus 8 (Kaposi's sarcoma-associated herpesvirus). J. Virol. 76:8252-8264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dou, S., X. Zeng, P. Cortes, H. Erdjument-Bromage, P. Tempst, T. Honjo, and L. D. Vales. 1994. The recombination signal sequence-binding protein RBP-2N functions as a transcriptional repressor. Mol. Cell. Biol. 14:3310-3319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Drexler, H., C. Uphoff, G. Gaidano, and A. Carbone. 1998. Lymphoma cell lines: in vitro models for the study of HHV-8+ primary effusion lymphomas (body cavity-based lymphomas). Leukemia 12:1507-1517. [DOI] [PubMed] [Google Scholar]
- 20.Eastman, D. S., R. Slee, E. Skoufos, L. Bangalore, S. Bray, and C. Delidakis. 1997. Synergy between suppressor of Hairless and Notch in regulation of Enhancer of split mγ and mδ expression. Mol. Cell. Biol. 17:5620-5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ellisen, L. W., J. Bird, D. C. West, A. L. Soreng, T. C. Reynolds, S. D. Smith, and J. Sklar. 1991. TAN-1, the human homolog of the Drosophila notch gene, is broken by chromosomal translocations in T lymphoblastic neoplasms. Cell 66:649-661. [DOI] [PubMed] [Google Scholar]
- 22.Fahraeus, R., A. Jansson, A. Ricksten, A. Sjoblom, and L. Rymo. 1990. Epstein-Barr virus-encoded nuclear antigen 2 activates the viral latent membrane protein promoter by modulating the activity of a negative regulatory element. Proc. Natl. Acad. Sci. USA 87:7390-7394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Feldman, B. J., T. Hampton, and M. L. Cleary. 2000. A carboxy-terminal deletion mutant of Notch1 accelerates lymphoid oncogenesis in E2A-PBX1 transgenic mice. Blood 96:1906-1913. [PubMed] [Google Scholar]
- 24.Fryer, C. J., E. Lamar, I. Turbachova, C. Kintner, and K. A. Jones. 2002. Mastermind mediates chromatin-specific transcription and turnover of the Notch enhancer complex. Genes Dev. 16:1397-1411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fryer, C. J., J. B. White, and K. A. Jones. 2004. Mastermind recruits CycC:CDK8 to phosphorylate the Notch ICD and coordinate activation with turnover. Mol. Cell 16:509-520. [DOI] [PubMed] [Google Scholar]
- 26.Girard, L., Z. Hanna, N. Beaulieu, C. D. Hoemann, C. Simard, C. A. Kozak, and P. Jolicoeur. 1996. Frequent provirus insertional mutagenesis of Notch1 in thymomas of MMTVD/myc transgenic mice suggests a collaboration of c-myc and Notch1 for oncogenesis. Genes Dev. 10:1930-1944. [DOI] [PubMed] [Google Scholar]
- 27.Gordadze, A. V., R. Peng, J. Tan, G. Liu, R. Sutton, B. Kempkes, G. W. Bornkamm, and P. D. Ling. 2001. Notch1IC partially replaces EBNA2 function in B cells immortalized by Epstein-Barr virus. J. Virol. 75:5899-5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gradoville, L., J. Gerlach, E. Grogan, D. Shedd, S. Nikiforow, C. Metroka, and G. Miller. 2000. Kaposi's sarcoma-associated herpesvirus open reading frame 50/Rta protein activates the entire lytic cycle in the HH-B2 primary effusion lymphoma cell line. J. Virol. 74:6207-6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gwack, Y., H. J. Baek, H. Nakamura, S. H. Lee, M. Meisterernst, R. G. Roeder, and J. U. Jung. 2003. Principal role of TRAP/mediator and SWI/SNF complexes in Kaposi's sarcoma-associated herpesvirus RTA-mediated lytic reactivation. Mol. Cell. Biol. 23:2055-2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hammerschmidt, W., and B. Sugden. 1989. Genetic analysis of immortalizing functions of Epstein-Barr virus in human B lymphocytes. Nature 340:393-397. [DOI] [PubMed] [Google Scholar]
- 31.Hofelmayr, H., L. J. Strobl, G. Marschall, G. W. Bornkamm, and U. Zimber-Strobl. 2001. Activated Notch1 can transiently substitute for EBNA2 in the maintenance of proliferation of LMP1-expressing immortalized B cells. J. Virol. 75:2033-2040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hsieh, J. J., and S. D. Hayward. 1995. Masking of the CBF1/RBPJ kappa transcriptional repression domain by Epstein-Barr virus EBNA2. Science 268:560-563. [DOI] [PubMed] [Google Scholar]
- 33.Hsieh, J. J., T. Henkel, P. Salmon, E. Robey, M. G. Peterson, and S. D. Hayward. 1996. Truncated mammalian Notch1 activates CBF1/RBPJk-repressed genes by a mechanism resembling that of Epstein-Barr virus EBNA2. Mol. Cell. Biol. 16:952-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsieh, J. J., S. Zhou, L. Chen, D. B. Young, and S. D. Hayward. 1999. CIR, a corepressor linking the DNA binding factor CBF1 to the histone deacetylase complex. Proc. Natl. Acad. Sci. USA 96:23-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jarriault, S., C. Brou, F. Logeat, E. H. Schroeter, R. Kopan, and A. Israel. 1995. Signalling downstream of activated mammalian Notch. Nature 377:355-358. [DOI] [PubMed] [Google Scholar]
- 36.Jeffries, S., D. J. Robbins, and A. J. Capobianco. 2002. Characterization of a high-molecular-weight Notch complex in the nucleus of Notch(ic)-transformed RKE cells and in a human T-cell leukemia cell line. Mol. Cell. Biol. 22:3927-3941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jin, X. W., and S. H. Speck. 1992. Identification of critical cis elements involved in mediating Epstein-Barr virus nuclear antigen 2-dependent activity of an enhancer located upstream of the viral BamHI C promoter. J. Virol. 66:2846-2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johannsen, E., E. Koh, G. Mosialos, X. Tong, E. Kieff, and S. R. Grossman. 1995. Epstein-Barr virus nuclear protein 2 transactivation of the latent membrane protein 1 promoter is mediated by Jκ and PU. 1. J. Virol. 69:253-262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kao, H. Y., P. Ordentlich, N. Koyano-Nakagawa, Z. Tang, M. Downes, C. R. Kintner, R. M. Evans, and T. Kadesch. 1998. A histone deacetylase corepressor complex regulates the Notch signal transduction pathway. Genes Dev. 12:2269-2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kato, H., T. Sakai, K. Tamura, S. Minoguchi, Y. Shirayoshi, Y. Hamada, Y. Tsujimoto, and T. Honjo. 1996. Functional conservation of mouse Notch receptor family members. FEBS Lett. 395:221-224. [DOI] [PubMed] [Google Scholar]
- 41.Kato, H., Y. Taniguchi, H. Kurooka, S. Minoguchi, T. Sakai, S. Nomura-Okazaki, K. Tamura, and T. Honjo. 1997. Involvement of RBP-J in biological functions of mouse Notch1 and its derivatives. Development 124:4133-4141. [DOI] [PubMed] [Google Scholar]
- 42.Kedes, D. H., and D. Ganem. 1997. Sensitivity of Kaposi's sarcoma-associated herpesvirus replication to antiviral drugs. Implications for potential therapy. J. Clin. Investig. 99:2082-2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kidd, S., T. Lieber, and M. W. Young. 1998. Ligand-induced cleavage and regulation of nuclear entry of Notch in Drosophila melanogaster embryos. Genes Dev. 12:3728-3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kitagawa, M., T. Oyama, T. Kawashima, B. Yedvobnick, A. Kumar, K. Matsuno, and K. Harigaya. 2001. A human protein with sequence similarity to Drosophila mastermind coordinates the nuclear form of Notch and a CSL protein to build a transcriptional activator complex on target promoters. Mol. Cell. Biol. 21:4337-4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kovall, R. A., and W. A. Hendrickson. 2004. Crystal structure of the nuclear effector of Notch signaling, CSL, bound to DNA. EMBO J. 23:3441-3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Krebs, L. T., Y. Xue, C. R. Norton, J. R. Shutter, M. Maguire, J. P. Sundberg, D. Gallahan, V. Closson, J. Kitajewski, R. Callahan, G. H. Smith, K. L. Stark, and T. Gridley. 2000. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 14:1343-1352. [PMC free article] [PubMed] [Google Scholar]
- 47.Kurooka, H., and T. Honjo. 2000. Functional interaction between the mouse notch1 intracellular region and histone acetyltransferases PCAF and GCN5. J. Biol. Chem. 275:17211-17220. [DOI] [PubMed] [Google Scholar]
- 48.Lai, E. C. 2004. Notch signaling: control of cell communication and cell fate. Development 131:965-973. [DOI] [PubMed] [Google Scholar]
- 49.Lan, K., D. A. Kuppers, and E. S. Robertson. 2005. Kaposi's sarcoma-associated herpesvirus reactivation is regulated by interaction of latency-associated nuclear antigen with recombination signal sequence-binding protein Jκ, the major downstream effector of the Notch signaling pathway. J. Virol. 79:3468-3478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Lawson, N. D., A. M. Vogel, and B. M. Weinstein. 2002. Sonic hedgehog and vascular endothelial growth factor act upstream of the Notch pathway during arterial endothelial differentiation. Dev. Cell 3:127-136. [DOI] [PubMed] [Google Scholar]
- 51.Lecourtois, M., and F. Schweisguth. 1995. The neurogenic suppressor of hairless DNA-binding protein mediates the transcriptional activation of the enhancer of split complex genes triggered by Notch signaling. Genes Dev. 9:2598-2608. [DOI] [PubMed] [Google Scholar]
- 52.Lee, K. A., A. Bindereif, and M. R. Green. 1988. A small-scale procedure for preparation of nuclear extracts that support efficient transcription and pre-mRNA splicing. Gene Anal. Tech. 5:22-31. [DOI] [PubMed] [Google Scholar]
- 53.Liang, Y., J. Chang, S. Lynch, D. M. Lukac, and D. Ganem. 2002. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jk, the target of the Notch signaling pathway. Genes Dev. 16:1977-1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liang, Y., and D. Ganem. 2003. Lytic but not latent infection by Kaposi's sarcoma-associated herpesvirus requires host CSL protein, the mediator of Notch signaling. Proc. Natl. Acad. Sci. USA 100:8490-8495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liang, Y., and D. Ganem. 2004. RBP-J (CSL) is essential for activation of the K14/vGPCR promoter of Kaposi's sarcoma-associated herpesvirus by the lytic switch protein RTA. J. Virol. 78:6818-6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liao, W., Y. Tang, Y. L. Kuo, B. Y. Liu, C. J. Xu, and C. Z. Giam. 2003. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 transcriptional activator Rta is an oligomeric DNA-binding protein that interacts with tandem arrays of phased A/T-trinucleotide motifs. J. Virol. 77:9399-9411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ling, P. D., and S. D. Hayward. 1995. Contribution of conserved amino acids in mediating the interaction between EBNA2 and CBF1/RBPJk. J. Virol. 69:1944-1950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ling, P. D., D. R. Rawlins, and S. D. Hayward. 1993. The Epstein-Barr virus immortalizing protein EBNA-2 is targeted to DNA by a cellular enhancer-binding protein. Proc. Natl. Acad. Sci. USA 90:9237-9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liu, Z.-J., T. Shirakawa, Y. Li, A. Soma, M. Oka, G. P. Dotto, R. M. Fairman, O. C. Velazquez, and M. Herlyn. 2003. Regulation of Notch1 and Dll4 by vascular endothelial growth factor in arterial endothelial cells: implications for modulating arteriogenesis and angiogenesis. Mol. Cell. Biol. 23:14-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
-
60.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the
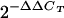 method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar] - 61.Lukac, D., L. Garibyan, J. Kirshner, D. Palmeri, and D. Ganem. 2001. DNA binding by the Kaposi's sarcoma-associated herpesvirus lytic switch protein is necessary for transcriptional activation of two viral delayed early promoters. J. Virol. 75:6786-6799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lukac, D. M., J. R. Kirshner, and D. Ganem. 1999. Transcriptional activation by the product of the open reading frame 50 of Kaposi's-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348-9361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lukac, D. M., J. R. Manuppello, and J. C. Alwine. 1994. Transcriptional activation by the human cytomegalovirus immediate-early proteins: requirements for simple promoter structures and interactions with multiple components of the transcription complex. J. Virol. 68:5184-5193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lukac, D. M., R. Renne, J. R. Kirshner, and D. Ganem. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304-312. [DOI] [PubMed] [Google Scholar]
- 65.Mailhos, C., U. Modlich, J. Lewis, A. Harris, R. Bicknell, and D. Ish-Horowicz. 2001. Delta4, an endothelial specific notch ligand expressed at sites of physiological and tumor angiogenesis. Differentiation 69:135-144. [DOI] [PubMed] [Google Scholar]
- 66.Maillard, I., T. Fang, and W. S. Pear. 2005. Regulation of lymphoid development, differentiation, and function by the Notch pathway. Annu. Rev. Immunol. 23:945-974. [DOI] [PubMed] [Google Scholar]
- 67.Marras, S. A., F. R. Kramer, and S. Tyagi. 2003. Genotyping SNPs with molecular beacons. Methods Mol. Biol. 212:111-128. [DOI] [PubMed] [Google Scholar]
- 68.Martin, D. F., B. D. Kuppermann, R. A. Wolitz, A. G. Palestine, H. Li, and C. A. Robinson for The Roche Ganciclovir Study Group. 1999. Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. N. Engl. J. Med. 340:1063-1070. [DOI] [PubMed] [Google Scholar]
- 69.Moore, P., and Y. Chang. 2001. Kaposi's sarcoma-associated herpesvirus, p. 2803-2834. In D. Knipe and P. Howley (ed.), Fields virology, 4th ed., vol. 2. Lippincott Williams and Wilkins, Philadelphia, Pa. [Google Scholar]
- 70.Nam, Y., A. P. Weng, J. C. Aster, and S. C. Blacklow. 2003. Structural requirements for assembly of the CSL · intracellular Notch1 · Mastermind-like 1 transcriptional activation complex. J. Biol. Chem. 278:21232-21239. [DOI] [PubMed] [Google Scholar]
- 71.Nellesen, D. T., E. C. Lai, and J. W. Posakony. 1999. Discrete enhancer elements mediate selective responsiveness of enhancer of split complex genes to common transcriptional activators. Dev. Biol. 213:33-53. [DOI] [PubMed] [Google Scholar]
- 72.Oswald, F., S. Liptay, G. Adler, and R. M. Schmid. 1998. NF-κB2 is a putative target gene of activated Notch-1 via RBP-Jκ. Mol. Cell. Biol. 18:2077-2088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pear, W. S., and J. C. Aster. 2004. T cell acute lymphoblastic leukemia/lymphoma: a human cancer commonly associated with aberrant NOTCH1 signaling. Curr. Opin. Hematol. 11:426-433. [DOI] [PubMed] [Google Scholar]
- 74.Pear, W. S., J. C. Aster, M. L. Scott, R. P. Hasserjian, B. Soffer, J. Sklar, and D. Baltimore. 1996. Exclusive development of T cell neoplasms in mice transplanted with bone marrow expressing activated Notch alleles. J. Exp. Med. 183:2283-2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rickinson, A. B., L. S. Young, and M. Rowe. 1987. Influence of the Epstein-Barr virus nuclear antigen EBNA 2 on the growth phenotype of virus-transformed B cells. J. Virol. 61:1310-1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sakakibara, S., K. Ueda, J. Chen, T. Okuno, and K. Yamanishi. 2001. Octamer-binding sequence is a key element for the autoregulation of Kaposi's sarcoma-associated herpesvirus ORF50/Lyta gene expression. J. Virol. 75:6894-6900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Schlee, M., T. Krug, O. Gires, R. Zeidler, W. Hammerschmidt, R. Mailhammer, G. Laux, G. Sauer, J. Lovric, and G. W. Bornkamm. 2004. Identification of Epstein-Barr virus (EBV) nuclear antigen 2 (EBNA2) target proteins by proteome analysis: activation of EBNA2 in conditionally immortalized B cells reflects early events after infection of primary B cells by EBV. J. Virol. 78:3941-3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schroeter, E. H., J. A. Kisslinger, and R. Kopan. 1998. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain. Nature 393:382-386. [DOI] [PubMed] [Google Scholar]
- 79.Song, M. J., X. Li, H. J. Brown, and R. Sun. 2002. Characterization of interactions between RTA and the promoter of polyadenylated nuclear RNA in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 76:5000-5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Struhl, G., and A. Adachi. 1998. Nuclear access and action of notch in vivo. Cell 93:649-660. [DOI] [PubMed] [Google Scholar]
- 81.Sun, R., S. F. Lin, L. Gradoville, Y. Yuan, F. Zhu, and G. Miller. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. USA 95:10866-10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Sung, N. S., S. Kenney, D. Gutsch, and J. S. Pagano. 1991. EBNA-2 transactivates a lymphoid-specific enhancer in the BamHI C promoter of Epstein-Barr virus. J. Virol. 65:2164-2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tanigaki, K., H. Han, N. Yamamoto, K. Tashiro, M. Ikegawa, K. Kuroda, A. Suzuki, T. Nakano, and T. Honjo. 2002. Notch-RBP-J signaling is involved in cell fate determination of marginal zone B cells. Nat. Immunol. 3:443-450. [DOI] [PubMed] [Google Scholar]
- 84.Tanigaki, K., M. Tsuji, N. Yamamoto, H. Han, J. Tsukada, H. Inoue, M. Kubo, and T. Honjo. 2004. Regulation of alphabeta/gammadelta T cell lineage commitment and peripheral T cell responses by Notch/RBP-J signaling. Immunity 20:611-622. [DOI] [PubMed] [Google Scholar]
- 85.Taniguchi, Y., T. Furukawa, T. Tun, H. Han, and T. Honjo. 1998. LIM protein KyoT2 negatively regulates transcription by association with the RBP-J DNA-binding protein. Mol. Cell. Biol. 18:644-654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Tsang, S. F., F. Wang, K. M. Izumi, and E. Kieff. 1991. Delineation of the cis-acting element mediating EBNA-2 transactivation of latent infection membrane protein expression. J. Virol. 65:6765-6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ueda, K., K. Ishikawa, K. Nishimura, S. Sakakibara, E. Do, and K. Yamanishi. 2002. Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) replication and transcription factor activates the K9 (vIRF) gene through two distinct cis elements by a non-DNA-binding mechanism. J. Virol. 76:12044-12054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wallberg, A. E., K. Pedersen, U. Lendahl, and R. G. Roeder. 2002. p300 and PCAF act cooperatively to mediate transcriptional activation from chromatin templates by Notch intracellular domains in vitro. Mol. Cell. Biol. 22:7812-7819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang, F., H. Kikutani, S. F. Tsang, T. Kishimoto, and E. Kieff. 1991. Epstein-Barr virus nuclear protein 2 transactivates a cis-acting CD23 DNA element. J. Virol. 65:4101-4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang, S. E., F. Y. Wu, H. Chen, M. Shamay, Q. Zheng, and G. S. Hayward. 2004. Early activation of the Kaposi's sarcoma-associated herpesvirus RTA, RAP, and MTA promoters by the tetradecanoyl phorbol acetate-induced AP1 pathway. J. Virol. 78:4248-4267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang, S. E., F. Y. Wu, M. Fujimuro, J. Zong, S. D. Hayward, and G. S. Hayward. 2003. Role of CCAAT/enhancer-binding protein alpha (C/EBPα) in activation of the Kaposi's sarcoma-associated herpesvirus (KSHV) lytic-cycle replication-associated protein (RAP) promoter in cooperation with the KSHV replication and transcription activator (RTA) and RAP. J. Virol. 77:600-623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang, S. E., F. Y. Wu, Y. Yu, and G. S. Hayward. 2003. CCAAT/enhancer-binding protein-alpha is induced during the early stages of Kaposi's sarcoma-associated herpesvirus (KSHV) lytic cycle reactivation and together with the KSHV replication and transcription activator (RTA) cooperatively stimulates the viral RTA, MTA, and PAN promoters. J. Virol. 77:9590-9612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Weng, A. P., A. A. Ferrando, W. Lee, J. P. Morris IV, L. B. Silverman, C. Sanchez-Irizarry, S. C. Blacklow, A. T. Look, and J. C. Aster. 2004. Activating mutations of NOTCH1 in human T cell acute lymphoblastic leukemia. Science 306:269-271. [DOI] [PubMed] [Google Scholar]
- 94.Whitby, D., M. R. Howard, M. Tenant-Flowers, N. S. Brink, A. Copas, C. Boshoff, T. Hatzioannou, F. E. Suggett, D. M. Aldam, A. S. Denton, et al. 1995. Detection of Kaposi sarcoma associated herpesvirus in peripheral blood of HIV-infected individuals and progression to Kaposi's sarcoma. Lancet 346:799-802. [DOI] [PubMed] [Google Scholar]
- 95.Wu, L., J. C. Aster, S. C. Blacklow, R. Lake, S. Artavanis-Tsakonas, and J. D. Griffin. 2000. MAML1, a human homologue of Drosophila mastermind, is a transcriptional co-activator for NOTCH receptors. Nat. Genet. 26:484-489. [DOI] [PubMed] [Google Scholar]
- 96.Xu, Y., D. P. AuCoin, A. R. Huete, S. A. Cei, L. J. Hanson, and G. S. Pari. 2005. A Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 79:3479-3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Xue, Y., X. Gao, C. E. Lindsell, C. R. Norton, B. Chang, C. Hicks, M. Gendron-Maguire, E. B. Rand, G. Weinmaster, and T. Gridley. 1999. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum. Mol. Genet. 8:723-730. [DOI] [PubMed] [Google Scholar]
- 98.Yu, Y., S. E. Wang, and G. S. Hayward. 2005. The KSHV immediate-early transcription factor RTA encodes ubiquitin E3 ligase activity that targets IRF7 for proteosome-mediated degradation. Immunity 22:59-70. [DOI] [PubMed] [Google Scholar]
- 99.Yuan, C. C., W. Miley, and D. Waters. 2001. A quantification of human cells using an ERV-3 real time PCR assay. J. Virol. Methods 91:109-117. [DOI] [PubMed] [Google Scholar]
- 100.Zeng, Q., S. Li, D. B. Chepeha, T. J. Giordano, J. Li, H. Zhang, P. J. Polverini, J. Nor, J. Kitajewski, and C. Y. Wang. 2005. Crosstalk between tumor and endothelial cells promotes tumor angiogenesis by MAPK activation of Notch signaling. Cancer Cell 8:13-23. [DOI] [PubMed] [Google Scholar]
- 101.Zhang, J., J. Wang, C. Wood, D. Xu, and L. Zhang. 2005. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 replication and transcription activator regulates viral and cellular genes via interferon-stimulated response elements. J. Virol. 79:5640-5652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Zhou, F. C., Y. J. Zhang, J. H. Deng, X. P. Wang, H. Y. Pan, E. Hettler, and S. J. Gao. 2002. Efficient infection by a recombinant Kaposi's sarcoma-associated herpesvirus cloned in a bacterial artificial chromosome: application for genetic analysis. J. Virol. 76:6185-6196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhou, S., M. Fujimuro, J. J. Hsieh, L. Chen, and S. D. Hayward. 2000. A role for SKIP in EBNA2 activation of CBF1-repressed promoters. J. Virol. 74:1939-1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhou, S., M. Fujimuro, J. J. Hsieh, L. Chen, A. Miyamoto, G. Weinmaster, and S. D. Hayward. 2000. SKIP, a CBF1-associated protein, interacts with the ankyrin repeat domain of NotchIC to facilitate NotchIC function. Mol. Cell. Biol. 20:2400-2410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhou, S., and S. D. Hayward. 2001. Nuclear localization of CBF1 is regulated by interactions with the SMRT corepressor complex. Mol. Cell. Biol. 21:6222-6232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Zimber-Strobl, U., E. Kremmer, F. Grasser, G. Marschall, G. Laux, and G. W. Bornkamm. 1993. The Epstein-Barr virus nuclear antigen 2 interacts with an EBNA2 responsive cis-element of the terminal protein 1 gene promoter. EMBO J. 12:167-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zimber-Strobl, U., and L. J. Strobl. 2001. EBNA2 and Notch signalling in Epstein-Barr virus mediated immortalization of B lymphocytes. Semin. Cancer Biol. 11:423-434. [DOI] [PubMed] [Google Scholar]