

Abstract
The cellular pathways that Epstein-Barr virus (EBV) manipulates in order to effect its lifelong persistence within hosts and facilitate its transmission between hosts are not well understood. The EBV nuclear antigen 3 (EBNA-3) family of latent infection proteins consists of transcriptional regulators that influence viral and cellular gene expression in EBV-infected cells. To identify EBNA-3B- and EBNA-3C-regulated cellular genes potentially important for virus infection in vivo, we studied a lymphoblastoid cell line (LCL) infected with an unusual EBV mutant, where a genetic manipulation to delete EBNA-3B also resulted in a significant decrease in EBNA-3C expression and slower than normal growth (3B−/3Clow). Transcriptional profiling was performed on the 3B−/3Clow LCLs, and comparison of mutant and wild-type LCL profiles resulted in a group of 21 probe sets representing 16 individual genes showing statistically significant differences in expression. Further quantitative reverse transcription-PCR analyses comparing 3B−/3Clow LCLs to a previously described EBNA-3B mutant (3B−) where EBNA-3C expression was normal revealed three potential EBNA-3B-repressed genes, three potential EBNA-3C-repressed genes, and two potential EBNA-3C-activated genes. The most highly EBNA-3C-repressed gene was Jagged1, a cell surface ligand and inducer of the Notch receptor signaling pathway that is usurped by EBV genes essential for B-cell immortalization. 3B−/3Clow LCLs expressed increased levels of Jagged1 protein and were able to more efficiently induce functional Notch signaling, and this signaling was dependent on Notch cleavage by γ-secretase. However, inhibiting γ-secretase-mediated Notch cleavage did not rescue 3B−/3Clow LCL growth, suggesting that EBNA-3C-mediated repression of this signaling pathway did not contribute to LCL growth in tissue culture. Similarly, expression of the chemokine receptor CXCR4 was reproducibly upregulated in EBNA-3B-null LCLs. Since deletion of EBNA-3B has no significant impact on B-cell immortalization in tissue culture, this finding suggested that EBNA-3B-mediated regulation of CXCR4 may be an important viral strategy for alteration of B-cell homing in the infected host. These studies identify two cellular genes that do not contribute to EBV-induced B-cell growth but whose expression levels are strongly EBNA-3 regulated in EBV-infected primary B cells. These EBV-manipulated cellular pathways may be important for virus survival or transmission in humans, and their independence from EBV-induced B-cell growth makes them potential targets for testing in vivo with the rhesus lymphocryptovirus animal model for EBV infection.
Epstein-Barr virus (EBV)-mediated immortalization of B cells in tissue culture provides a model system for dissecting cellular pathways important for virus entry, virus replication, and virus-induced B-cell-growth transformation. Thus, EBV genes essential for these processes have been defined and studied intensively. However, defining additional cellular pathways important for virus infection of a complete organism is more difficult to accomplish using in vitro models of B-cell infection and immortalization. The EBV genome contains the capacity to express at least 80 gene products, and only a fraction of these genes are essential for virus propagation in tissue culture. The ways in which many viral genes contribute to virus infection, persistence, immune evasion, and transmission in the context of the complete organismic host remain untested or largely unknown.
In recent years, infection of rhesus macaques with the closely related rhesus lymphocryptovirus (rhLCV) has emerged as a viable model for EBV infection to test the roles of individual viral genes in vivo (51). The overall biology of rhLCV is similar to that of EBV, and infection of naïve rhesus macaques with rhLCV appears to reproduce all the crucial aspects of human EBV infection, including the establishment and maintenance of long-term persistence (32). Lymphoblastoid cell lines (LCLs) generated from rhLCV-infected rhesus B cells express a panel of latent infection gene products identical to those of EBV-infected LCLs (51), and sequencing of the complete rhLCV genome has provided genetic validation of rhLCV as an accurate animal model for EBV infection (41).
The Epstein-Barr virus nuclear antigen 3 (EBNA-3) proteins are a set of three related nuclear proteins (EBNA-3A, -3B, and -3C) expressed in EBV-immortalized B cells and are believed to function as transcriptional regulators. EBNA-3A and -3C are essential for EBV-induced B-cell immortalization in tissue culture (47), whereas EBNA-3B is completely dispensable (2, 46). However, the EBNA-3B gene is conserved in LCV naturally infecting Old World primates (18), suggesting that this gene is important for LCV infection in the host despite its nonessential role in tissue culture. This makes EBNA-3B an especially attractive candidate for animal model studies, as any effects on the establishment or maintenance of persistent infection are separated from overtly adverse effects on B-cell growth or survival. Similarly, EBNA-3A and EBNA-3C may have additional, non-growth-related functions that make important contributions to the successful LCV infection of a primate host. However, these pathways may be more difficult to dissect in the context of in vivo infection unless they can be separated from their growth-related functions, since growth-compromising mutations are likely to have strongly attenuating phenotypes early during primary infection that will dominate and mask the effects on other cellular and immunological pathways.
It is not obvious how EBNA-3B, and any non-growth-related effects of EBNA-3A and -3C, might contribute to successful virus infection in vivo. All the EBNA-3s are immunodominant targets for EBV-specific immune responses (10, 19, 34), but it is likely that EBNA-3B does not function solely as a target for cytotoxic T cells. All three nuclear proteins have both activating and repressive activities on viral promoters (3, 24, 28, 39, 55), they interact with a variety of cellular corepressor proteins (4, 15, 23, 40, 48), and the expression of one or more of the EBNA-3s in the context of EBV-negative B cells has been shown to influence the expression of several cellular genes (20, 43, 52). Their ability to modulate the expression of both viral and cellular genes makes them probable candidates for viral proteins that actively manipulate the infected cell in vivo to enable successful infection of the host. Thus, we analyzed an EBV mutant which was completely deleted for EBNA-3B but also exhibited decreased EBNA-3C expression (3B−/3Clow) to identify EBNA-3B- or -3C-regulated cellular pathways that could be readily tested in the rhesus LCV animal model.
MATERIALS AND METHODS
Generation of mutant EBV BACs and LCLs.
The generation of wild-type and 3B− bacterial artificial chromosomes (BACs) has been described previously (2). The 3B−/3Clow BAC was derived from EBV BAC clone 2-6 by inserting a kanamycin resistance cassette derived from the pKD4 plasmid (6) in place of the EBNA-3B open reading frame (ORF). In brief, this was performed by first transfecting the pKD119 plasmid into Escherichia coli containing the EBV BAC and inducing transient lambda red recombinase protein expression at 30°C with 1 mM arabinose (6). Competent E. coli cells were then transformed with PCR-amplified DNA containing 50 nucleotides 5′ of the EBNA-3B start site (nucleotides 95281 to 95330), a kanamycin resistance marker flanked by two FLP recombinase target (FRT) sites, and 50 nucleotides lying 3′ of the EBNA-3B translational stop site (nucleotides 98248 to 98297). PCR primers used for amplification of the kanamycin resistance cassette are as follows: 5′-CATGAGAGAGACCTCGCATATTTGCAGAAGGGTCACTGAAACATCTTATCGTGTAGGCTGGAGCTGCTTC-3′ and 5′-CGCAGTCTGTTGCCCCAGGGTTCATCCCAGTTCTTGTTACATGGGCGAAACATATGAATATCCTCCTTA-3′. Recombinants were selected by growth in the presence of kanamycin, and replacement of EBNA-3B with the kanamycin resistance cassette was confirmed by Southern blotting. The 3B−/3Clow BAC was then transformed into BM2710 E. coli (12) and transduced into 293 cells, and virus replication was induced from puromycin-resistant 293 cells as described previously (2). BAC-derived LCLs were maintained in RPMI containing either 10% fetal calf serum (FCS) (3B−/3Clow LCLs) or 10% Neugem serum (wild-type and 3B− LCLs) (Gemini), with 4 μg/ml puromycin.
Growth curves.
LCLs were seeded at a concentration of 4 × 105 cells/ml in 24-well plates at day 0. Cell counts were taken over a period of several days, using a hemocytometer and averaging a total of three counts per cell line per day. For cell counts in the presence of compound E (Calbiochem), cells were resuspended in fresh media with or without 1 μM compound E at day 0 and again at day 3.
Western blotting.
Whole-cell lysates were prepared by solubilizing LCLs in sodium dodecyl sulfate sample buffer, sonicating for 90 seconds, and boiling for 5 min. Lysates from 2 × 105 cells were loaded per lane. Samples were separated on 8% sodium dodecyl sulfate-polyacrylamide gel electrophoresis gels, transferred to 0.45-μm-pore-sized nitrocellulose membranes (Schleicher & Schuell), blocked with phosphate-buffered saline (PBS) containing 5% milk, and probed with primary antibodies. EBV-immune human serum was used at a dilution of 1:100 in PBS containing 2.5% milk; the A10 monoclonal antibody directed against EBNA-3C was used undiluted from tissue culture supernatant (30); the JF186 antibody against EBNA-LP was used at a dilution of 1:3,000 in PBS containing 5% milk (9), and the S12 antibody against LMP-1 was used at a dilution of 1:5,000 in PBS containing 5% milk (27). The H-114 antibody against Jagged1 (Santa Cruz) was used at a dilution of 1:200 in PBS containing 5% milk. Blots were subsequently incubated with horseradish peroxidase-conjugated anti-human, anti-mouse, or anti-rabbit immunoglobulin G antibodies (Jackson) at dilutions of 1:5,000 in PBS containing 5% milk, and immunoreactive proteins were visualized using a chemiluminescent reagent (Perkin Elmer).
RNA isolation and transcriptional profiling.
Total cellular RNA was isolated from LCLs infected with B95-8, recombinant P3HR1, wild-type BAC-derived, and 3B−/3Clow BAC-derived viruses. Cells (2 × 106) were lysed in 1.2 ml RNA-Bee reagent (Tel-Test). RNA was extracted by the addition of 200 μl chloroform, recovered by isopropanol precipitation, washed with 70% ethanol, and resuspended in diethyl pyrocarbonate-treated water at a concentration of 1 μg/μl. RNA quality was verified on a 1% denaturing agarose gel. Transcriptional profiling was performed by the Harvard Medical School Partners Healthcare Center for Genetics and Genomics Core Facility, using the Affymetrix GeneChip System. RNA was hybridized to Affymetrix HG-U133A chips (Affymetrix) containing 22,283 probe sets representing at least 14,500 well-characterized human genes, according to the manufacturer's specifications. The microarrays were scanned using a GeneChip Scanner 3000 (Affymetrix), and the data were prescaled to a target intensity of 500 before being imported into GeneSpring 6.0 software.
Analysis of microarray data.
Gene expression data were imported into GeneSpring 6.0 software and were normalized in three ways. Measurements of less than 0.01 were set to 0.01. Per-chip normalization was accomplished by dividing each measurement on a chip by the 50th percentile of all values on that chip. On a per-gene basis, signal strength was normalized to the median measurement of that gene across all samples. Normalized data from B95-8 and recombinant P3HR1 LCLs and those from wild-type BAC-derived LCLs were then compared. Differences in gene expression were considered significant if there was a >2-fold difference in signal intensities between the two groups of samples, if the signal was flagged as being either present or marginal in at least half the samples under consideration, and if the samples passed a one-way analysis of variance with a P value of 0.05. The Benjamini and Hochberg false discovery rate for multiple testing correction was incorporated into the analysis of variance. The same statistical analysis was then expanded to include data obtained from the 3B−/3Clow-derived LCLs, and these LCLs were compared to all wild-type samples (B95-8, recombinant P3HR1, and wild-type BAC-derived LCLs) grouped together.
Quantitative RT-PCR.
RNA was isolated from wild-type BAC-derived, 3B−/3Clow BAC-derived, and 3B− BAC-derived LCLs as described above. A one-step quantitative reverse transcription (RT)-PCR assay, with SYBR green as a detection reagent, was used to determine relative RNA quantities (QuantitTect SYBR green RT-PCR kit; QIAGEN). Primers used for amplification of specific targets were designed using Primer Express and were as follows: GAPDH forward (5′-GATTCCACCCATGGCAAATTC-3′) and reverse (5′-TGGGATTTCCATTGATGACAAG-3′); Jagged1 forward (5′-TCTCACTCGGGCATGATCAAC-3′) and reverse (5′-CAGGTCACGCGGATCTGATAC-3′); Neurocalcin D forward (5′-CCATGCCTAGAGGGCAGATG-3′) and reverse (5′-TGGTCCCCTGTCATACATGTTAAC-3′); Aldo-keto reductase family, member C1, forward (5′-GGAGGACCCAGTCCTTTGTG-3′) and reverse (5′-TGGTAGCGCAGGGCAATC-3′); Uronyl-2-sulfotransferase forward (5′-TGAGGTGGGAGGATCACTTGA-3′) and reverse (5′-TGGAGTGCAGTGGTGTGATCA-3′); Carbohydrate sulfotransferase 4 forward (5′-CGCTGGTCTTTGCCCTATGA-3′) and reverse (5′-CGGTAGCCCAGCAAATTCA-3′); chemokine (C-X-C motif) receptor 4 forward (5′-TCTGGAGAACCAGCGGTTAC-3′) and reverse (5′-TCCTCGGTGTAGTTATCTG-3′); Enthoprotin forward (5′-CTTCAACCAAGCCCCATCA-3′) and reverse (5′-GCTGTGAGGCACTGCCAAA-3′); Absent in melanoma 1 forward (5′-TCACCTGCAGAAGTACCTAATTGGTA-3′) and reverse (5′-ATGAATAACCCTGTTGCTTTGTTTC-3′); phosphoinositide-3-kinase, regulatory subunit, polypeptide p101, forward (5′-ACCTGACAGAAGTGGTGAAAAGG-3′) and reverse (5′-TCCACTTTGATCTGCGATGTG-3′); Transcription termination factor, RNA polymerase II (pol II), forward (5′-TGCTTCTGCCACAGGACAAG-3′) and reverse (5′-GCGTTTCCCCTCTGCCTTAC-3′); Vitamin A responsive forward (5′-TCCATCTATTCAAGTGTGTTTCTAATTCT-3′) and reverse (5′-GGTATGCACAAAAGGCCTAAAATC-3′); Filamin A forward (5′-GAAGATGCACCGCAAGCA-3′) and reverse (5′-CTCGCGGTCCAGGAACTC-3′); Pyridoxal kinase forward (5′-ACCGGAGCTTCATCTAGTGATTG-3′) and reverse (5′-GCTTTGATGCCTCCGAAGAG-3′); Integrin α4 forward (5′-CTGGAGCAATCGAATCGGTTA-3′) and reverse (5′-AGCAGCCTCTCAGGCTGAAC-3′); Ecotropic viral integration site 2A forward (5′-ATACCCGTCTGTGGGCTAACA-3′) and reverse (5′-CATTTTGGTTTCTGCCTGTCTTG-3′); and T-cell leukemia/lymphoma 1A forward (5′-GCCAAGGAGGATTCCATTTG-3′) and reverse (5′-GAAAAGCCGAGGCACAGGTA-3′). Amplification of each target gene was performed in triplicate for each RNA sample and repeated at least twice. SYBR green fluorescence was detected using an ABI GeneAmp 5700 cycler and accompanying sequence detection system software. Dissociation curves were generated after every reaction to verify specific amplification. Average cycle threshold (CT) values for each target gene were normalized to CT values for GAPDH (glyceraldehyde-3-phosphate dehydrogenase) RNA. RNA expression levels in 3B−/3Clow and 3B− LCLs relative to wild-type LCL levels were calculated using the 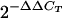 equation (25).
equation (25).
Notch activation assays.
U2OS cells in six-well plates were transfected in triplicate with 1 μg of an artificial Notch-sensitive firefly luciferase reporter plasmid (17) and 20 ng of pRL-TK, an internal-control plasmid that expresses Renilla luciferase from a thymidine kinase promoter element. Transfections were performed using Lipofectamine Plus (Invitrogen). Cells were washed posttransfection and allowed to recover in Dulbecco's modified Eagle's medium (DMEM) containing 10% FCS overnight. Twenty-four hours posttransfection, cells were refed with 2 ml of DMEM plus 10% FCS containing (i) no addition, (ii) 1 μM compound E, (iii) 1 × 106 cells from wild-type LCL, (iv) 1 × 106 cells from wild-type LCL plus 1 μM compound E, (v) 1 × 106 cells from 3B−/3Clow LCL, or (vi) 1 × 106 cells from 3B−/3Clow LCL plus 1 μM compound E. Cells were spun for 5 min at 200 × g to enhance contact between LCLs and U2OS cells. Forty-eight hours posttransfection, cells were washed with PBS and cell lysates prepared, and dual luciferase assays (dual luciferase assay kit; Promega) were read using a luminometer (Turner Systems). The Renilla luciferase activity was used to normalize the firefly luciferase activity for transfection efficiencies. Normalized activities were expressed relative to those for the DMEM plus those for the 10% FCS controls, the mean activity of which was set to 1.
Flow cytometry.
Wild-type and 3B− LCLs were stained for CXCR4 expression using a phycoerythrin (PE)-conjugated monoclonal antibody directed against CXCR4 (1D9, BD Pharmingen). Briefly, 5 × 105 cells from LCL were washed with PBS containing 1% FCS and 0.1% sodium azide and incubated with 0.5 μg PE-conjugated 1D9 antibody for 30 min in the dark at 4°C. A PE-conjugated, isotype-matched antibody was used as a negative control. Cells were then washed with PBS containing 1% FCS and 0.1% sodium azide, and the cell surface fluorescence of live cells was detected and analyzed using a FACSCalibur system and CellQuest software (Becton Dickinson).
Migration assays.
Migration of wild-type and 3B− LCLs was assessed in 24-well plates by using Transwell inserts of a 6.5-mm diameter and 5.0-μm pore size (Costar). Cells from LCL (3 × 105) were resuspended in 100 μl RPMI plus 0.2% bovine serum albumin and loaded into the upper chamber of a Transwell insert. To examine LCL migration in response to CXCL12, 600 μl RPMI plus 0.2% bovine serum albumin containing 100 ng/ml CXCL12 (SDF-1α, R & D Systems) was loaded into the lower compartment, and cells were allowed to migrate for 4 h at 37°C. Migrated cells were then collected from the lower chamber and counted. Percent migration was calculated as the fraction of total cells migrating across the Transwell filter in response to ligand, after having subtracted the number of cells migrating to medium alone in the absence of ligand.
RESULTS
LCLs infected with virus derived from an EBNA-3B-deleted BAC mutant containing a kanamycin resistance cassette grow slowly and are hypomorphic for EBNA-3C expression.
We have previously reported that complete deletion of the EBNA-3B ORF from an EBV genome cloned as a BAC resulted in viruses (3B−) that immortalized B cells as efficiently as the wild-type genome (2). In the process of making this mutant, we had created an intermediate EBNA-3B BAC mutant containing the kanamycin resistance cassette flanked by FRT sites in place of the EBNA-3B ORF (Fig. 1). The FLP recombinase was then used to remove the kanamycin resistance gene to produce the previously described EBNA-3B deletion mutant (3B−). When peripheral blood mononuclear cells (PBMC) were infected with viruses generated from the intermediate EBNA-3B BAC mutant containing the kanamycin resistance cassette, we were surprised to find that while growing clumps of cells were present in every well, the cells grew much more slowly than either wild-type or 3B− virus-immortalized B cells. In order to expand the cell numbers, cells from multiple wells had to be combined and maintained at higher densities. This suggested that the dominant phenotype was a lower rate of growth at low cell density. These experiments did not rule out a potential difference in the initial B-cell immortalization efficiency of viruses produced from the kanamycin resistance cassette-containing EBNA-3B BAC mutant. Once larger quantities of cells were available, the difference in cell growth rate compared to that of wild-type LCLs could be demonstrated (Fig. 2A).
FIG. 1.

Schematic of EBNA-3B mutations made in the EBV BAC. F plasmid sequences for prokaryotic replication were inserted into the EBV internal repeat 1 (IR1) region. The wild-type BAC contains the full complement of EBNA-3A, -3B, and -3C genes. EBNA-3B was knocked out in the 3B−/3Clow BAC by replacement of the entire EBNA-3B open reading frame with a kanamycin resistance cassette (Kanr) flanked by FRT sites. The 3B− BAC has been described previously and contains a single FRT site in place of the EBNA-3B open reading frame (2).
FIG. 2.

3B−/3Clow LCLs are impaired for cell growth and express low levels of EBNA-3C protein. (A) Growth curves for wild-type and 3B−/3Clow LCLs. Wild-type and two independently derived 3B−/3Clow LCLs (3B−/3Clow A1 and A2) were seeded at an initial density of 4 × 105 cells per ml in a 24-well plate at day 0, and viable cell counts were taken over a period of 7 days. Data are means ± standard deviations for three replicates. (B) Expression of EBV latent proteins in 3B−/3Clow LCLs. Whole-cell lysates were analyzed for expression of EBV latent proteins by Western blotting. EBV-immune human serum was used to detect EBNA-1, -2, -3A, -3B, and -3C; EBNA-3C alone was detected using the A10 monoclonal antibody (30); EBNA-LP was detected using the JF186 monoclonal antibody (9); and LMP-1 was detected using the S12 monoclonal antibody (27). Controls included an EBV-negative Burkitt lymphoma line (BJAB), an LCL infected with the B95-8 virus (B95-8), and an LCL derived from B95-8-immortalized umbilical-cord B cells (IB4) (21) which lacks full-length EBNA-3B protein expression (38) but is normal for expression of all the other latent proteins. An LCL infected with the wild-type BAC-derived virus (wild-type) and two independently derived 3B−/3Clow LCLs are shown.
EBV latent gene expression was evaluated by immunoblotting to determine whether the lower growth rate of these LCLs was associated with a defect in latent infection protein expression. We have previously reported that the 3B− LCL expressed normal levels of EBV latent infection proteins except for the absence of EBNA-3B (2). As expected, there was no detectable EBNA-3B expression in LCLs infected with viruses derived from the kanamycin resistance cassette-containing EBNA-3B BAC mutant, but surprisingly, there was a marked reduction in EBNA-3C expression (3B−/3Clow cells) (Fig. 2B). EBNA-3C was not detectable in these LCLs by immunoblotting with human sera, but immunoblotting with an EBNA-3C monoclonal antibody detected a low level of EBNA-3C expression. In repeat immunoblots, the EBNA-3C expression levels were reproducibly <20% of wild-type EBNA-3C levels. Thus, LCLs derived from the kanamycin resistance cassette-containing EBNA-3B mutant are referred to as 3B−/3Clow LCLs. EBNA-3A levels in 3B−/3Clow LCLs were comparable to or slightly elevated over those in wild-type cells. Other latent infection protein expression levels (EBNA-1, -2, -LP, and LMP-1) were unchanged compared to those in wild-type cells.
Thus, the presence of a kanamycin resistance cassette in place of the EBNA-3B ORF in 3B−/3Clow LCLs was associated with decreased EBNA-3C expression and a decreased growth rate compared to that of either wild-type LCLs or 3B− LCLs where the kanamycin resistance cassette had been removed. Since small amounts of full-length EBNA-3C could be detected, we speculated that the reduced level of EBNA-3C expression in the 3B−/3Clow LCLs was due to an adverse effect of the kanamycin resistance cassette on the proper slicing of mature EBNA-3C transcripts. Evidence supporting the hypothesis that the kanamycin resistance ORF was interfering with normal EBNA-3C splicing came from the emergence of a more rapidly growing cell line with normal EBNA-3C expression when one of the 3B−/3Clow LCLs was propagated in culture for a prolonged period (Fig. 3A). When the DNA region between EBNA-3A and EBNA-3C was PCR amplified and sequenced from this revertant, we found that the 3′ end of the kanamycin resistance gene was still present at the precise insertion site of the EBNA-3B ORF, but the majority of the kanamycin resistance cassette had been deleted, in addition to much of the genomic sequence between the EBNA-3A ORF and the kanamycin resistance cassette insertion site (Fig. 3B). This apparently spontaneous deletion was consistent with the hypothesis that sequences in the kanamycin resistance cassette conferring an inhibitory effect on EBNA-3C splicing had been removed, thereby restoring EBNA-3C gene expression and a more normal growth rate.
FIG. 3.

Spontaneous restoration of normal EBNA-3C expression in a 3B−/3Clow LCL after several months in culture. (A) Expression of the EBNA-3 proteins in a 3B−/3Clow LCL after prolonged culture. Whole-cell lysates were analyzed for expression of EBNA-3 proteins by Western blotting. EBV-immune serum was used to detect all the EBNA-3s, and the A10 monoclonal antibody was used to detect EBNA-3C alone. (B) Schematic of the mutation in the EBNA-3C restored 3B−/3Clow LCL. The DNA region between the EBNA-3A and EBNA-3C open reading frames was PCR amplified and sequenced. KanamycinR/KanR represents the kanamycin resistance cassette inserted in the place of the EBNA-3B ORF; FRT represents the FLP recombinase target sites flanking the Kanr cassette; a and b represent nonviral, unique flanking sequences on either side of the FRT sites. The numbers of EBV nucleotides present between the nonviral insert and the EBNA-3A stop codon or EBNA-3C start codon are indicated by the numbered base pairs (bp).
Transcriptional profiling of 3B−/3Clow LCLs revealed several EBNA-3-repressed and -activated genes.
Thus, the 3B−/3Clow LCLs provided a system for identifying candidate cellular genes regulated by either EBNA-3B or EBNA-3C in the context of virus-immortalized peripheral blood B cells. We performed transcriptional profiling, using RNA prepared from three independently derived 3B−/3Clow LCLs. These were compared to RNA profiles generated in duplicate from two independent LCLs derived from wild-type BACs. To increase the power of analysis, RNA was also isolated from two LCLs immortalized with B95-8 EBV and three LCLs immortalized with a P3HR1 virus with EBNA-LP and EBNA-2 restored. RNA was hybridized to Affymetrix HG-U133A chips containing 22,283 probe sets representing at least 14,500 human genes.
Data were analyzed using GeneSpring 6.0 software, and normalized signal intensities were used to compare gene expression levels between different cell lines. First, the B95-8/P3HR1 LCLs were compared to wild-type LCLs. Under a fixed set of statistical conditions, expression levels from only 7 out of 22,283 probe sets were found to be statistically different between B95-8/P3HR1- and wild-type LCL-derived RNA. Three of these probe sets corresponded to major histocompatibility complex DQ alleles and likely reflected genetic polymorphisms between B-cell donors. Due to the very small number of statistically significant differences in gene expression between B95-8/P3HR1 virus-infected and wild-type LCLs, we grouped all these samples together for the purposes of subsequent analyses.
A comparison was then made between 3B−/3Clow LCLs and all wild-type LCLs under the same set of statistical conditions. Under these conditions, expression levels from 21 out of 22,283 probe sets were found to be significantly different between all wild-type and 3B−/3Clow LCLs, corresponding to 18 gene products, 2 of which represent unknown genes. Of the 16 known genes, 12 were found to be upregulated in 3B−/3Clow LCLs relative to what was found for wild-type LCLs, and 4 were found to be downregulated (Fig. 4 and Table 1).
FIG. 4.

Altered cellular gene expression in 3B−/3Clow BAC-infected LCLs. Shown are genes displaying significant changes in expression levels in 3B−/3Clow LCLs relative to expression levels in all wild-type LCLs grouped together (B95-8, P3HR1, and wild-type BAC-derived LCLs). Vertical columns represent data obtained for each individual cell line: for two LCLs immortalized with B95-8 virus (B95-8 clones 1 and 2), three LCLs derived by homologous recombination of EBNA-LP and EBNA-2 in P3HR1 EBV (P3HR1 clones 1, 2, and 3), two LCLs derived from the wild-type EBV BAC (wild-type A1 and A2), and three LCLs derived from the 3B−/3Clow BAC (3B−/3Clow A1, A2, and B1). Horizontal rows represent data obtained for each individual gene across all cell lines. The color of each data point represents the normalized signal-to-control ratio for a given gene in a specific cell line. Identities of individual genes are as follows: JAG1, jagged1; NCALD, neurocalcin D; AKR1C1, aldoketoreductase family, member C1; UST, uronyl-2-sulfotransferase; CHST4, carbohydrate sulfotransferase 4; CXCR4, chemokine (C-X-C motif) receptor 4; ENTH, enthoprotin; AIM1, absent in melanoma 1; P101-PI3K, phosphoinositide-3-kinase, regulatory subunit, polypeptide p101; TTF2, transcription termination factor, RNA polymerase II; JWA, vitamin A responsive; FLNA, filamin A, alpha; PDXK, pyridoxal kinase; ITGA4, integrin α4; EVI2A, ecotropic viral integration site 2A; TCL1A, T-cell leukemia/lymphoma 1A. GenBank accession numbers for the two unknown genes are shown.
TABLE 1.
Changes in RNA expression levels in 3B−/3Clow LCLs relative to expression levels in wild-type LCLs
| Gene | Fold change in RNA levela as determined by:
|
|
|---|---|---|
| Transcriptional array | Quantitative RT-PCR | |
| Jagged1 | 10.61 | 3.57 |
| Neurocalcin D | 6.54 | 5.38 |
| Aldo-keto reductase family, member C1 | 3.33 | 1.47 |
| Uronyl-2-sulfotransferase | 2.71 | 1.21 |
| Carbohydrate sulfotransferase 4 | 2.83 | −1.37 |
| CXCR4 | 2.57 | 12.39 |
| Enthoprotin | 2.09 | 6.78 |
| Absent in melanoma 1 | 1.99 | −1.18 |
| PI3 kinase, p101 subunit | 1.99 | 1.56 |
| Transcription termination factor, RNA pol II | 2.48 | 2.99 |
| Vitamin A responsive | 2.18 | 1.25 |
| Filamin A | 2.31 | 2.24 |
| Pyridoxal kinase | −2.80 | −1.10 |
| Integrin α4 | −4.24 | −5.01 |
| Ecotropic viral integration site 2A | −4.55 | 1.04 |
| T-cell leukemia/lymphoma 1A | −8.84 | −1,170.59 |
Average change (n-fold) in RNA expression levels in 3B−/3Clow LCLs relative to expression levels in grouped wild-type (B95-8, P3HR1, and wild-type BAC-derived) LCLs.
Identification of potential EBNA-3B- and EBNA-3C-regulated genes by quantitative reverse-transcription PCR.
Quantitative RT-PCR experiments were carried out to verify the microarray data for the 16 known genes. RNAs from wild-type and 3B−/3Clow LCLs from the same donor were compared. At least two independent quantitative RT-PCR experiments were carried out for each gene, and each experiment was performed in triplicate. GAPDH controls were included to confirm that equal amounts of total RNA were used in each experiment, and the GAPDH CT values were used to normalize CT values for each gene in different samples. The average levels of gene expression in 3B−/3Clow LCLs compared to those in wild-type LCLs for the 16 genes of interest are shown in Table 1. Changes in cellular gene expression were considered significant if there was a >2-fold change exhibited by both microarray and RT-PCR analysis, and 8 of the 16 genes met this criteria (Jagged1, Neurocalcin D, CXCR4, Enthoprotin, Transcription termination factor RNA pol II, Filamin A, Integrin α4, and T-cell leukemia/lymphoma 1A).
In order to evaluate whether the expression of these eight genes was regulated by EBNA-3B or EBNA-3C, additional quantitative RT-PCR was performed on RNA isolated from an LCL lacking EBNA-3B expression only (3B−) (Fig. 5). In this case, if gene expression in 3B− LCLs, where EBNA-3C expression was normal, was detected at levels comparable to those for wild-type LCLs, then the gene was likely EBNA-3C regulated. On the other hand, if gene expression remained different in the 3B− LCLs, then the gene was probably regulated by EBNA-3B, since this was the only defect common to both the 3B−/3Clow and 3B− LCLs. Three genes (JAG1, NCALD, and FLNA) were potentially repressed by either EBNA-3B or EBNA-3C since they were expressed at higher levels in the 3B−/3Clow cells than in wild-type cells. However, the expression levels of these three genes in wild-type and 3B− LCLs were comparable, suggesting that they were repressed by EBNA-3C since the lack of normal EBNA-3C expression in 3B−/3Clow LCLs was associated with increased levels of gene expression. Three other genes (CXCR4, TTF2, and ENTH) were expressed at high levels in 3B−/3Clow cells but either remained relatively unchanged (TTF2) or were expressed at even higher levels (CXCR4 and ENTH) in 3B− cells. Thus, the presence of normal EBNA-3C levels had no effect on the expression of these genes, suggesting that they were EBNA-3B repressed. The last two cell genes (ITGA4 and TCL1A) were found to be downregulated in 3B−/3Clow LCLs compared to what was found for wild-type LCLs, and the expression levels of both were increased closer to wild-type levels in the 3B− LCLs, suggesting that ITGA4 and TCL1A expression were induced by EBNA-3C. Thus, this analysis identified three potential EBNA-3C-repressed genes (JAG1, NCALD, and FLNA), three potential EBNA-3B-repressed genes (CXCR4, ENTH, and TTF2), and two possible EBNA-3C-activated genes (ITGA4 and TCL1A).
FIG. 5.

Quantitative RT-PCR analysis of cellular gene expression levels in 3B−/3Clow and 3B− BAC-derived LCLs. Quantitative RT-PCR was performed on RNA derived from wild-type, 3B−/3Clow, and 3B− LCLs. Average CT values for each gene were normalized to GAPDH levels, and data displayed are changes (n-fold) in expression levels of 3B−/3Clow and 3B− LCLs relative to expression levels in wild-type LCLs. Data are means ± standard deviations for two independent experiments performed in triplicate on the same RNA preparation.
The Notch ligand Jagged1 is upregulated in 3B−/3Clow LCLs, and its expression is regulated by EBNA-3C.
The high level of Jagged1 expression in 3B−/3Clow LCLs was noteworthy because Jagged1 is a Notch ligand and the Notch signaling pathway is important for EBV-induced cell growth (11, 13, 14). Jagged1 was the most highly upregulated cell gene in the 3B−/3Clow LCLs by transcriptional array analysis (Fig. 4). The RT-PCR studies also showed elevated Jagged1 expression levels in 3B−/3Clow LCLs relative to those in wild-type LCLs but not in 3B− LCLs, suggesting that it was an EBNA-3C-repressed gene (Fig. 5). Protein expression levels were evaluated by immunoblotting with a polyclonal antibody directed against Jagged1. As shown in Fig. 6, Jagged1 protein expression was relatively abundant in 3B−/3Clow LCLs but was virtually undetectable in LCLs where EBNA-3C expression was normal (i.e., 3B− LCLs, wild-type LCLs, and the revertant 3B−/3Clow LCL, where a spontaneous deletion in the kanamycin resistance cassette restored normal EBNA-3C expression). These results were consistent with EBNA-3C-mediated repression of Jagged1 protein expression in EBV-immortalized B cells.
FIG. 6.

Jagged1 protein expression is increased in 3B−/3Clow BAC-derived LCLs. (A) Whole-cell lysates from wild-type, 3B−/3Clow, and 3B− BAC-derived LCLs were analyzed for EBNA-3 and Jagged1 expression by Western blotting. EBV-immune human serum was used to detect the EBNA-3 proteins; Jagged1 protein was detected using the H-114 polyclonal antibody. (B) Downregulation of Jagged1 expression in 3B−/3Clow LCLs that are restored for EBNA-3C expression. Whole-cell lysates from the 3B−/3Clow LCL restored for normal EBNA-3C expression were analyzed for Jagged1 expression by Western blotting. The bottom two panels are reproduced from Fig. 3A, for reference and loading control purposes.
Increased Jagged1 expression in 3B−/3Clow LCLs can induce functional Notch signaling but is not responsible for their slow-growth phenotype.
To test whether the increased Jagged1 expression on the surfaces of cells from 3B−/3Clow LCLs had the potential to induce Notch signaling, we employed a coculture assay in which U2OS cells that naturally express the Notch1 receptor were transfected with a Notch reporter construct containing four RBP-Jκ (RBPSUH [recombining binding protein suppressor of hairless]) binding sites in a promoter controlling luciferase expression (17). U2OS cells transfected with the RBP-Jκ responsive reporter construct were then incubated for 24 h with either wild-type or 3B−/3Clow LCLs to test whether these cells differ in their capacity to activate Notch signaling. Incubation of reporter U2OS cells with 1 × 106 3B−/3Clow LCL cells resulted in a 3.1-fold increase in luciferase activity over that in the medium control, whereas reporter cells cocultured with 1 × 106 wild-type LCL cells exhibited only a 1.7-fold increase in reporter activity (Fig. 7A). In comparison, the maximal stimulation levels seen when U2OS cells were cocultivated with 3T3 cells expressing Jagged1 were approximately fourfold higher than those in the medium control (data not shown). To confirm that increased reporter activity was due specifically to Notch-induced signaling, the coculture experiment was repeated in the presence of compound E, a γ-secretase inhibitor capable of inhibiting the cleavage of all four isoforms of membrane-associated Notch after interaction with ligand (42) (Fig. 7B). Adding compound E to the medium of reporter cells cocultured with wild-type LCLs had no effect on luciferase activity. However, compound E neutralized the increased luciferase activity in reporter cells cocultured with 3B−/3Clow LCLs, indicating that the presence of increased Jagged1 expression in 3B−/3Clow LCLs was associated with increased Notch activation in neighboring cells.
FIG. 7.

3B−/3Clow LCLs are able to induce Notch signaling. (A) Notch-induced luciferase activity in U2OS cells stimulated with wild-type or 3B−/3Clow LCLs. U2OS cells were transfected with a Notch luciferase reporter construct containing four RBP-Jκ binding sites in its promoter (17). Twenty-four hours posttransfection, cells were overlaid with either wild-type or 3B−/3Clow LCLs at different concentrations. Forty-eight hours posttransfection, cells were harvested and lysates assayed for luciferase activity. Data shown are stimulation levels (n-fold) relative to those in unstimulated controls and represent the means ± standard deviations for three replicates. (B) Inhibition of 3B−/3Clow LCL-stimulated, Notch-induced luciferase activity by the γ-secretase inhibitor compound E. U2OS cells were transfected with luciferase reporter constructs and treated with 1 μM compound E. Twenty-four hours posttransfection, cells were overlaid with either wild-type or 3B−/3Clow LCLs at a density of 1 × 106 cells/well. Forty-eight hours posttransfection, cells were harvested and lysates assayed for luciferase activity. Data shown are stimulation levels (n-fold) relative to those in untreated, unstimulated controls and represent the means ± standard deviations for three replicates.
Since Notch signaling has been associated with apoptosis in B cells (33), we examined whether the increased Jagged1 expression and potential autocrine Notch signaling in the 3B−/3Clow LCLs might be an important EBNA-3C-regulated pathway contributing to the slow growth of these cells. As shown in Fig. 8, the growth of either wild-type or 3B−/3Clow cells was not significantly affected by the presence of compound E, suggesting that autocrine Notch signaling was not a major mechanism responsible for the slow-growth phenotype of the 3B−/3Clow LCLs.
FIG. 8.

Notch signaling induced by 3B−/3Clow BAC-derived LCLs is not responsible for their slow growth. Wild-type and 3B−/3Clow LCLs (3B−/3Clow A1 and 3B−/3Clow B1) were treated with either dimethyl sulfoxide or 1 μM compound E (+E) at day 0. Cells were seeded at an initial density of 4 × 105 cells per ml in a 24-well plate at day 0, and cell counts were taken over a period of 6 days. Data shown are means ± standard deviations for three replicates.
The chemokine receptor CXCR4 is reproducibly upregulated in 3B− LCLs, and its expression is regulated by EBNA-3B.
By transcriptional array analysis, the expression of the chemokine receptor CXCR4 was upregulated approximately 2.5-fold in 3B−/3Clow LCLs compared with that in wild-type LCLs (Fig. 4 and Table 1). By quantitative RT-PCR analysis, CXCR4 RNA expression in both 3B−/3Clow and 3B− LCLs was upregulated at least 10-fold over that in wild-type LCLs (Fig. 5), suggesting that this gene was repressed by EBNA-3B. CXCR4 is an important mediator of B-cell trafficking (5, 49); thus, it is possible that repression of CXCR4 by EBNA-3B could have effects on the migratory behavior of EBV-infected cells in humans.
In order to investigate the reproducibility of CXCR4 repression by EBNA-3B, matched pairs of wild-type and 3B− LCLs were generated from four different genetic donors (referred to as D9, D11, D12, and D13; the original quantitative RT-PCR experiments were performed on D9 LCLs). Quantitative RT-PCR was performed on RNA isolated from these cells to assess levels of CXCR4 mRNA. Figure 9A shows the average increases in CXCR4 expression levels for multiple 3B−/wild-type LCL pairs from each donor. For three out of four donors (D9, D11, and D13), average CXCR4 mRNA levels in 3B− LCLs were elevated at least 15-fold over those in wild-type LCLs, suggesting that CXCR4 was being strongly repressed by EBNA-3B in the wild-type LCLs. In 3B− LCLs from the fourth donor (D12), there was only a 1.5-fold average increase in CXCR4 mRNA levels, consistent with either minimal repression of CXCR4 expression by EBNA-3B in wild-type LCLs generated from this donor or a donor-specific defect in CXCR4 expression. However, levels of CXCR4 mRNA in the D12 wild-type LCLs were elevated over those in other wild-type LCLs (data not shown), suggesting that EBNA-3B was largely unable to repress CXCR4 expression in wild-type LCLs from this donor. Thus, CXCR4 mRNA levels were significantly increased in the majority of 3B− LCLs tested, relative to those in genetically matched wild-type LCLs, but some polymorphisms that affect CXCR4 responsiveness to repression by EBNA-3B may exist among different individuals.
FIG. 9.

RNA and cell surface protein expression of CXCR4 in 3B− LCLs from multiple donors. (A) Average expression levels of CXCR4 mRNA in 3B− LCLs, relative to expression levels in wild-type LCLs. Multiple independent wild-type and 3B− LCLs were generated from four different donors, and quantitative RT-PCR for CXCR4 was performed on RNA isolated from these cells. Average CT values for CXCR4 were normalized to GAPDH levels, and data displayed are changes (n-fold) in expression levels in 3B− LCLs relative to expression levels in wild-type LCLs. Data are means ± standard deviations for independent experiments performed in triplicate on independently derived 3B− LCLs. (B) Cell surface expression of CXCR4 protein in wild-type (wt) and 3B− LCLs from different donors. Wild-type and 3B− LCLs were stained for CXCR4 expression by using a PE-conjugated antibody directed against CXCR4 (clone 1D9). A PE-conjugated, isotype-matched antibody was used as a control. The shaded area shows CXCR4 staining of wild-type LCLs, and the bold line represents staining of 3B− LCLs. The mean fluorescence intensities (MFI) of each sample are indicated on their respective graphs.
To examine whether the increased CXCR4 mRNA expression in 3B− LCLs translated into increased CXCR4 protein levels, cell surface CXCR4 expression was assessed for wild-type and 3B− LCL pairs. For those donors for whom CXCR4 mRNA was significantly upregulated in 3B− LCLs (D9, D11, and D13), cell surface CXCR4 protein expression was also significantly increased in 3B− LCLs relative to that in wild-type LCLs (Fig. 9B). In the matched LCL pair from donor 12, in whom CXCR4 mRNA expression was unchanged between wild type and 3B− LCLs, there was also no difference in cell surface CXCR4 protein expression levels. In this case, there was high-level cell surface CXCR4 expression in both wild type and 3B− LCLs, consistent with a lack of EBNA-3B-mediated repression of CXCR4 expression in D12 cells. Thus, levels of CXCR4 protein expression correlated tightly with the amounts of CXCR4 mRNA detected in various wild-type and 3B− LCL pairs.
Increased CXCR4 expression results in an increase in the ability of 3B− LCLs to migrate in response to chemokine.
To determine whether increased CXCR4 protein expression in the 3B− LCLs translated to an increased ability of the cells to migrate in response to chemokine, wild-type and 3B− LCLs were tested for chemotaxis in response to CXCL12 (SDF-1), the only known chemokine ligand for CXCR4. A Transwell assay was employed to assess CXCL12-induced chemotaxis, and the results are shown in Fig. 10. While the 3B− LCLs from donors 9 and 13, which expressed high levels of CXCR4, were able to migrate across the Transwell membrane in response to CXCL12, wild-type LCLs in which CXCR4 expression was repressed did not migrate.
FIG. 10.

Increased CXCR4 expression on 3B− LCLs restores their ability to migrate in response to ligand. Wild-type and 3B− LCLs from two different donors (D9 and D13) were loaded into the upper chamber of a Transwell insert, and their chemotactic responses to 100 ng/ml CXCL12 (SDF-1α) were compared after 4 h. Percent migration was calculated as the fraction of total cells migrating across the Transwell filter, after subtracting the number of cells migrating in response to medium alone.
DISCUSSION
In this study, we have identified a number of potential EBNA-3B- and EBNA-3C-regulated genes in the context of EBV-infected LCLs. In particular, we have identified two cellular proteins, Jagged1 and CXCR4, whose repression does not appear to be required for in vitro B-cell growth, suggesting that EBV-mediated regulation of these cell pathways may instead be important for persistence of virus-infected cells within the host or viral transmission between hosts. These studies also demonstrate that repression of Jagged1 and CXCR4 by EBNA-3C and EBNA-3B, respectively, occur in the natural target cells for virus infection and that loss or decreased expression of a single EBV latent infection protein, in cells that otherwise express the full array of latent proteins, is sufficient for the lack of repression. This experimental approach is very different from those in which EBV-regulated effects are demonstrated by the transfection of single EBV genes into EBV-negative B-lymphoma cells or by cotransfection with reporter constructs. Furthermore, the current studies show that EBNA-3-mediated repression of these cellular targets is potent enough to provide a significant phenotypic and functional difference in virus-infected cells when the repression is disrupted. Thus, the deregulation of Jagged1 in the 3B−/3Clow LCLs resulted in a significant increase in cell surface Jagged1 expression, along with the functional potential to induce signaling through the Notch receptor pathway. Similarly, the loss of EBNA-3B resulted in a large increase in cell surface CXCR4 expression that led to a marked functional change in the migratory potential of EBV-infected B cells.
Recent studies have indicated that Notch signaling has a role in controlling the antigen-dependent development of peripheral CD4+ T-cell subsets, including Th1, Th2, and regulatory T cells (Treg) (31). Of relevance to our work, LCLs overexpressing Jagged1 via an adenovirus construct have been reported to induce a Treg population that repressed T-cell responses to alloantigens in a mixed-lymphocyte reaction as well as antigen/EBV-specific T-cell proliferation and EBV-specific cytotoxic T-cell responses (50, 54). Thus, one might predict that inducing Treg against viral antigens would be a desirable immune evasion strategy. However, it appears that EBV pursues the opposite strategy, repressing Jagged1 expression and potentially preventing the induction of excessive Treg activity. This might be more advantageous for the virus in the long run since a competent EBV-specific immune response is required to control B-cell proliferation and prevent the premature death of the host before virus transmission to a new generation of EBV-susceptible hosts can occur. Thus, EBNA-3C-mediated repression of Jagged1 expression in EBV-infected cells may facilitate long-term virus survival by serving as an immunity enhancement mechanism.
Since EBNA-3C is absolutely essential for B-cell immortalization in vitro, and the 3B−/3Clow LCLs expressing low EBNA-3C levels are impaired for growth, assessing the effects of increased Jagged1 expression on EBNA-3C mutant-infected cells in the rhesus LCV animal model system may be difficult. Simply deleting EBNA-3C from rhesus LCV will likely result in an immortalization-incompetent virus, and any phenotype observed in macaques could be due to defects in B-cell growth/survival rather than growth-unrelated phenomena, such as the lack of Jagged1 repression. Ideally, it would be preferable to specifically mutate the EBNA-3C domain responsible for Jagged1 repression and leave the EBNA-3C domains important for LCL growth intact. However, if the EBNA-3C domains required for Jagged1 repression are the same as those required for efficient B-cell immortalization, it may be impossible to separate EBNA-3C-mediated Jagged1 repression from other growth-related effects during in vivo infection.
On the other hand, EBNA-3B is completely dispensable for B-cell immortalization in vitro; thus, any consequences of an EBNA-3B deletion on the outcome of virus infection in vivo should be independent of cellular requirements for B-cell growth or survival. The discovery that EBNA-3B expression is closely linked to CXCR4 downregulation in EBV-infected B cells may now provide a strategy for testing the role of CXCR4 for EBV infection in vivo. Both CXCR4 and its only known ligand, CXCL12, are absolutely essential for B-cell lymphopoiesis (26, 35, 44, 56), and CXCR4 expression can be found on all B-cell subsets (8, 16). During B-cell development, CXCR4/CXCL12 signaling in the bone marrow is responsible for the retention of B-cell precursors, and inactivation of CXCR4 in developing B cells results in their premature migration to the periphery and spleen (37). Regulation of CXCR4 expression appears to be a common feature of herpesvirus infection, as EBV, human herpesvirus 6, and human herpesvirus 7 have all been shown to downmodulate CXCR4 expression in infected B and T cells, respectively (36, 53). In addition, EBV infection of primary tonsillar B cells results in downregulation of CXCR4 expression within 48 h, with a corresponding decrease in the ability of infected cells to respond to CXCL12 (7). Although the precise mechanism of EBNA-3B-mediated CXCR4 regulation remains to be defined, our preliminary studies suggest that EBNA-3B alone cannot repress CXCR4 expression in T-cell lines and that either the presence of other EBV genes or the context of an LCL background may be required for EBNA-3B-mediated modulation of CXCR4 expression.
Given the role of CXCR4 in retaining B cells in the bone marrow during development, it is possible that EBNA-3B-mediated downregulation of CXCR4 in infected cells might be important for allowing EBV-infected cells to gain access to the periphery. When and where EBNA-3B is expressed during human EBV infection are still somewhat controversial. EBNA-3B and other latent infection proteins associated with type III latency are proposed to be important for driving the differentiation of newly infected, naïve B cells to memory B cells in primary EBV infection (1), and Tierney et al. have reported detection of type III latency mRNAs in PBMC from infectious-mononucleosis patients (45). Thus, EBNA-3B-mediated repression of CXCR4 may aid virus dissemination during primary EBV infection, by helping virus-infected cells gain access to the peripheral circulation. Alternatively, EBNA-3B-associated CXCR4 downregulation may be important during persistent infection. Although EBNA-3B and other type III latency transcripts are not detected in PBMC from persistently infected hosts, the long-term maintenance of EBNA-3-specific cytotoxic T lymphocytes at high precursor frequencies in the peripheral blood suggests that the EBNA-3s may be periodically expressed during persistent infection, thereby restimulating EBNA-3-specific cytotoxic T lymphocytes. In this case, the persistence and survival of EBV-infected B cells might require more than the simple circulation of type I latency-expressing cells in the peripheral blood. Persistence may be a more dynamic process, where EBV-infected B cells periodically circulate through other body compartments besides the peripheral blood, where they are subject to apoptotic stress or where survival may require the expression of type III latency genes. In this case, EBNA-3B-mediated repression of CXCR4 may be important for allowing B-cell escape from a central compartment where type III latency might be transiently expressed. Thus, these studies provide the basis for specific hypotheses that can be evaluated during experimental rhesus macaque infection with a rhLCV deleted for rhEBNA-3B.
One of our wild-type LCLs, derived from D12, did not exhibit any EBNA-3B-mediated repression of CXCR4. Studies of a larger population will be required to establish a more precise frequency for this phenotype. In D12, there was likely a lack of EBNA-3B repressive function, as opposed to a defect in CXCR4 expression, because CXCR4 expression levels in the wild-type LCL of D12 remained high, similar to the levels seen in the 3B− LCLs from other donors. The observed lack of CXCR4 repression in D12 may be due to either (i) a polymorphism in an EBNA-3B-responsive element in the CXCR4 promoter or (ii) a polymorphism in an EBNA-3B-interacting protein important for EBNA-3B-mediated transcriptional repression. Studies are under way to test these hypotheses, and this may be a situation where a host genetic polymorphism can influence the outcome of EBV infection.
The low EBNA-3C expression levels in 3B−/3Clow LCLs were most likely responsible for their poor growth in tissue culture, since a spontaneous, partial deletion of the kanamycin resistance cassette in one of our 3B−/3Clow LCLs resulted in the outgrowth of a cell line restored for both wild-type EBNA-3C expression levels and normal growth. Since EBNA-3C-mediated repression of Jagged1 does not appear to be important for LCL growth, one or more of the other EBNA-3C-regulated genes identified by transcriptional array analysis may be involved in cell growth. One potential candidate gene is T-cell leukemia/lymphoma 1A (TCL1A), an EBNA-3C-activated oncogene whose expression is drastically downmodulated in 3B−/3Clow LCLs. TCL1A expression is upregulated in various T- and B-cell lymphomas, has been associated with EBV infection in virus-infected Burkitt lymphoma cell lines, and is highly expressed in LCLs (22).
In general, we were surprised to find that the expression of so few cellular genes was significantly changed in the 3B−/3Clow LCLs compared to what was found for wild-type LCLs, despite such a large difference in growth phenotypes. However, while the 3B−/3Clow LCLs grew at a significantly lower rate, they were able to sustain cell proliferation, suggesting that all the critical cellular processes involved in virus-induced B-cell immortalization were still intact and functional. Thus, it might be expected that overall cellular gene expression would not be significantly affected since large changes in the expression levels of essential genes would likely result in the failure of the cells to survive. There may exist several subtle changes in gene expression that act synergistically to produce the drastic growth phenotype, which may not have been detected by our relatively stringent statistical analysis.
Our transcriptional array and RT-PCR results also showed that EBNA-3B and EBNA-3C, while being related EBV proteins, were able to differentially regulate the expression of cellular genes. It was previously known that the EBNA-3s all have distinct functions, as neither EBNA-3B nor EBNA-3C was able to complement LCL growth arrest mediated by EBNA-3A withdrawal (29). Additionally, the EBNA-3s are able to differentially regulate viral promoters, as only EBNA-3C, and not EBNA-3A or EBNA-3B, was able to coactivate reporter expression from the LMP-1 promoter with EBNA-2 (55). Stable transfectants of one or more EBNA-3 proteins into EBV-negative cells exhibited different phenotypes (20, 43, 52), implying that the EBNA-3s have distinct cellular targets, but differential regulation of cellular genes by the EBNA-3s in the context of virus-infected cells has not been observed. Our results demonstrate that EBNA-3B and EBNA-3C target different subsets of cellular genes and that at least EBNA-3C has both activating and repressive functions. We have identified only EBNA-3B-repressed genes, but the possibility that it also possesses activating properties still exists.
Acknowledgments
This work was supported by a grant from the U.S. Public Health Service (CA68051).
We thank the Harvard Medical School Partners Healthcare Center for Genetics and Genomics Core Facility for performing the transcriptional profiling and Samuel Pattle for critical reading of the manuscript.
REFERENCES
- 1.Babcock, G. J., L. L. Decker, M. Volk, and D. A. Thorley-Lawson. 1998. EBV persistence in memory B cells in vivo. Immunity 9:395-404. [DOI] [PubMed] [Google Scholar]
- 2.Chen, A., M. Divisconte, X. Jiang, C. Quink, and F. Wang. 2005. Epstein-Barr virus with the latent infection nuclear antigen 3B completely deleted is still competent for B-cell growth transformation in vitro. J. Virol. 79:4506-4509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cludts, I., and P. J. Farrell. 1998. Multiple functions within the Epstein-Barr virus EBNA-3A protein. 72:1862-1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cotter, M. A., II, and E. S. Robertson. 2000. Modulation of histone acetyltransferase activity through interaction of Epstein-Barr nuclear antigen 3C with prothymosin alpha. Mol. Cell. Biol. 20:5722-5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.D'Apuzzo, M., A. Rolink, M. Loetscher, J. A. Hoxie, I. Clark-Lewis, F. Melchers, M. Baggiolini, and B. Moser. 1997. The chemokine SDF-1, stromal cell-derived factor 1, attracts early stage B cell precursors via the chemokine receptor CXCR4. Eur. J. Immunol. 27:1788-1793. [DOI] [PubMed] [Google Scholar]
- 6.Datsenko, K. A., and B. L. Wanner. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. USA 97:6640-6645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ehlin-Henriksson, B., F. Mowafi, G. Klein, and A. Nilsson. 2006. Epstein-Barr virus infection negatively impacts the CXCR4-dependent migration of tonsillar B cells. Immunology 117:379-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fedyk, E. R., D. H. Ryyan, I. Ritterman, and T. A. Springer. 1999. Maturation decreases responsiveness of human bone marrow B lineage cells to stromal-derived factor 1 (SDF-1). J. Leukoc. Biol. 66:667-673. [DOI] [PubMed] [Google Scholar]
- 9.Finke, J., M. Rowe, B. Kallin, I. Ernberg, A. Rosen, J. Dillner, and G. Klein. 1987. Monoclonal and polyclonal antibodies against Epstein-Barr virus nuclear antigen 5 (EBNA-5) detect multiple protein species in Burkitt's lymphoma and lymphoblastoid cell lines. J. Virol. 61:3870-3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gavioli, R., P. O. De Campos-Lima, M. G. Kurilla, E. Kieff, G. Klein, and M. G. Masucci. 1992. Recognition of the Epstein-Barr virus-encoded nuclear antigens EBNA-4 and EBNA-6 by HLA-A11-restricted cytotoxic T lymphocytes: implications for down-regulation of HLA-A11 in Burkitt lymphoma. Proc. Natl. Acad. Sci. USA 89:5862-5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordadze, A. V., R. Peng, J. Tan, G. Liu, R. Sutton, B. Kempkes, G. W. Bornkamm, and P. D. Ling. 2001. Notch1IC partially replaces EBNA2 function in B cells immortalized by Epstein-Barr virus. J. Virol. 75:5899-5912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grillot-Courvalin, C., S. Goussard, F. Huetz, D. M. Ojcius, and P. Courvalin. 1998. Functional gene transfer from intracellular bacteria to mammalian cells. Nat. Biotechnol. 16:862-866. [DOI] [PubMed] [Google Scholar]
- 13.Grossman, S. R., E. Johannsen, X. Tong, R. Yalamanchili, and E. Kieff. 1994. The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein. Proc. Natl. Acad. Sci. USA 91:7568-7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Henkel, T., P. D. Ling, S. D. Hayward, and M. G. Peterson. 1994. Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa. Science 265:92-95. [DOI] [PubMed] [Google Scholar]
- 15.Hickabottom, M., G. A. Parker, P. Freemont, T. Crook, and M. J. Allday. 2002. Two nonconsensus sites in the Epstein-Barr virus oncoprotein EBNA3A cooperate to bind the co-repressor carboxyl-terminal-binding protein (CtBP). J. Biol. Chem. 277:47197-47204. [DOI] [PubMed] [Google Scholar]
- 16.Honczarenko, M., R. S. Douglas, C. Mathias, B. Lee, M. Z. Ratajczak, and L. E. Silberstein. 1999. SDF-1 responsiveness does not correlate with CXCR4 expression levels of developing human bone marrow B cells. Blood 94:2990-2998. [PubMed] [Google Scholar]
- 17.Hsieh, J. J., T. Henkel, P. Salmon, E. Robey, M. G. Peterson, and S. D. Hayward. 1996. Truncated mammalian Notch1 activates CBF1/RBPJk-repressed genes by a mechanism resembling that of Epstein-Barr virus EBNA2. Mol. Cell. Biol. 16:952-959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jiang, H., Y. G. Cho, and F. Wang. 2000. Structural, functional, and genetic comparisons of Epstein-Barr virus nuclear antigen 3A, 3B, and 3C homologues encoded by the rhesus lymphocryptovirus. J. Virol. 74:5921-5932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khanna, R., S. R. Burrows, M. G. Kurilla, C. A. Jacob, I. S. Misko, T. B. Sculley, E. Kieff, and D. J. Moss. 1992. Localization of Epstein-Barr virus cytotoxic T cell epitopes using recombinant vaccinia: implications for vaccine development. J. Exp. Med. 176:169-176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kienzle, N., D. Young, S. L. Silins, and T. B. Sculley. 1996. Induction of pleckstrin by the Epstein-Barr virus nuclear antigen 3 family. Virology 224:167-174. [DOI] [PubMed] [Google Scholar]
- 21.King, W., A. L. Thomas-Powell, N. Raab-Traub, M. Hawke, and E. Kieff. 1980. Epstein-Barr virus RNA. V. Viral RNA in a restringently infected, growth-transformed cell line. J. Virol. 36:506-518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kiss, C., J. Nishikawa, K. Takada, P. Trivedi, G. Klein, and L. Szekely. 2003. T cell leukemia I oncogene expression depends on the presence of Epstein-Barr virus in the virus-carrying Burkitt lymphoma lines. Proc. Natl. Acad. Sci. USA 100:4813-4818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Knight, J. S., K. Lan, C. Subramanian, and E. S. Robertson. 2003. Epstein-Barr virus nuclear antigen 3C recruits histone deacetylase activity and associates with the corepressors mSin3A and NCoR in human B-cell lines. J. Virol. 77:4261-4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin, J., E. Johannsen, E. Robertson, and E. Kieff. 2002. Epstein-Barr virus nuclear antigen 3C putative repression domain mediates coactivation of the LMP1 promoter with EBNA-2. J. Virol. 76:232-242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Livak, K. J., and T. D. Schmittgen. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25:402-408. [DOI] [PubMed] [Google Scholar]
- 26.Ma, Q., D. Jones, P. R. Borghesani, R. A. Segal, T. Nagasawa, T. Kishimoto, R. T. Bronson, and T. A. Springer. 1998. Impaired B-lymphopoiesis, myelopoiesis, and derailed cerebellar neuron migration in CXCR4- and SDF-1-deficient mice. Proc. Natl. Acad. Sci. USA 95:9448-9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mann, K. P., D. Staunton, and D. A. Thorley-Lawson. 1985. Epstein-Barr virus-encoded protein found in plasma membranes of transformed cells. J. Virol. 55:710-720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Marshall, D., and C. Sample. 1995. Epstein-Barr virus nuclear antigen 3C is a transcriptional regulator. J. Virol. 69:3624-3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Maruo, S., E. Johannsen, D. Illanes, A. Cooper, and E. Kieff. 2003. Epstein-Barr Virus nuclear protein EBNA3A is critical for maintaining lymphoblastoid cell line growth. J. Virol. 77:10437-10447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maunders, M. J., L. Petti, and M. Rowe. 1994. Precipitation of the Epstein-Barr virus protein EBNA 2 by an EBNA 3c-specific monoclonal antibody. J. Gen. Virol. 75:769-778. [DOI] [PubMed] [Google Scholar]
- 31.McKenzie, G. J., M. Khan, E. Briend, Y. Stallwood, and B. R. Champion. 2005. Notch: a unique therapeutic target for immunomodulation. Expert Opin. Ther. Targets 9:395-410. [DOI] [PubMed] [Google Scholar]
- 32.Moghaddam, A., M. Rosenzweig, D. Lee-Parritz, B. Annis, R. P. Johnson, and F. Wang. 1997. An animal model for acute and persistent Epstein-Barr virus infection. Science 276:2030-2033. [DOI] [PubMed] [Google Scholar]
- 33.Morimura, T., R. Goitsuka, Y. Zhang, I. Saito, M. Reth, and D. Kitamura. 2000. Cell cycle arrest and apoptosis induced by Notch1 in B cells. J. Biol. Chem. 275:36523-36531. [DOI] [PubMed] [Google Scholar]
- 34.Murray, R. J., M. G. Kurilla, J. M. Brooks, W. A. Thomas, M. Rowe, E. Kieff, and A. B. Rickinson. 1992. Identification of target antigens for the human cytotoxic T cell response to Epstein-Barr virus (EBV): implications for the immune control of EBV-positive malignancies. J. Exp. Med. 176:157-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nagasawa, T., T. Nakajima, K. Tachibana, H. Iizasa, C. C. Bleul, O. Yoshie, K. Matsushima, N. Yoshida, T. A. Springer, and T. Kishimoto. 1996. Molecular cloning and characterization of a murine pre-B-cell growth-stimulating factor/stromal cell-derived factor 1 receptor, a murine homolog of the human immunodeficiency virus 1 entry coreceptor fusin. Proc. Natl. Acad. Sci. USA 93:14726-14729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nakayama, T., R. Fujisawa, D. Izawa, K. Hieshima, K. Takada, and O. Yoshie. 2002. Human B cells immortalized with Epstein-Barr virus upregulate CCR6 and CCR10 and downregulate CXCR4 and CXCR5. J. Virol. 76:3072-3077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nie, Y., J. Waite, F. Brewer, M. J. Sunshine, D. R. Littman, and Y. R. Zou. 2004. The role of CXCR4 in maintaining peripheral B cell compartments and humoral immunity. J. Exp. Med. 200:1145-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petti, L., and E. Kieff. 1988. A sixth Epstein-Barr virus nuclear protein (EBNA3B) is expressed in latently infected growth-transformed lymphocytes. J. Virol. 62:2173-2178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Radkov, S. A., M. Bain, P. J. Farrell, M. West, M. Rowe, and M. J. Allday. 1997. Epstein-Barr virus EBNA3C represses Cp, the major promoter for EBNA expression, but has no effect on the promoter of the cell gene CD21. J. Virol. 71:8552-8562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radkov, S. A., R. Touitou, A. Brehm, M. Rowe, M. West, T. Kouzarides, and M. J. Allday. 1999. Epstein-Barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription. J. Virol. 73:5688-5697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Rivailler, P., H. Jiang, Y. G. Cho, C. Quink, and F. Wang. 2002. Complete nucleotide sequence of the rhesus lymphocryptovirus: genetic validation for an Epstein-Barr virus animal model. J. Virol. 76:421-426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Seiffert, D., J. D. Bradley, C. M. Rominger, D. H. Rominger, F. Yang, J. E. Meredith, Jr., Q. Wang, A. H. Roach, L. A. Thompson, S. M. Spitz, J. N. Higaki, S. R. Prakash, A. P. Combs, R. A. Copeland, S. P. Arneric, P. R. Hartig, D. W. Robertson, B. Cordell, A. M. Stern, R. E. Olson, and R. Zaczek. 2000. Presenilin-1 and -2 are molecular targets for gamma-secretase inhibitors. J. Biol. Chem. 275:34086-34091. [DOI] [PubMed] [Google Scholar]
- 43.Silins, S. L., and T. B. Sculley. 1994. Modulation of vimentin, the CD40 activation antigen and Burkitt's lymphoma antigen (CD77) by the Epstein-Barr virus nuclear antigen EBNA-4. Virology 202:16-24. [DOI] [PubMed] [Google Scholar]
- 44.Tachibana, K., S. Hirota, H. Iizasa, H. Yoshida, K. Kawabata, Y. Kataoka, Y. Kitamura, K. Matsushima, N. Yoshida, S. Nishikawa, T. Kishimoto, and T. Nagasawa. 1998. The chemokine receptor CXCR4 is essential for vascularization of the gastrointestinal tract. Nature 393:591-594. [DOI] [PubMed] [Google Scholar]
- 45.Tierney, R. J., N. Steven, L. S. Young, and A. B. Rickinson. 1994. Epstein-Barr virus latency in blood mononuclear cells: analysis of viral gene transcription during primary infection and in the carrier state. J. Virol. 68:7374-7385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tomkinson, B., and E. Kieff. 1992. Use of second-site homologous recombination to demonstrate that Epstein-Barr virus nuclear protein 3B is not important for lymphocyte infection or growth transformation in vitro. J. Virol. 66:2893-2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tomkinson, B., E. Robertson, and E. Kieff. 1993. Epstein-Barr virus nuclear proteins EBNA-3A and EBNA-3C are essential for B-lymphocyte growth transformation. J. Virol. 67:2014-2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Touitou, R., M. Hickabottom, G. Parker, T. Crook, and M. J. Allday. 2001. Physical and functional interactions between the corepressor CtBP and the Epstein-Barr virus nuclear antigen EBNA3C. J. Virol. 75:7749-7755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vicente-Manzanares, M., M. C. Montoya, M. Mellado, J. M. Frade, M. A. del Pozo, M. Nieto, M. O. de Landazuri, A. C. Martinez, and F. Sanchez-Madrid. 1998. The chemokine SDF-1alpha triggers a chemotactic response and induces cell polarization in human B lymphocytes. Eur. J. Immunol. 28:2197-2207. [DOI] [PubMed] [Google Scholar]
- 50.Vigouroux, S., E. Yvon, H. J. Wagner, E. Biagi, G. Dotti, U. Sili, C. Lira, C. M. Rooney, and M. K. Brenner. 2003. Induction of antigen-specific regulatory T cells following overexpression of a Notch ligand by human B lymphocytes. J. Virol. 77:10872-10880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wang, F. 2001. A new animal model for Epstein-Barr virus pathogenesis. Curr. Top. Microbiol. Immunol. 258:201-219. [DOI] [PubMed] [Google Scholar]
- 52.Wang, F., C. Gregory, C. Sample, M. Rowe, D. Liebowitz, R. Murray, A. Rickinson, and E. Kieff. 1990. Epstein-Barr virus latent membrane protein (LMP1) and nuclear proteins 2 and 3C are effectors of phenotypic changes in B lymphocytes: EBNA-2 and LMP1 cooperatively induce CD23. J. Virol. 64:2309-2318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yasukawa, M., A. Hasegawa, I. Sakai, H. Ohminami, J. Arai, S. Kaneko, Y. Yakushijin, K. Maeyama, H. Nakashima, R. Arakaki, and S. Fujita. 1999. Down-regulation of CXCR4 by human herpesvirus 6 (HHV-6) and HHV-7. J. Immunol. 162:5417-5422. [PubMed] [Google Scholar]
- 54.Yvon, E. S., S. Vigouroux, R. F. Rousseau, E. Biagi, P. Amrolia, G. Dotti, H. J. Wagner, and M. K. Brenner. 2003. Overexpression of the Notch ligand, Jagged-1, induces alloantigen-specific human regulatory T cells. Blood 102:3815-3821. [DOI] [PubMed] [Google Scholar]
- 55.Zhao, B., and C. E. Sample. 2000. Epstein-Barr virus nuclear antigen 3C activates the latent membrane protein 1 promoter in the presence of Epstein-Barr virus nuclear antigen 2 through sequences encompassing an Spi-1/Spi-B binding site. J. Virol. 74:5151-5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zou, Y. R., A. H. Kottmann, M. Kuroda, I. Taniuchi, and D. R. Littman. 1998. Function of the chemokine receptor CXCR4 in haematopoiesis and in cerebellar development. Nature 393:595-599. [DOI] [PubMed] [Google Scholar]