

Abstract
The p52/p100 nuclear factor kappa B (NF-κB) subunit (NF-κB2) is aberrantly expressed in many tumour types and has been implicated as a regulator of cell proliferation. Here, we demonstrate that endogenous p52 is a direct regulator of Cyclin D1 expression. However, stimulation of Cyclin D1 expression alone cannot account for all the cell cycle effects of p52/p100 and we also find that p52 represses expression of the Cyclin-dependent kinase inhibitor p21WAF/CIP1. Significantly, this latter effect is dependent upon basal levels of the tumour suppressor p53. By contrast, p52 cooperates with p53 to regulate other known p53 target genes such as PUMA, DR5, Gadd45α and Chk1. p52 associates directly with these p53-regulated promoters where it regulates coactivator and corepressor binding. Moreover, recruitment of p52 is p53 dependent and does not require p52-DNA-binding activity. These results reveal a complex role for p52 as regulator of cell proliferation and p53 transcriptional activity. Furthermore, they imply that in some cell types, p52 can regulate p53 function and influence p53-regulated decision-making following DNA damage and oncogene activation.
Keywords: apoptosis, cell cycle, NF-κB2, p53, p21WAF1
Introduction
In mammalian cells, there are five members of the nuclear factor kappa B (NF-κB) family of transcriptional regulators, RelA(p65), RelB, c-Rel, p50/p105 (NF-κB1) and p52/p100 (NF-κB2), all of which share an N-terminal DNA-binding and dimerization domain, termed the Rel homology domain (RHD). Biochemical and genetic approaches have demonstrated that these subunits have both overlapping and distinct functions (Gerondakis et al, 1999; Hoffmann et al, 2003; Hayden and Ghosh, 2004). The p50 and p52 subunits are synthesized as longer precursor proteins, p105 and p100 that contain ankyrin repeats in their C-termini, similar to those possessed by the inhibitor of NF-κB (IκB) family of proteins (Hayden and Ghosh, 2004). Like IκB proteins, the C-termini of p100 and p105 also act as inhibitory motifs, resulting in cytoplasmic retention of both these proteins and their dimer partners (Hayden and Ghosh, 2004). Most inducers of NF-κB stimulate IκB kinase (IKK) β activity, resulting in the phosphorylation and degradation of IκB proteins (Hayden and Ghosh, 2004). However, a subset of NF-κB inducers also activate IKKα, which phosphorylates p100, leading to its partial proteolytic degradation by the proteasome and the generation of p52 in a process termed the ‘noncanonical' or ‘alternative' pathway (Bonizzi and Karin, 2004; Hayden and Ghosh, 2004).
One consequence of the activation of the noncanonical pathway is a different spectrum of nuclear NF-κB complexes containing p52. Although these are often depicted as p52/RelB heterodimers, p52/RelA complexes have also been observed (Hoffmann et al, 2003; Marinari et al, 2004; Lo et al, 2006) and, in addition to the presence of p52 homodimers, it is likely that p52 also complexes with the other NF-κB subunits. p52 complexes demonstrate relatively weak binding to the consensus κB elements generally used to analyse NF-κB DNA-binding activity (Duckett et al, 1993). Consistent with this analysis, p52/RelB heterodimers have recently been shown to recognize a distinct κB element, resulting in the regulation of specific p52/RelB target genes (Bonizzi et al, 2004). These differences in DNA-binding affinity and specificity may have led to p52 activity being overlooked in many circumstances.
A number of observations have suggested an important role for p52 NF-κB in cancer. Most obviously, the p52/p100 gene is found rearranged in some B- and T-cell lymphomas, resulting in C-terminally truncated and constitutively nuclear forms of the protein (Fracchiolla et al, 1993; Thakur et al, 1994; Chang et al, 1995). Significantly, these tumour-associated truncations have transforming effects in murine fibroblasts (Ciana et al, 1997). Furthermore, p52 has been found aberrantly nuclear in human breast and mouse skin tumours (Budunova et al, 1999; Cogswell et al, 2000) and its processing is also induced by the viral onco-proteins Tax and LMP1 (Kanno et al, 1994; Lanoix et al, 1994; Eliopoulos et al, 2003). In addition, the p52 coactivator Bcl-3, has also been described as a regulator of cell proliferation and tumorigenesis (Westerheide et al, 2001; Viatour et al, 2004; Massoumi et al, 2006).
NF-κB complexes can contribute towards tumorigenesis in many ways, including effects on cell survival, angiogenesis, metastasis and cell proliferation (Greten and Karin, 2004; Perkins and Gilmore, 2006). p52 may contribute to many of these processes but there is growing evidence to suggest that p52 can function as a regulator of cell proliferation. At least in part, this results from regulation of the Cyclin D1 promoter (Westerheide et al, 2001; Rocha et al, 2003b). Furthermore, targeted deletion of the C-terminus of the nfkb2 gene in mice, to produce constitutively active p52, results in animals that appear normal at birth but exhibit hyperplasia in some tissues and consequently develop multiple pathologies (Ishikawa et al, 1997). Although this result is consistent with a proproliferative role for p52, these mice die postnatally preventing longer-term studies on cancer development. This result is in contrast to mice entirely lacking p52/p100, which develop normally, with the major defect being a disruption of splenic and lymph node architecture (Gerondakis et al, 1999; Weih and Caamano, 2003). A more dramatic effect is seen with nfkb1−/− nfkb2−/− (p105 and p100) double knockout mice relative to either single gene knockout, suggesting compensatory mechanisms between these subunits. These mice are phenotypically indistinguishable from control littermates at birth but soon exhibit growth retardation and craniofacial abnormalities, the latter being a result of bone thickening because of osteopetrosis (Franzoso et al, 1997; Iotsova et al, 1997). Furthermore, osteoclast numbers are markedly reduced in the double-mutant mice and nfkb1−/− nfkb2−/− osteoclast progenitors cannot differentiate in vitro (Franzoso et al, 1997; Iotsova et al, 1997). B-cell development is also blocked in nfkb1−/− nfkb2−/− double-mutant mice at the immature IgM+IgD− stage (Franzoso et al, 1997; Iotsova et al, 1997). Whether any of these effects are related to a p52-dependent proliferative defect is not known but these observations suggest that its aberrant activation might contribute to tumorigenesis in a wider range of cell types.
Here, we have investigated the ability of endogenous p52 protein to function as a regulator of cell proliferation. We find that in addition to regulating Cyclin D1 expression, p52 can have much wider effects on cell growth through modulation of p53 tumour suppressor activity and the regulation of p53-target genes.
Results
The p52/p100 NF-κB subunit regulates U-2 OS cell proliferation and Cyclin D1 expression
To investigate p52/100 effects on cell proliferation and the cell cycle, three distinct short interfering RNAs (siRNAs) were designed targeting this NF-κB subunit. U-2 OS human osteosarcoma cells were initially used in these studies because we have found that they possess a significant basal level of nuclear p52 NF-κB, which allows the study of its function in the absence of cell stimulation and induction of the noncanonical pathway. This high level of p52 is dependent upon serum in the growth media: withdrawal of serum strongly inhibits p100 processing that is, in turn, stimulated by its readdition (data not shown). All siRNAs, either individually or in combination, efficiently downregulated p52/p100 mRNA and protein levels in U-2 OS cells (Figure 1A and B and data not shown). Significantly, knockdown of p52/p100 by all siRNAs resulted in a significant decrease in cell proliferation (Figure 1C).
Figure 1.
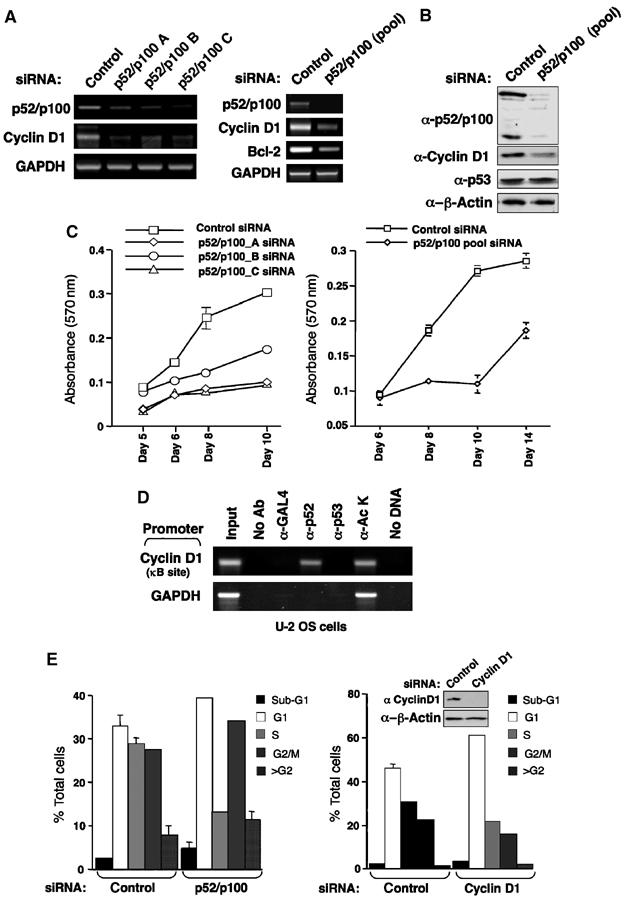
Regulation of Cyclin D1 expression and cell proliferation by the p52/p100 NF-κB subunit. (A, B) siRNAs targeting p52/p100 inhibit Cyclin D1 expression. U-2 OS cells were transfected with either individual or pooled siRNAs targeting p52/p100. mRNA (A) and protein (B) expression was analysed as shown. In these and subsequent experiments, cells were transfected with siRNA on day 1. On day 2, the cells were split and transfected with siRNA again on day 3. On day 4, cells were rested before being harvested on day 5. (C) p52/p100 regulates cell proliferation. U-2 OS cells were transfected with individual or pooled p52/p100 siRNAs as indicated and alamar blue assays were performed. Times are from the day of the first siRNA transfection. (D) p52 binds the Cyclin D1 promoter. ChIP analysis of the Cyclin D1 and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (control) promoters, using the indicated antibodies, was performed in U-2 OS as shown. (E) Cell cycle analysis of p52/p100 and Cyclin D1 siRNA-treated cells. U-2 OS cells were transfected with pooled p52/p100 and Cyclin D1 siRNAs as indicated and on day 5 were harvested, stained with propidium iodide and subjected to fluorescence-activated cell sorting (FACS) analysis. Also shown is Western blot analysis of Cyclin D1 levels following treatment with the Cyclin D1 siRNA.
Reports from a number of laboratories have indicated that p52 regulates the Cyclin D1 promoter (Westerheide et al, 2001; Romieu-Mourez et al, 2003; Rocha et al, 2003b). However, definitive evidence of endogenous p52 directly regulating endogenous Cyclin D1 expression had not been clearly established. Here, we found that loss of endogenous p52/p100 in U-2 OS cells does lead to downregulation of Cyclin D1 mRNA and protein levels (Figure 1A and B). In addition, downregulation of Bcl-2 expression, which has been previously described as a p52 target gene (Viatour et al, 2003), was also observed. To establish that Cyclin D1 is a direct target of p52 in these cells, chromatin immunoprecipitation (ChIP) analysis was performed. This demonstrated that endogenous p52 directly binds in the region of the previously identified NF-κB responsive region of the Cyclin D1 promoter in U-2 OS cells (Figure 1D).
We next examined the cell cycle distribution of cells treated with p52/p100 siRNAs. This analysis indicated that the effects seen on proliferation were primarily associated with a decrease in the number of S-phase cells, together with concomitant increases in G1 and G2 phase cells. The absence of a decrease in G2 phase cells was surprising, suggesting that p52 had effects on proliferation independently of Cyclin D1, loss of which would be expected to lead to a G1 phase arrest only. Indeed, when U-2 OS cells were treated with a Cyclin D1 siRNA, cells accumulated only in G1 phase (Figure 1E). mRNA and protein analysis confirmed the efficacy of this siRNA (Figure 1E and data not shown). Therefore, although Cyclin D1 can exhibit a number of Cyclin-dependent kinase (CDK)-independent effects and has been described as a regulator of the p21 promoter (Bienvenu et al, 2005), regulation of Cyclin D1 expression cannot account for all the effects on proliferation seen upon loss of p52/p100 expression in U-2 OS cells.
Regulation of p21WAF1/CIP1 expression by p52/p100
Further analysis of cells transfected with siRNAs targeting p52/p100 revealed a significant upregulation of the CDK inhibitor p21WAF1/CIP1 (Figure 2A and B). Moreover, transfection of p52 expression plasmids inhibited the activity of a p21 promoter luciferase reporter plasmid (Figure 2C). By contrast, full-length p100, which would be expected to inhibit endogenous p52 and other NF-κB subunits, activated the p21 promoter (Figure 2C). Similarly, cotransfection of p52/p100 siRNA-expressing plasmids also stimulated the p21 promoter (Figure 2D). This latter effect required the distal promoter region containing the p53-binding sites. As expression of p21 will exert multiple effects on the cell cycle, these observations indicated that in U-2 OS cells, the p52 NF-κB subunit can stimulate cell proliferation through a combination of inducing and repressing the expression of Cyclin D1 and p21, respectively.
Figure 2.
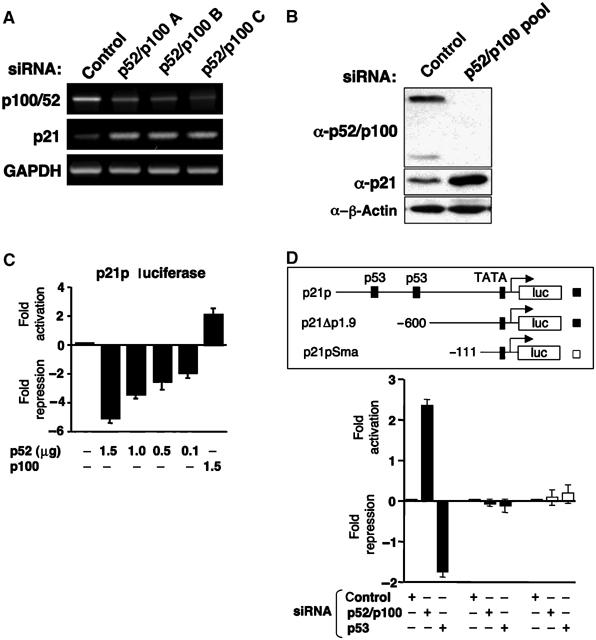
p52 is a repressor of the p21WAF1/CIP1 promoter. (A, B) siRNAs targeting p52/p100 induce p21WAF1/CIP1 expression. U-2 OS cells were transfected with either individual or pooled siRNAs targeting p52/p100 as indicated. mRNA (A) and protein (B) expression was analysed as shown. (C) Regulation of the p21 promoter by overexpressed p52 and p100. U-2 OS cells were transfected with 1.5 μg of the full-length p21 promoter luciferase reporter plasmid together with the indicated levels of RSV p52 and p100 expression plasmids. RSV control plasmid was included such that all condition contained equivalent levels of plasmid DNA. Results are expressed as fold activation or repression relative to levels seen in untreated controls. Results are normalized such that no change in luciferase activity has a value of 0. (D) Deletion analysis of the p21 promoter. U-2 OS cells were transfected with 1.5 μg of the full-length p21 promoter luciferase reporter plasmid together with 1.5 μg of p52/p100, p53 or control siRNA expression plasmids. A schematic diagram of the plasmids used, showing the p53 binding sites, is shown.
Regulation of Cyclin D1 and p21 is a specific effect of p52 NF-κB
The genes of many NF-κB subunits, including p52/p100, contain κB sites in their promoters and are subject to autoregulation (Hayden and Ghosh, 2004). Therefore, it was possible that the effects of the p52/p100 siRNAs might result from indirect effects on other NF-κB subunits. However, further analysis demonstrated that depletion of p52/p100 mRNA and protein did not significantly affect the expression of other NF-κB subunits, although slight decreases in the protein levels of RelB, c-Rel and Bcl-3, a p52 coactivator (Bours et al, 1993), were observed (Figure 3A and B). Furthermore, depletion of RelB and p50/p105 did not affect Cyclin D1 and p21 levels (Figure 3C). Interestingly, whereas depletion of Bcl-3 did reduce Cyclin D1 levels, consistent with our and others observations (Westerheide et al, 2001; Rocha et al, 2003b), this did not affect p21 levels, indicating distinct mechanisms of p52-dependent regulation (Figure 3C). Moreover, when considered together with the absence of significant effects on either p53 and Hdm2 protein levels (Figures 1B and 3D), this indicates that the effects we see with p52 are distinct from the recently described Bcl-3-mediated effects on p53 stability through regulation of the Hdm2/Mdm2 promoter (Kashatus et al, 2006). The effects of RelA and c-Rel siRNAs on Cyclin D1 and p21 are not shown as they significantly regulate p52/p100 expression levels, making interpretation of any results difficult (data not shown).
Figure 3.
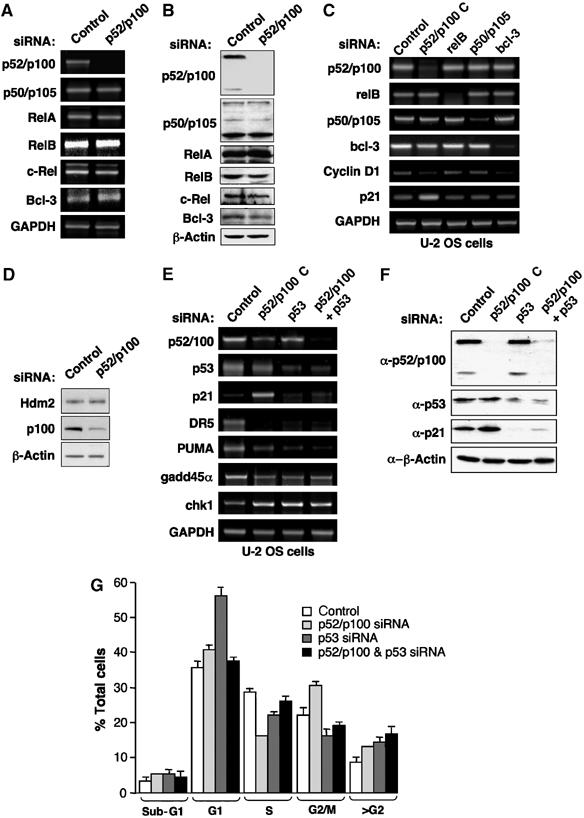
The p52/p100 NF-κB subunit specifically regulates p53 target genes. (A, B) Depletion of p52/p100 does not significantly affect the expression of other NF-κB subunits. U-2 OS cells were transfected with pooled p52/p100 or control siRNAs and mRNA (A) and protein (B) expression for the NF-κB subunits and the p52 coactivator Bcl-3 was analysed as shown. (C) Regulation of Cyclin D1 and p21 expression in U-2 OS cells is p52/p100 specific. U-2 OS cells were transfected with p52/p100 (version C), p50/p105, RelB, Bcl-3 or control siRNAs and mRNA levels for the NF-κB subunits, Cyclin D1 and p21 together with a GAPDH control were analysed as shown. (D) Depletion of p52/p100 does not significantly affect the expression of Hdm2. U-2 OS cells were transfected as in (A) and analysed for expression of Hdm2 and p100 expression as shown. (E) Regulation of multiple p53 target genes in U-2 OS cells by p52/p100. U-2 OS cells were transfected with p52/p100 (version C), p53 or control siRNAs as shown and mRNA levels for the indicated p53 target genes were analysed. (F) Regulation of p21 protein levels. U-2 OS cells were transfected with p52/p100 (pooled), p53 or control siRNAs as shown and Western blotted for the indicated proteins. (G) Cell cycle analysis of p52/p100 and p53 siRNA-treated cells. U-2 OS cells were transfected with pooled p52/p100 and p53 siRNAs as indicated and treated as in Figure 1E.
p53-dependent effects of p52 NF-κB
The p21 promoter is an established target of the p53 tumour suppressor, although it can also be regulated by many p53-independent mechanisms (Gartel and Tyner, 1999). Our earlier luciferase assay data had suggested that p52 regulation of p21 expression might be p53 dependent (Figure 2D and E). Therefore, to determine whether this was the case, we analysed the effect of depleting p53, either alone or in conjunction with p52/p100. No evidence for cross-regulation of p53 and p52/p100 mRNA levels was observed but significantly, induction of p21 protein and mRNA expression upon p52/p100 depletion was lost in the absence of p53 (Figure 3E and F). Moreover, the reduction in S phase cells seen upon depletion of p52 (Figure 1E) was also p53 dependent (Figure 3G). Interestingly, depletion of p53 alone resulted in an increase in G1 phase cells, which was lost upon co-depletion of p52. Taken together, these results demonstrate that the p52 NF-κB subunit is a p53-dependent regulator of p21 expression and the cell cycle in unstimulated U-2 OS cells.
This result suggested that p52 might regulate the expression of other p53 target genes. Analysis of the Death Receptor (DR) 5, PUMA, Gadd45α and Chk1 genes, all of which have been shown to be directly regulated by p53 (Takimoto and El-Deiry, 2000; Nakano and Vousden, 2001; Espinosa et al, 2003; An et al, 2004; Kho et al, 2004), demonstrated that all were also regulated by p52 NF-κB (Figure 3E). However, whereas p52 opposes p53 function at the p21 promoter, with these genes, the effect of p52 depletion mirrored the effect of loss of p53. This occurred regardless of whether the gene is dependent upon p53 for its expression or, as is the case with Chk1, is repressed by p53. The p53 dependence of these genes in the absence of overt DNA damage was surprising, although this may reflect a partial activation of p53 resulting from transfection or the stress of cell manipulation. Alternatively, other reports have indicated that p53 contributes towards the basal level expression of many of its target genes in both U-2 OS and other cell types (Espinosa et al, 2003; An et al, 2004; Bates et al, 2005).
Loss of p52 affects inducible p53 function
We next investigated whether p52 also regulates inducible p53 function and whether the effects are similar or distinct to those seen in untreated cells. Following knockdown of p52, p53 was still significantly induced by UV-C treatment, albeit at a slightly reduced level (Figure 4A). A reduction in p53 serine 15 phosphorylation was also observed. This effect may result from p52 effects on the cell cycle, which could affect the function of checkpoint kinases such as ATR. Nonetheless, effects on p53 function could be observed. Interestingly, loss of p52 inhibited any further induction of p21 mRNA from its higher basal level in p52 siRNA-treated cells whereas inducible DR5 expression was significantly repressed following UV-C treatment (Figure 4B and Supplementary Figure 1A). Furthermore, depletion of p52 significantly inhibited UV-C-induced caspase 3 activity (Figure 4C).
Figure 4.
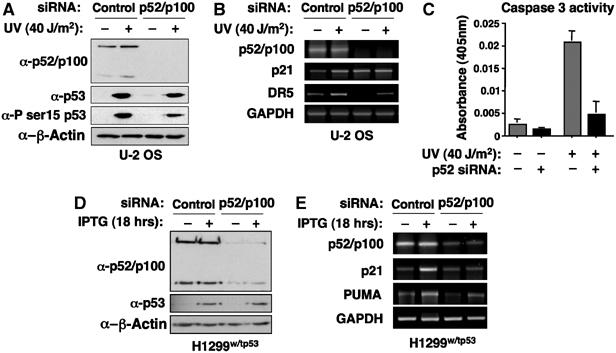
p52 also regulates inducible p53-dependent gene expression. (A) Induction of p53 in UV-C and p52 siRNA-treated cells. Cell lysates were prepared from U-2 OS cells transfected with control or pooled p52/p100 siRNAs, with and without UV-C treatment for 6 h, and analysed for p52/p100, p53, phospho-p53 serine 15 and β-actin (control) expression by Western blot. (B) Analysis of p53 target gene expression following UV treatment. U-2 OS cells were transfected with control or pooled p52/p100 siRNAs and UV-C treated for 6 h. mRNA was collected and analysed for p52/p100, p21, DR5 and GAPDH (control) expression. (C) Depletion of p52 inhibits UV-C-induced apoptosis. U-2 OS cells were transfected with control or pooled p52/p100 siRNAs, with and without UV-C treatment for 6 h, and analysed for caspase 3 activity as indicated. (D) Analysis of H1299 cells with an IPTG-inducible p53 expression plasmid. Cell lysates were prepared from H1299w/tp53 cells, transfected with control or pooled p52/p100 siRNAs, with and without IPTG treatment for 18 h. Lysates were analysed for p52/p100, p53 and β-actin (control) expression by Western blot. (E) Analysis of p53 target gene expression in p53 in H1299w/tp53 cells. p53-inducible H1299w/tp53 cells were transfected with control or pooled p52/p100 siRNAs and treated for 18 h. mRNA was collected and analysed for p52/p100, p21, PUMA and GAPDH (control) expression.
Because of the complications that may be associated with UV-C treatment in U-2 OS cells, we next examined the effect of p52 in H1299 cells containing an IPTG-inducible p53 expression plasmid (H1299w/tp53 cells) (Rocha et al, 2003b). Depletion of p52/p100 did not significantly affect IPTG-inducible p53 levels in H1299 cells (Figure 4D). Importantly, loss of p52 also significantly compromised p53-induced expression of p21 and PUMA in these cells (Figure 4E and Supplementary Figure 1B and C). This result is in contrast to the repression of p21 expression seen in unstimulated U-2 OS cells but confirmed that p52 can be required for p53-inducible gene expression.
The p52 NF-κB subunit is associated with the promoters of p53-regulated genes
To determine whether p52 directly regulates the promoters of p53-regulated genes or whether more indirect mechanisms are involved, further ChIP assays were performed. Interestingly, p52 was found to be associated with the regions bound by p53 at all the p53-regulated promoters examined (Figure 5A). No binding of p53 or p52 was seen to control regions derived from the same promoters (Figure 5A). Furthermore, re-ChIP analysis demonstrated that p53 and p52 simultaneously associate with the promoter DNA (Figure 5B). These results indicated that in U-2 OS cells, p53 and p52 directly coregulate the expression of many genes.
Figure 5.
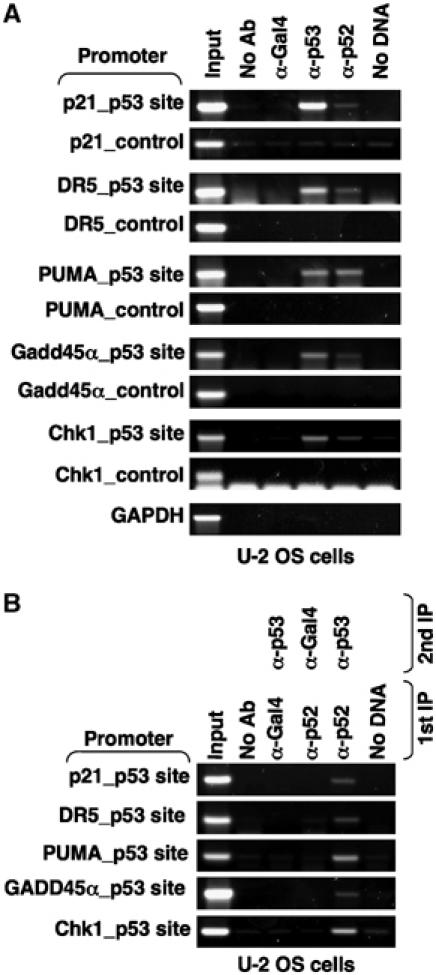
The p52 NF-κB subunit is recruited to p53 target gene promoters. (A) Association of p52 with p53 target gene promoters. ChIP analysis of the p21, DR5, PUMA, Gadd45α, Chk1 and GAPDH (control) promoters, using the indicated antibodies, was performed in U-2 OS cells as shown. To ensure sensitivity and specificity, each of the control primer sets in this and all later experiments were subjected to five more cycles of PCR than the primers associated with p53-binding sites. (B) p52 and p53 are simultaneously bound to the same promoter regions. Re-ChIP analysis of p53 target gene promoters was performed using the indicated antibodies.
p52 regulates cell proliferation and p53 target gene expression in many cell types
To learn more about the specificity of these effects, we next investigated the effect of depleting p52/100 in different cell lines. Interestingly, in nontransformed MCF10A breast epithelial cells (Soule et al, 1990) and human foreskin fibroblasts (HFFs), loss of p52/p100 also inhibited cell proliferation. By contrast, loss of p52/p100 in H1299 human non-small-cell lung carcinoma cells had a less pronounced effect whereas no significant decrease was seen in Saos2 osteosarcoma or MCF7 breast cancer cells (Figure 6).
Figure 6.
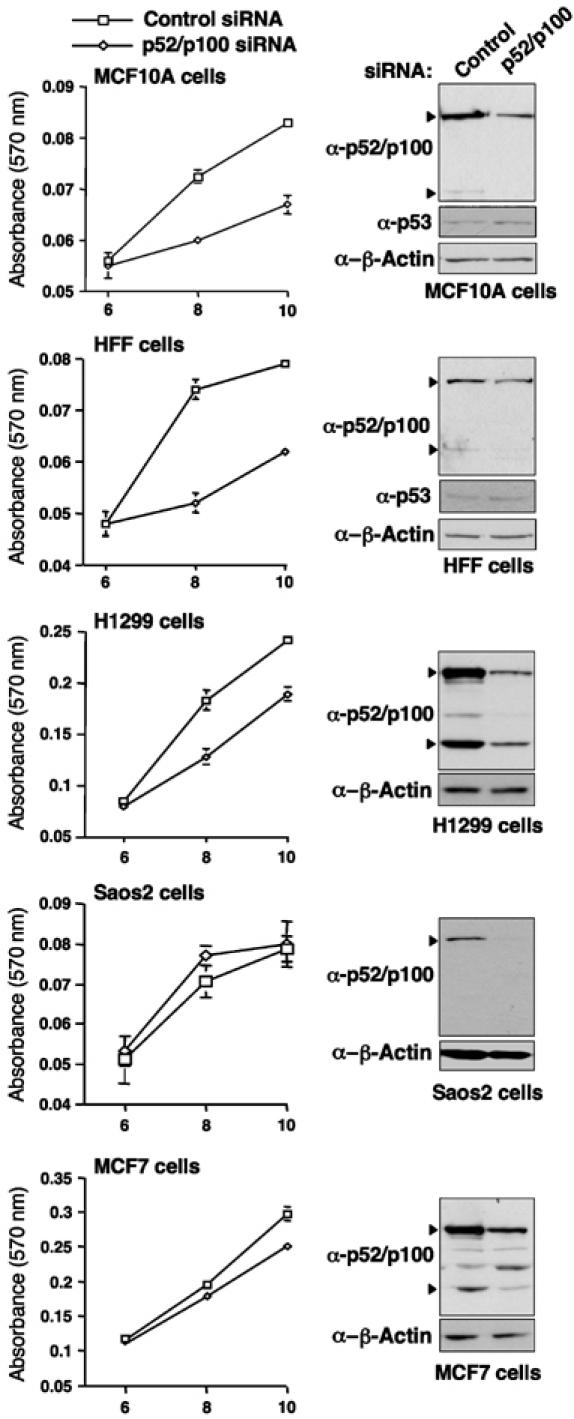
Cell type specificity of p52/p100 effects on cell proliferation. MCF10A, HFF, H1299, Saos2 and MCF7 cells were transfected with pooled p52/p100 siRNAs as indicated and alamar blue assays were performed. Times are from the day of the first siRNA transfection. Also shown are Western blot analyses showing the reduction of p52/p100 protein levels seen at the 6-day time point, after siRNA treatment in each cell line.
Gene expression analysis revealed that in both MCF10A and HFF cells, loss of p52 resulted in similar changes to those seen to U-2 OS cells: Cyclin D1, DR5 and PUMA expression decreased whereas p21 expression increased (Figure 7A). Moreover, ChIP analysis confirmed that p52, although present at lower levels in these cells, was bound to the Cyclin D1, DR5, PUMA and p21 promoters (Figure 7B and C). Similar to Figure 1D, no binding of p53 to the Cyclin D1 promoter was detected (data not shown). Confirming the specificity of these results, no binding of p52 to these p53-regulated promoters was seen in the p53-null Saos2 or H1299 cells (Figures 7D and 8D).
Figure 7.
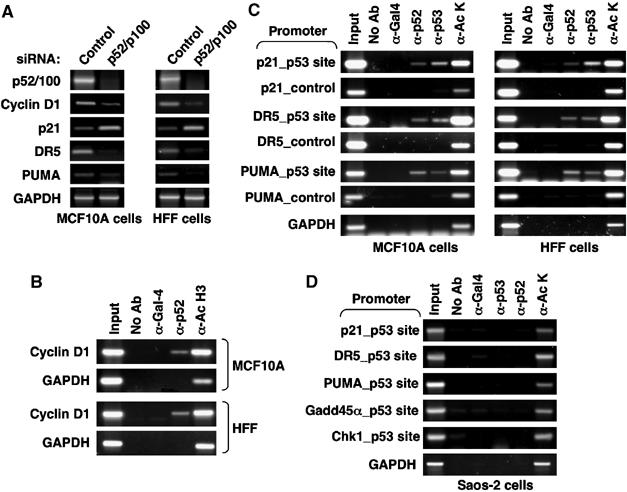
Regulation of Cyclin D1 and p53 target gene expression by p52 in MCF10A and HFF cells. (A) Regulation of Cyclin D1 and multiple p53 target genes in MCF10A and HFF cells by p52/p100. Cells were transfected with p52/p100 (pooled) or control siRNAs as shown and mRNA levels for the indicated genes were analysed as shown. (B, C) p52 promoter binding in MCF10A and HFF cells. ChIP analysis of the Cyclin D1 (B) or p21, DR5 and PUMA (C) promoters. Note, two-fold more material was required to produce a detectable signal compared to results shown in Figure 5. (D) Analysis of promoter binding in Saos2 cells. ChIP analysis was performed as in Figure 5A but using p53-null Saos2 cells.
Figure 8.
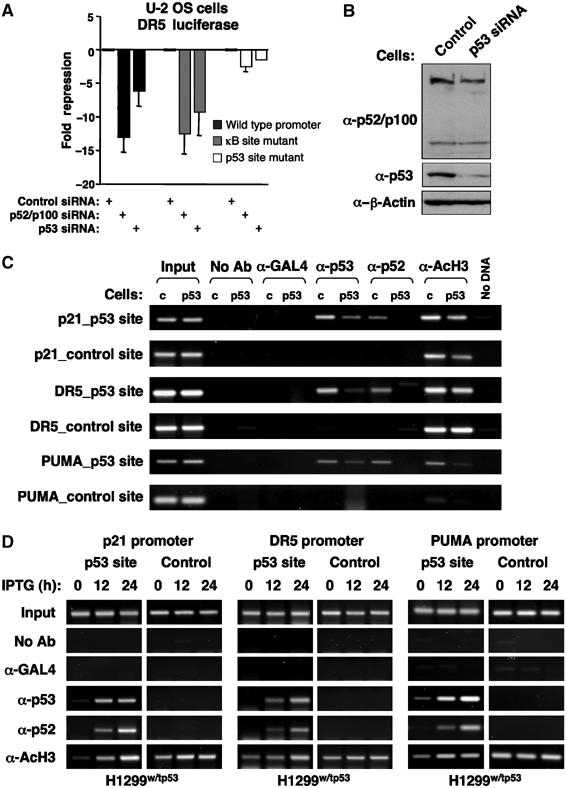
p53-dependent recruitment of p52 NF-κB to target gene promoters. (A) Regulation of the DR5 promoter by p52 is not dependent upon the κB site. U-2 OS cells were transfected with 1.5 μg of the full-length and κB element mutated DR5 promoter luciferase reporter plasmids together with 1.5 μg of p52/p100 or control siRNA expression plasmids. (B) Analysis of U-2 OS (p53 siRNA) cells. U-2 OS cells were stably transfected with either p53 or control siRNA expression plasmids. Cell lysates were prepared and analysed for p52/p100, p53 and β-actin (control) expression by Western blot. (C) Inhibition of p52 binding to the promoters of p53 target genes in p53 siRNA-transfected U-2 OS cells. ChIP analysis of the p21, DR5 and PUMA promoters, using the indicated antibodies, was performed in U-2 OS stably transfected with either p53 or control siRNA expression plasmids (indicated as p53 or c, respectively). (D) p53-dependent recruitment to p53 target genes in H1299w/tp53 cells. ChIP analysis of the p21, DR5 and PUMA promoters, using the indicated antibodies, was performed in H1299w/tp53 cells with and without IPTG treatment for the indicated times.
Binding of p52 to p53-regulated promoters is dependent upon p53
Although the ChIP data indicated co-occupancy of these promoters by p52 and p53, it was unclear whether this results from p53 recruitment of p52 or whether p52 binds independently. Of these genes, DR5 has previously been determined to also be an NF-κB target and to possess a κB site in its promoter (Shetty et al, 2005). We therefore analysed p52 regulation of the DR5 promoter in more detail. Consistent with expression of the endogenous gene, siRNA-mediated knockdown of both p52 and p53 inhibited the activity of a DR5 promoter luciferase reporter plasmid (Figure 8A). Significantly, only mutation of the p53 binding site and not the known κB site affected p52-dependent regulation. This suggested that recruitment of p52 to these promoters is p53 dependent. To investigate this hypothesis, a U-2 OS cell line stably expressing a p53 siRNA was created (Figure 8B). Knockdown of p53 did not affect p52/p100 levels and did not appear to significantly affect cell proliferation (Figure 8B and data not shown). ChIP analysis was then performed to determine whether loss of p53 would affect recruitment of p52 to target promoters. Significantly, depletion of p53 inhibited recruitment of p52 to the p21, DR5 and PUMA promoters (Figure 8C). To extend these results, ChIP assays were performed using the H1299w/tp53 cells. Using these cells, no binding of p52 to the promoters of p53-regulated genes is seen until p53 is induced by IPTG treatment, thus confirming that recruitment of p52 to these promoters is p53 dependent. In all cases, binding of p52 and p53 to these promoters was associated with an increase in histone H3 acetylation, suggesting cooperative recruitment of coactivators. Consistent with these effects, we find that p53 interacts with p52 but not with RelA or p50 in vitro (Supplementary Figure 2A). Moreover, this interaction requires the C-terminal regulatory domain of p53 (Supplementary Figure 2B). We also observed weak binding of overexpressed GFP-p52 to endogenous p53 (Supplementary Figure 2C). However, it should be noted that we consider it unlikely that the endogenous proteins will interact efficiently in solution. Indeed, only minimal colocalization of p53 and p52 is seen by immunofluorescence (data not shown) suggesting that any interaction in cells is likely to occur at the promoters of the genes they regulate.
Recruitment of p52 to p53-regulated promoters is independent of p52 DNA binding
Although the known κB site of the DR5 promoter was not required for p52 regulation (Figure 8A), it remained possible that p52 was binding other, uncharacterized or cryptic, κB elements in the promoters under study. To address this issue, a mutant, HA-tagged, version of p52 was created (p52-DBM), in which two arginines (R52 and R54), known to be critical for DNA binding (Cramer et al, 1997), were mutated to alanine (Figure 9A). In contrast to wild-type p52, p52-DBM does not bind the palindromic H2 κB element in vitro (Figure 9B) while ChIP analysis demonstrated that it does not bind the Cyclin D1 promoter in cells (Figure 9C). Significantly, ChIP analysis demonstrated that both wild-type p52 and p52-DBM were recruited, at equivalent levels, to the p21, DR5 and PUMA promoters (Figure 9D). Therefore, p53 recruitment of p52 to its target genes is independent of p52 DNA binding activity.
Figure 9.
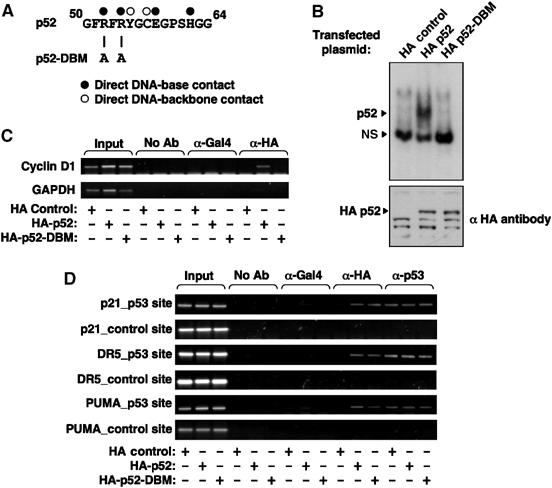
Recruitment of p52 to p53-regulated promoters is independent of p52 DNA binding. (A) Schematic diagram showing the R52A/R54A p52 DNA-binding mutation. Information shown about base contacts was obtained from (Cramer et al, 1997). (B) EMSA analysis of HA-tagged wild-type p52 and p52-DBM. Nuclear protein lysates prepared from U-2 OS cells transfected with either HA control plasmid, HA-tagged p52 or HA-tagged p52 DBM were analysed by EMSA using a 32P labeled H2 κB element probe (upper panel). Western blot analysis of nuclear protein extracts demonstrates equivalent expression of p52 and p52-DBM (lower panel). Nonspecific bands recognized by the HA antibody indicate equivalent loading. (C, D) The p52 DNA-binding mutant does not bind the Cyclin D1 promoter but is still recruited to p53 target genes. ChIP analysis of the Cyclin D1 (C) or p21, DR5 and PUMA (D) promoters in U-2 OS cells transfected with either HA control plasmid, HA-tagged p52 or HA-tagged p52 DBM. The antibodies used are indicated.
p52 regulates coactivator and corepressor recruitment to p53 target genes
To further investigate the role of p52 at these promoters, a U-2 OS cell line stably expressing a p52 siRNA was also created (Figure 10A). Owing to the effects of p52 loss in U-2 OS cells described above, these cells proliferate poorly and have a limited survival time in culture. Nonetheless, knockdown of p52 did not affect p53 levels and it was possible to perform ChIP analysis in these cells. Significantly, loss of p52 did not affect p53 binding to the p21, DR5 and PUMA promoters. Together with the results of Figures 8 and 9, this demonstrates that p53 recruits p52 to the promoters of these genes and that cooperative DNA binding is not occurring. Significantly, loss of p52 had opposing effects on coactivator and corepressor recruitment to these promoters: at the p21 promoter loss of HDAC1 recruitment was observed with no effect on p300 binding whereas at the DR5 and PUMA promoters, a strong increase in HDAC1 binding and loss of p300 recruitment was seen (Figure 10B and Supplementary Figure 3). These effects are consistent with the effects of p52 loss on the expression of these genes described above. p52 therefore regulates coactivator and corepressor recruitment to p53 target genes in U-2 OS cells.
Figure 10.
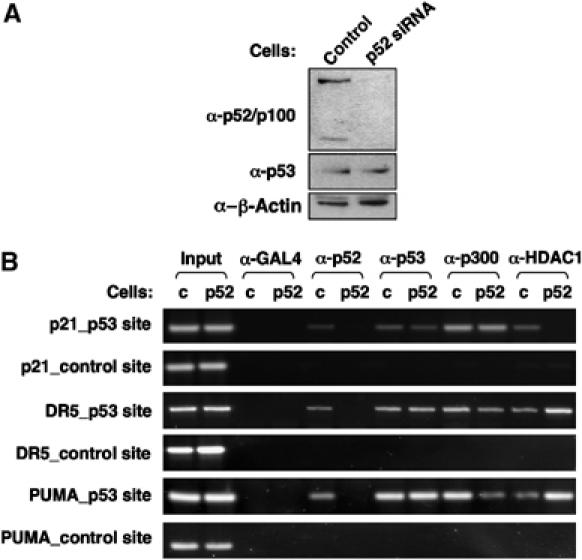
p52-dependent recruitment of HDAC1 and p300 to p53 target gene promoters. (A) Analysis of U-2 OS (p52 siRNA) cells. U-2 OS cells were stably transfected with either p52 or control siRNA expression plasmids. Cell lysates were prepared and analysed for p52/p100, p53 and β-actin (control) expression by Western blot. (B) Analysis of p53, HDAC1 and p300 recruitment to the promoters of p53 target genes in p52 siRNA-transfected U-2 OS cells. ChIP analysis of the p21, DR5 and PUMA promoters, using the indicated antibodies, was performed in U-2 OS stably transfected with either p52 or control siRNA expression plasmids (indicated as p52 or c, respectively).
Discussion
In this report, we reveal a complex regulatory network, dependent upon the p52 NF-κB subunit, that controls cell proliferation, Cyclin D1 expression and p53 tumour suppressor function. These results have important implications for the role of p52 in tumorigenesis and cancer therapy. Furthermore, a direct role for p52 as a regulator of p53 and its target genes has not been defined previously. Indeed, although there is a growing body of literature linking the NF-κB family and its regulators to the p53 pathway, these reports do not demonstrate the direct coregulation of p53 target genes and p53-dependent recruitment of NF-κB subunits to the promoters of these genes that we establish here (Webster and Perkins, 1999; Ryan et al, 2000; Gu et al, 2002; Tergaonkar et al, 2002; Culmsee et al, 2003; Rocha et al, 2003b; Aleyasin et al, 2004; Komarova et al, 2005; Perkins and Gilmore, 2006). Possible exceptions to this have been reported with the DR5 and COX-2 promoters, where binding of RelA has been shown to be p53-dependent (Shetty et al, 2005; Benoit et al, 2006). However, regulation of DR5 by RelA was κB site dependent and here we find recruitment of p52 to the p21, DR5 and PUMA promoters is independent of p52 DNA binding (Figure 9). Taken together, our data support a model of κB site and DNA-binding independent recruitment of p52 to the p53-regulated promoters we have studied (Figure 11). Interestingly, a similar model of κB site-independent recruitment of a RelA/HDAC4 complex to the Kruppel-like factor 2 promoter by MEF2 has recently been proposed (Kumar et al, 2005), suggesting that this might be a more common mechanism through which NF-κB subunits regulate transcription than previously realized.
Figure 11.
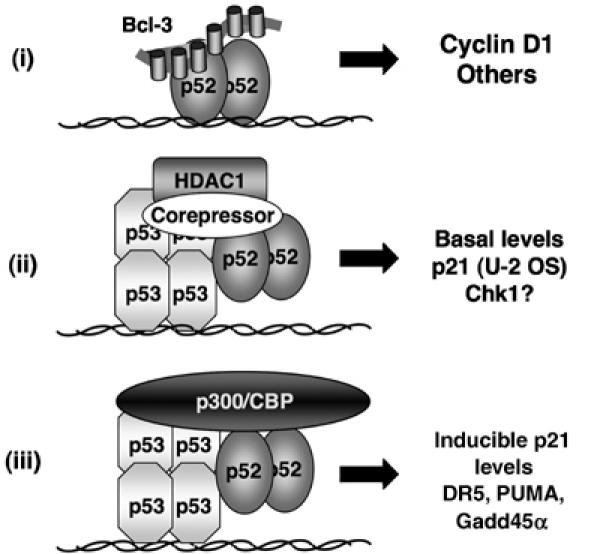
Model depicting different classes of p52-regulated target genes in U-2 OS cells. Some genes, such as Cyclin D1, represent direct targets of p52 and its coactivator Bcl-3. By contrast, p52 can be recruited by p53 to the promoters of its target genes independently of its ability to bind DNA or κB elements. At some promoters, p52 is required for the recruitment of HDAC1, resulting in repression of transcription whereas at other promoters, it contributes towards p300/CBP coactivator recruitment and stimulation of transcription.
These effects appear specific to p52 (Figure 3C, Supplementary Figure 2A), although we have not definitively demonstrated that similar effects cannot occur with the other NF-κB subunits. Interestingly, subunit-specific interactions involving the RHD are possible and have been predicted for p52 (Cramer et al, 1997). In particular, the ‘insert region', which lies between the two subdomains of the RHD, is strikingly different in p52 (Cramer et al, 1997) and represents a candidate motif that might mediate specific and DNA-binding independent interactions with p53 and other proteins.
p52 can appear antagonistic to p53, by repressing p21 expression in unstimulated U-2 OS cells, while cooperating with p53 at other promoters and following DNA damage. This behaviour is reminiscent of the Tip60 and p400 coactivator proteins, which have antagonistic roles regulating p21 expression in unstressed cells but cooperate to induce apoptosis following UV-induced DNA damage (Tyteca et al, 2006). p52 is therefore a modulator of p53 function, rather than strictly a coactivator or corepressor suggesting that in some contexts it will have a role in p53-regulated decision-making and the selectivity of p53-dependent gene expression. As we have previously demonstrated that induction of p53 can result in the replacement of p52/Bcl-3 complexes with p52/HDAC1 complexes and the subsequent repression of the Cyclin D1 promoter (Rocha et al, 2003b), there is clearly two-way feedback between these pathways. However, in our experiments, p53 does not appear to be recruited directly to the NF-κB responsive Cyclin D1 promoter (Figure 1D and data not shown), indicating that whereas p53 may influence p52 function, it is unlikely to be in the same direct manner we see with the p21, DR5 and PUMA genes.
Although we see p52-dependent effects on proliferation and p53-target gene expression in multiple cell lines, including nontransformed MCF10A and HFF cells, crosstalk between p52 and p53 is probably more restricted in vivo because cell culture conditions result in higher basal levels of NF-κB and p53 activity. In a living organism, there will be limited circumstances, in the absence of infection and genotoxic damage, when both transcription factors will be active in the same cells. Indeed, nfkb2−/− mice do not demonstrate any overt growth defects or cancer phenotypes, although this may reflect functional redundancy between p52 and other NF-κB subunits or heterologous proteins. Such redundancy is clearly demonstrated with regulation of Cyclin D1, which although p52 dependent in many of the cell types we analysed, can be regulated by other NF-κB subunits and heterologous transcription factors in other contexts (Guttridge et al, 1999; Romieu-Mourez et al, 2003, Albanese et al, 2003, Park et al, 2005). Importantly, numerous other lines of evidence do point to a widespread role for p52 as regulator of cell proliferation and tumorigenesis in vivo. These include the proliferative defects exhibited by mice with a C-terminal deletion of the nfkb2 gene, the aberrant activation of p52 seen in breast and skin cancers and the translocations of the NFKB2 gene observed in human T and B-cell lymphomas (Fracchiolla et al, 1993; Thakur et al, 1994; Chang et al, 1995; Ishikawa et al, 1997; Budunova et al, 1999; Cogswell et al, 2000). Whether these oncogenic effects of p52 involve the inhibition of specific p53 functions or whether they first require the inactivation of p53 will require further investigation.
Despite this complexity, the role of p52 as a regulator of cell proliferation implies that in some tumour types, inhibitors of IKKα activity, which would prevent processing of p100 to p52, might possess useful anticancer properties. In addition, the likely efficacy of such inhibitors is enhanced by the NF-κB-independent effects of IKKα on cell proliferation, such as regulation of oestrogen receptor function in breast cancer cells (Park et al, 2005), phosphorylation of the SMRT corepressor (Hoberg et al, 2004) and NF-κB-independent regulation of Cyclin D1 expression (Albanese et al, 2003). However, our discovery that p52 can be both a critical regulator of the cell cycle and p53 function suggests that, under some circumstances, such inhibitors might stimulate tumorigenesis. Therefore, further investigation of the p52/p100 pathway could lead to the development of useful diagnostic and prognostic indicators of tumour cell growth and malignancy.
Materials and methods
Cell growth, antibodies, plasmids, oligonucleotides and ChIP assays
Information on cell growth conditions, antibodies, plasmids, oligonucleotides, flow cytometric analysis of cell cycle distribution, caspase 3 assays and ChIP assays can be found in Supplementary data.
Other assays
Transient transfections, luciferase assays, alamar blue assays, GST pull-down assays, electrophoretic mobility shift assays (EMSAs), siRNA knockdown, RNA extraction, semiquantitative reverse transcription–polymerase chain reaction, protein extracts and Western blots were all performed as described previously (Rocha et al, 2003a; Campbell et al, 2004; Roche et al, 2004). All transfections were performed a minimum of three times before calculating means and standard deviations as shown in figures and contained appropriate levels of control plasmid such that each dish received the same amount of DNA.
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Online Information
Acknowledgments
We thank Rosie Clarke for help with FACs analysis, Adel Ibrahim and Mark Peggie from the SLS cloning service for plasmid construction, Michelle Garrett for supplying MCF10A cells and Toshiyuki Sakai, David Gillespie, Spencer Gibson and David Lane for supplying plasmids. We also thank all the members of the NDP laboratory and the Division of Gene Regulation and Expression at the University of Dundee for their help. We thank Kirsteen Campbell and David Meek for reading this manuscript and their helpful comments. KS is funded by a Wellcome Trust 4-year PhD studentship and SR received funding from a Cancer Research UK programme grant (C1443/A5435) and an EPSRC career development fellowship.
References
- Albanese C, Wu K, D'Amico M, Jarrett C, Joyce D, Hughes J, Hulit J, Sakamaki T, Fu M, Ben-Ze'ev A, Bromberg JF, Lamberti C, Verma U, Gaynor RB, Byers SW, Pestell RG (2003) IKKα regulates mitogenic signaling through transcriptional induction of cyclin D1 via Tcf. Mol Biol Cell 14: 585–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleyasin H, Cregan SP, Iyirhiaro G, O'Hare MJ, Callaghan SM, Slack RS, Park DS (2004) Nuclear factor-κB modulates the p53 response in neurons exposed to DNA damage. J Neurosci 24: 2963–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- An W, Kim J, Roeder RG (2004) Ordered cooperative functions of PRMT1, p300, and CARM1 in transcriptional activation by p53. Cell 117: 735–748 [DOI] [PubMed] [Google Scholar]
- Bates GJ, Nicol SM, Wilson BJ, Jacobs AM, Bourdon JC, Wardrop J, Gregory DJ, Lane DP, Perkins ND, Fuller-Pace FV (2005) The DEAD box protein p68: a novel transcriptional coactivator of the p53 tumour suppressor. EMBO J 24: 543–553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benoit V, de Moraes E, Dar NA, Taranchon E, Bours V, Hautefeuille A, Taniere P, Chariot A, Scoazec JY, de Moura Gallo CV, Merville MP, Hainaut P (2006) Transcriptional activation of cyclooxygenase-2 by tumor suppressor p53 requires nuclear factor-κB. Oncogene, May 8 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- Bienvenu F, Barre B, Giraud S, Avril S, Coqueret O (2005) Transcriptional regulation by a DNA-associated form of cyclin D1. Mol Biol Cell 16: 1850–1858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzi G, Bebien M, Otero DC, Johnson-Vroom KE, Cao Y, Vu D, Jegga AG, Aronow BJ, Ghosh G, Rickert RC, Karin M (2004) Activation of IKKα target genes depends on recognition of specific κB binding sites by RelB:p52 dimers. EMBO J 23: 4202–4210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonizzi G, Karin M (2004) The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol 25: 280–288 [DOI] [PubMed] [Google Scholar]
- Bours V, Franzoso G, Azarenko V, Park S, Kanno T, Brown K, Siebenlist U (1993) The oncoprotein Bcl-3 directly transactivates through κB motifs via association with DNA-binding p50B homodimers. Cell 72: 729–739 [DOI] [PubMed] [Google Scholar]
- Budunova IV, Perez P, Vaden VR, Spiegelman VS, Slaga TJ, Jorcano JL (1999) Increased expression of p50-NF-κB and constitutive activation of NF-κB transcription factors during mouse skin carcinogenesis. Oncogene 18: 7423–7431 [DOI] [PubMed] [Google Scholar]
- Campbell KJ, Rocha S, Perkins ND (2004) Active repression of antiapoptotic gene expression by ReIA(p65) NF-κB. Mol Cell 13: 853–865 [DOI] [PubMed] [Google Scholar]
- Chang CC, Zhang JD, Lombardi L, Neri A, Dallafavera R (1995) Rearranged NFKB-2 genes in lymphoid neoplasms code for constitutively active nuclear transactivators. Mol Cell Biol 15: 5180–5187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciana P, Neri A, Cappellini C, Cavallo F, Pomati M, Chang CC, Maiolo AT, Lombardi L (1997) Constitutive expression of lymphoma-associated NFKB-2/Lyt-10 proteins is tumorigenic in murine fibroblasts. Oncogene 14: 1805–1810 [DOI] [PubMed] [Google Scholar]
- Cogswell PC, Guttridge DC, Funkhouser WK, Baldwin AS (2000) Selective activation of NF-κB subunits in human breast cancer: potential roles for NF-κB2/p52 and for Bcl-3. Oncogene 19: 1123–1131 [DOI] [PubMed] [Google Scholar]
- Cramer P, Larson CJ, Verdine GL, Muller CW (1997) Structure of the human NF-κB p52 homodimer-DNA complex at 2.1 angstrom resolution. EMBO J 16: 7078–7090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee C, Siewe J, Junker V, Retiounskaia M, Schwarz S, Camandola S, El-Metainy S, Behnke H, Mattson MP, Krieglstein J (2003) Reciprocal inhibition of p53 and nuclear factor-κB transcriptional activities determines cell survival or death in neurons. J Neurosci 23: 8586–8595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckett CS, Perkins ND, Kowalik TF, Schmid RM, Huang ES, Baldwin AS Jr, Nabel GJ (1993) Dimerization of NF-KB2 with RelA(p65) regulates DNA binding, transcriptional activation, and inhibition by an IκB-α (MAD-3). Mol Cell Biol 13: 1315–1322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eliopoulos AG, Caamano JH, Flavell J, Reynolds GM, Murray PG, Poyet JL, Young LS (2003) Epstein–Barr virus-encoded latent infection membrane protein 1 regulates the processing of p100 NF-κB2 to p52 via an IKK γ/NEMO-independent signalling pathway. Oncogene 22: 7557–7569 [DOI] [PubMed] [Google Scholar]
- Espinosa JM, Verdun RE, Emerson BM (2003) p53 functions through stress- and promoter-specific recruitment of transcription initiation components before and after DNA damage. Mol Cell 12: 1015–1027 [DOI] [PubMed] [Google Scholar]
- Fracchiolla NS, Lombardi L, Salina M, Migliazza A, Baldini L, Berti E, Cro L, Polli E, Maiolo AT, Neri A (1993) Structural alterations of the NF-κB transcription factor Lyt-10 in lymphoid malignancies. Oncogene 8: 2839–2845 [PubMed] [Google Scholar]
- Franzoso G, Carlson L, Xing LP, Poljak L, Shores EW, Brown KD, Leonardi A, Tran T, Boyce BF, Siebenlist U (1997) Requirement for NF-κB in osteoclast and B-cell development. Genes Dev 11: 3482–3496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gartel AL, Tyner AL (1999) Transcriptional regulation of the p21((WAF1/CIP1)) gene. Exp Cell Res 246: 280–289 [DOI] [PubMed] [Google Scholar]
- Gerondakis S, Grossmann M, Nakamura Y, Pohl T, Grumont R (1999) Genetic approaches in mice to understand Rel/NF-κB and IκB function: transgenics and knockouts. Oncogene 18: 6888–6895 [DOI] [PubMed] [Google Scholar]
- Greten FR, Karin M (2004) The IKK/NF-κB activation pathway—a target for prevention and treatment of cancer. Cancer Lett 206: 193–199 [DOI] [PubMed] [Google Scholar]
- Gu LB, Findley HW, Zhou MX (2002) MDM2 induces NF-κB/p65 expression transcriptionally through Sp1-binding sites: a novel, p53-independent role of MDM2 in doxorubicin resistance in acute lymphoblastic leukemia. Blood 99: 3367–3375 [DOI] [PubMed] [Google Scholar]
- Guttridge DC, Albanese C, Reuther JY, Pestell RG, Baldwin AS (1999) NF-κB controls cell growth and differentiation through transcriptional regulation of cyclin D1. Mol Cell Biol 19: 5785–5799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden MS, Ghosh S (2004) Signaling to NF-κB. Genes Dev 18: 2195–2224 [DOI] [PubMed] [Google Scholar]
- Hoberg JE, Yeung F, Mayo MW (2004) SMRT derepression by the IκB kinase α: a prerequisite to NF-κB transcription and survival. Mol Cell 16: 245–255 [DOI] [PubMed] [Google Scholar]
- Hoffmann A, Leung TH, Baltimore D (2003) Genetic analysis of NF-κB/Rel transcription factors defines functional specificities. EMBO J 22: 5530–5539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iotsova V, Caamano J, Loy J, Yang Y, Lewin A, Bravo R (1997) Osteopetrosis in mice lacking NF-κB1 and NF-κB2. Nat Med 3: 1285–1289 [DOI] [PubMed] [Google Scholar]
- Ishikawa H, Carrasco D, Claudio E, Ryseck RP, Bravo R (1997) Gastric hyperplasia and increased proliferative responses of lymphocytes in mice lacking the COOH-terminal ankyrin domain of NF-κB2. J Exp Med 186: 999–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanno T, Franzoso G, Siebenlist U (1994) Human T-Cell Leukemia-Virus Type-I Tax-Protein-Mediated Activation of NF-κB From p100 (NF-κB2)-Inhibited Cytoplasmic Reservoirs. Proc Natl Acad Sci USA 91: 12634–12638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashatus D, Cogswell P, Baldwin AS (2006) Expression of the Bcl-3 proto-oncogene suppresses p53 activation. Genes Dev 20: 225–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kho PS, Wang Z, Zhuang L, Li Y, Chew JL, Ng HH, Liu ET, Yu Q (2004) p53-regulated transcriptional program associated with genotoxic stress-induced apoptosis. J Biol Chem 279: 21183–21192 [DOI] [PubMed] [Google Scholar]
- Komarova EA, Krivokrysenko V, Wang K, Neznanov N, Chernov MV, Komarov PG, Brennan ML, Golovkina TV, Rokhlin OW, Kuprash DV, Nedospasov SA, Hazen SL, Feinstein E, Gudkov AV (2005) p53 is a suppressor of inflammatory response in mice. FASEB J 19: 1030–1032 [DOI] [PubMed] [Google Scholar]
- Kumar A, Lin Z, SenBanerjee S, Jain MK (2005) Tumor necrosis factor α-mediated reduction of KLF2 is due to inhibition of MEF2 by NF-κB and histone deacetylases. Mol Cell Biol 25: 5893–5903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanoix J, Lacoste J, Pepin N, Rice N, Hiscott J (1994) Overproduction of NFκB2(Lyt-10) and c-Rel—a mechanism for HTLV-I Tax-mediated transactivation via the NF-κB signaling pathway. Oncogene 9: 841–852 [PubMed] [Google Scholar]
- Lo JC, Basak S, James ES, Quiambo RS, Kinsella MC, Alegre ML, Weih F, Franzoso G, Hoffmann A, Fu YX (2006) Coordination between NF-κB family members p50 and p52 is essential for mediating LTβR signals in the development and organization of secondary lymphoid tissues. Blood 107: 1048–1055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinari B, Costanzo A, Marzano V, Piccolella E, Tuosto L (2004) CD28 delivers a unique signal leading to the selective recruitment of RelA and p52 NF-κB subunits on IL-8 and Bcl-xL gene promoters. Proc Natl Acad Sci USA 101: 6098–6103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massoumi R, Chmielarska K, Hennecke K, Pfeifer A, Fassler R (2006) Cyld inhibits tumor cell proliferation by blocking bcl-3-dependent NF-κB signaling. Cell 125: 665–677 [DOI] [PubMed] [Google Scholar]
- Nakano K, Vousden KH (2001) PUMA, a novel proapoptotic gene, is induced by p53. Mol Cell 7: 683–694 [DOI] [PubMed] [Google Scholar]
- Park KJ, Krishnan V, O'Malley BW, Yamamoto Y, Gaynor RB (2005) Formation of an IKKα-dependent transcription complex is required for estrogen receptor-mediated gene activation. Mol Cell 18: 71–82 [DOI] [PubMed] [Google Scholar]
- Perkins ND, Gilmore TD (2006) Good cop, bad cop: the different faces of NF-κB. Cell Death Differ 13: 759–772 [DOI] [PubMed] [Google Scholar]
- Rocha S, Campbell KJ, Perkins ND (2003a) p53- and Mdm2-independent repression of NF-κB transactivation by the ARF tumor suppressor. Mol Cell 12: 15–25 [DOI] [PubMed] [Google Scholar]
- Rocha S, Martin AM, Meek DW, Perkins ND (2003b) p53 Represses cyclin D1 transcription through down regulation of Bcl-3 and inducing increased association of the p52 NF-κB subunit with histone deacetylase 1. Mol Cell Biol 23: 4713–4727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roche KC, Wiechens N, Owen-Hughes T, Perkins ND (2004) The FHA domain protein SNIP1 is a regulator of the cell cycle and cyclin D1 expression. Oncogene 23: 8185–8195 [DOI] [PubMed] [Google Scholar]
- Romieu-Mourez R, Kim DW, Shin SM, Demicco EG, Landesman-Bollag E, Seldin DC, Cardiff RD, Sonenshein GE (2003) Mouse mammary tumor virus c-rel transgenic mice develop mammary tumors. Mol Cell Biol 23: 5738–5754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan KM, Ernst MK, Rice NR, Vousden KH (2000) Role of NF-κB in p53-mediated programmed cell death. Nature 404: 892–897 [DOI] [PubMed] [Google Scholar]
- Shetty S, Graham BA, Brown JG, Hu X, Vegh-Yarema N, Harding G, Paul JT, Gibson SB (2005) Transcription factor NF-κB differentially regulates death receptor 5 expression involving histone deacetylase 1. Mol Cell Biol 25: 5404–5416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soule HD, Maloney TM, Wolman SR, Peterson WD Jr, Brenz R, McGrath CM, Russo J, Pauley RJ, Jones RF, Brooks SC (1990) Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res 50:6075–6086 [PubMed] [Google Scholar]
- Takimoto R, El-Deiry WS (2000) Wild-type p53 transactivates the KILLER/DR5 gene through an intronic sequence-specific DNA-binding site. Oncogene 19: 1735–1743 [DOI] [PubMed] [Google Scholar]
- Tergaonkar V, Pando M, Vafa O, Wahl G, Verma I (2002) p53 stabilization is decreased upon NFκB activation: a role for NFκB in acquisition of resistance to chemotherapy. Cancer Cell 1: 493–503 [DOI] [PubMed] [Google Scholar]
- Thakur S, Lin H, Tseng W, Kumar S, Bravo R, Foss F, Gelinas C, Rabson AB (1994) Rearrangement and altered expression of the NFKB-2 gene in human cutaneous T-lymphoma cells. Oncogene 9: 2335–2344 [PubMed] [Google Scholar]
- Tyteca S, Vandromme M, Legube G, Chevillard-Briet M, Trouche D (2006) Tip60 and p400 are both required for UV-induced apoptosis but play antagonistic roles in cell cycle progression. EMBO J 25: 1680–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viatour P, Bentires-Alj M, Chariot A, Deregowski V, de Leval L, Merville MP, Bours V (2003) NF-κB2/p100 induces Bcl-2 expression. Leukemia 17: 1349–1356 [DOI] [PubMed] [Google Scholar]
- Viatour P, Dejardin E, Warnier M, Lair F, Claudio E, Bureau F, Marine JC, Merville MP, Maurer U, Green D, Piette J, Siebenlist U, Bours V, Chariot A (2004) GSK3-mediated BCL-3 phosphorylation modulates its degradation and its oncogenicity. Mol Cell 16: 35–45 [DOI] [PubMed] [Google Scholar]
- Webster GA, Perkins ND (1999) Transcriptional cross talk between NF-κB and p53. Mol Cell Biol 19: 3485–3495 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weih F, Caamano J (2003) Regulation of secondary lymphoid organ development by the nuclear factor-κB signal transduction pathway. Immunological Rev 195: 91–105 [DOI] [PubMed] [Google Scholar]
- Westerheide SD, Mayo MW, Anest V, Hanson JL, Baldwin AS (2001) The putative oncoprotein Bcl-3 induces cyclin D1 to stimulate G(1) transition. Mol Cell Biol 21: 8428–8436 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Online Information