

Abstract
In response to harmful stresses, cells induce programmed cell death (PCD) or apoptosis. Seizures can induce neural damage and activate biochemical pathways associated with PCD. Since seizures trigger intracellular calcium overload, it has been presumed that the intrinsic cell death pathway mediated by mitochondrial dysfunction would modulate cell death following seizures. However, previous work suggests that the extrinsic cell death pathway may initiate the damage program. Here we investigate intrinsic vs. extrinsic cell death pathway activation using caspase cleavage as a marker for activation of these pathways in a rat in vitro model of seizures. Hippocampal cells, chronically treated with kynurenic acid, had kynurenic acid withdrawn to induce seizure-like activity for 40 minutes. Subjecting rat hippocampal cultures to seizures increased cell death and apoptosis-like DNA fragmentation using TUNEL staining. Seizure-induced cell death was blocked by both MK801 (10 μM) and CNQX (40 μM), which suggests multiple glutamate receptors regulate seizure-induced cell death. Cleavage of the initiator caspases, caspase 8 and 12 were increased 4 hours following seizure, and cleavage of the quintessential executioner caspase, caspase 3 was increased 4 hours following seizure. In contrast, caspase 9 cleavage only increased 24 hours following seizure. Using an affinity labeling approach to trap activated caspases in situ, we show that caspase 8 is the apical caspase activated following seizures. Finally, we show that the caspase 8 inhibitor Ac-IETD-CHO was more effective at blocking seizure-induced cell death than the caspase 9 inhibitor Ac-LEHD-CHO. Taken together, our data suggests the extrinsic cell death pathway-associated caspase 8 is activated following seizures in vitro.
Keywords: Epilepsy, kynurenic acid, NMDA, apoptosis, brain
1. Introduction
Epilepsy is a common, chronic neurological disorder that affects approximately 1% of the population. Status epilepticus (SE), is a clinical emergency associated with profound neuronal damage in brain and subsequent cognitive dysfunction, and is also a significant risk factor for developing epilepsy (Coulter and DeLorenzo 1999, Hermann, et al. 2002, Hesdorffer and Verity 1997).
Initiation of programmed/active cell death has been broadly separated into processes involving the extrinsic pathways, such as those mediated by death receptors of the tumor necrosis factor (TNF) family (TNFR1, Fas, DR3), and the intrinsic pathways, which are initiated by disturbances to intracellular organelle homeostasis particularly at the mitochondrial membrane (Ashkenazi and Dixit 1998, Liou, et al. 2003, Reed 1998). Both pathways are regulated and executed by the Bcl-2 family of pro- and anti-cell death genes, and the caspase family of cell death proteases, along with several ancillary molecules such as the apoptotic protease-activating factor 1 (APAF-1), and death receptor adaptor proteins such as FADD (Fas associated protein with death domain) (Eldadah and Faden 2000, Liou, et al. 2003, Yuan and Yankner 2000).
Activation of intrinsic, mitochondria-dependent cell death pathways after seizures would be predicted based on the assumed significance of glutamate excitotoxicity and mitochondrial dysfunction due to calcium (Ca2+) loading (Griffiths, et al. 1984, Sztriha, et al. 1985, Wasterlain, et al. 1993). Indeed, cell death in several other “excitotoxic” neurological diseases (e.g. stroke), involves prominent, mitochondria-originated death pathways (Cao, et al. 2001, Yan, et al. 2000, Yin, et al. 2002). However, relatively weak, late or even absent roles for mitochondrial pathways have been described after seizures in degenerating neuronal populations (Fujikawa, et al. 2002, Puig and Ferrer 2002). Neuroprotection is also less pronounced when mitochondrially-activated caspase-9 is blocked after seizures (Henshall, et al. 2001a, Narkilahti, et al. 2003).
A series of studies by our group suggests the extrinsic/death receptor pathway may initiate seizure-induced neuronal death. Seizures activate extrinsic pathway caspases 2 & 8 before mitochondrial dysfunction is detected (Henshall, et al. 2001b, Henshall, et al. 2000a, Henshall, et al. 2001c). Caspase 8 activation involves recruitment to the TNFR1 signaling complex, which includes death domain adaptor proteins TRADD (TNF receptor-associated death domain protein) and FADD and the apoptosis signal-regulating kinase 1 (ASK1)(Shinoda, et al. 2003). Finally, neuroprotection in vivo is most robust when the extrinsic pathways are disrupted (Henshall, et al. 2003, Henshall, et al. 2001b, Henshall, et al. 2001c, Shinoda, et al. 2003).
We recently described an in vitro model to investigate programmed cell death pathway activation following kynurenic acid withdrawal- induced seizures in hippocampal neurons (Meller, et al. 2003), but a comparison of the intrinsic vs. the extrinsic cell death pathway was not undertaken. In this study, we have refined this model, by shortening the seizure duration from continuous seizure to a 40 min seizure and our data suggest that activation of the caspase 8 pathway predominates over caspase 9 and as such may offer a more effective neuroprotective target for epilepsy.
2. Materials and methods
2.1 Hippocampal culture
Hippocampal neuronal cultures were prepared from 1-2 day old Sprague Dawley rat pups. Briefly, rat pups were anaesthetized using isoflurane, decapitated and hippocampi were dissected and dissociated with papain (Worthington Biochemicals, Lakewood, NJ) and plated out in Neurobasal-A/B27 media (Invitrogen, Carlsbad, CA) supplemented with 10 mM kynurenic acid and 5 mM MgCl2; this leads to chronic neurotransmission blockade (Furshpan 1989, Furshpan and Potter 1989). After 5 days, 1 μM cytosine β-D-arabinofuranoside (Sigma, St Louis, MO) was added to the media. Cells were used after 3-4 weeks in culture and media replenished with fresh kynurenic acid/ Mg2+ every 2-3 days. Seizure-like activity was induced by incubating the cells in Neurobasal-A/ B27 media without kynurenic acid or magnesium for 40 min. Some cells were treated with the caspase inhibitors Ac IETD-CHO or Ac LEHD-CHO (Biomol, PA) during the 40 min seizure period and for 24 hours following seizure.
2.2. Electrophysiological recordings
For electrophysiology experiments, cells were plated out on poly-D-lysine coated glass cover slips at a density of 400,000 cell/coverslip. Patch-clamp recordings were performed as described previously (Xiong, et al. 1997). Patch electrodes were constructed from thin-walled borosilicate glass (1.5 mm diameter, WPI, Sarasota, FL) on a two-stage puller (PP83, Narishige, Tokyo, Japan). The tips of the electrodes were heat polished on a Narishige microforge (Scientific Instruments Laboratory, Tokyo, Japan, Model MF-83) to a final diameter of 1-2 μm. The patch electrodes had resistance between 3 to 5 MΩ when filled with the intracellular solution (140 mM KF, 2.0 mM MgCl2, 1.0 mM CaCl2, 10 mM Hepes, 11 mM EGTA, 4 mM MgATP, pH 7.3, using NaOH (the final Na+ concentration is about 10 mM), 300 mOsm). Membrane potentials were recorded in current-clamp mode using Axopatch 1-D amplifiers (Axon Instruments, Foster City, CA). Data were filtered at 2 kHz and digitized on-line using Digidata 1320A DAC units (Axon Instruments). The on-line acquisition was done using pClamp software (versin 8.0, Axon Instruments).
Cells were maintained in an extracellular solution(140 mM NaCl, 5.4 mM KCl, 25 mM Hepes, 33 mM Glucose, 1.3 mM CaCl2, 1.0 mM MgCl2, 10 mM kynurenic acid, pH 7.4 using NaOH; 320 - 335 mOsm). All electrophysiological experiments were done at room temperature (22-24 °C). Once cells were patched, a multi-barrel perfusion system (SF-77B, Warner Instrument Co.) was employed to achieve a rapid exchange of solutions containing kynurenic / Mg2+ to a kynurenic acid/ Mg2+ free solution.
2.3. Cell death assay
For cell death assays cells were grown in 24 well plates at a density of approximately 200,000 cell/well. Following withdrawal of kynurenic acid, lactate dehydrogenase (LDH) release from hippocampal cells was determined using a cytotoxicity detection kit (Roche, CA). Briefly, 100 μl of media was reacted with 100 μl reagent, incubated at room temp for 30 min and read on a spectrophotometer at 405 nm and 692 nm.
2.4. DNA fragmentation analysis
DNA fragmentation was assessed using a fluorescein TUNEL kit according to manufacturers instructions (Roche, CA). Briefly, coverslips were incubated with a reaction mixture containing terminal deoxynucleotidyl transferase and fluorescein conjugated nucleotides for 1 hour at 37°C. Coverslips were washed and mounted with 4′, 6 diamidino-2-phenylindole (DAPI) to stain for nuclei, as previously described (Meller, et al. 2003).
2.5. Immuno blotting
Immunoblotting was performed as previously described (Meller, et al. 2005). Tissue samples were lysed in a non-denaturing buffer containing protease inhibitors (100 μg/ml phenylmethylsulfonylfluoride, 1 μg/ml aprotinin, 1 μg/ml leupeptin,1 μg/ml pepstatin, 50 mM NaF, 2 mM Na3VO4 and phosphatase cocktail inhibitor (Sigma, St Louis, MO)). Protein concentration was determined by Bradford reagent spectrophotometrically at A595. Protein samples (50 μg) were denatured in a gel-loading buffer at 100 °C for 5 min and then loaded onto 12% SDS-polyacrylamide gels to detect full length and cleaved caspases. Proteins were transferred to polyvinylodene difluoride membranes and incubated with primary antibodies at 4 °C overnight; caspase 2, caspase 3, cleaved-caspase 3, caspase 6, caspase 7, caspase 9, cleaved-caspase 9, caspase 12 (Cell Signaling, Beverley, MA); cleaved-caspase 8 (Santa Cruz CA); BAP-31 (a gift from Gordon Shore, McGill University, Canada). Membranes were incubated with anti-rabbit IgG conjugated to horseradish peroxidase (Cell Signaling Technology, Beverly, MA) followed by chemiluminesense detection (NEN Life Science Products, Boston, MA) and then exposed to film. Images were captured using a Dage 72 camera.
2.6. Sub cellular fractionation
Sub-cellular fractions were prepared as previously described (Schindler, et al. 2004). Briefly, samples were homogenized in 1 × M-SHE buffer (210 mM Mannitol, 70 mM Sucrose, 10 mM HEPES-KOH pH 7.4, 1 mM EDTA, 1 mM EGTA and protease inhibitor cocktail) and then centrifuged twice at 1200 × g for 10 min. The post-nuclear supernatant was then centrifuged twice at 10,000 × g for 15 min and the resulting mitochondrial pellet was resuspended in a sucrose buffer (395 mM Sucrose, 0.1 mM EGTA, 10 mM HEPES-KOH pH 7.4) and purified through a percoll bilayer in gradient buffer (1.28 M Sucrose, 0.4 mM EGTA, 40 mM HEPES-KOH, pH 7.4) by centrifugation at 41,000 × g for 30 min. The crude cytosolic fraction was then centrifuged at 100,000 × g for 1 hour to separate the microsomal and cytosolic fractions.
2.7. In situ caspase trapping assay
Cells were plated out on 10 cm dishes at 5 million cell/dish. Cells were loaded with biotin-zVAD-fmk for 1 hour prior to 40 min seizure. Cells were harvested 4 hours following seizures and 500 μg total protein was incubated with 1 μg streptavidin-agarose beads (Vector Labs, CA) for 1 hour at 4 °C. Samples were washed using an immunoprecipitation kit (IP50, Sigma). The proteins were denatured at 95°C for 10 min and then loaded onto 12 % SDS-polyacrylamide gels. Blots were subject to immunoblotting as above.
2.8. Data Analysis
Data are presented as mean ± standard error of the mean (S.E.M.) of n determinations. Data were analyzed using one way analysis of variance (ANOVA) with appropriate post-hoc tests (Graphpad Prism). Significance was accepted at p<0.05.
3. Results
3.1. Withdrawal of chronic glutamate receptor blockade induces seizure-like activity leading to a time-dependent increase in cell death
Seizures were elicited in vitro by washout of kynurenic acid, which had been incubated with the cells to cause chronic glutamate receptor blockade (Furshpan and Potter 1989). Using patch clamp techniques, seizure-like activity was observed in hippocampal cultures following removal of kynurenic acid in accordance with our previous study (Meller, et al. 2003) and others (Furshpan 1989, Furshpan 1991, Furshpan and Potter 1989). Following the removal of kynurenic acid we observed an increase in firing and a depolarization of the cell (Fig. 1a), which we previously attributed to activation of AMPA and NMDA receptors, respectively (Meller, et al. 2003).
Figure 1.
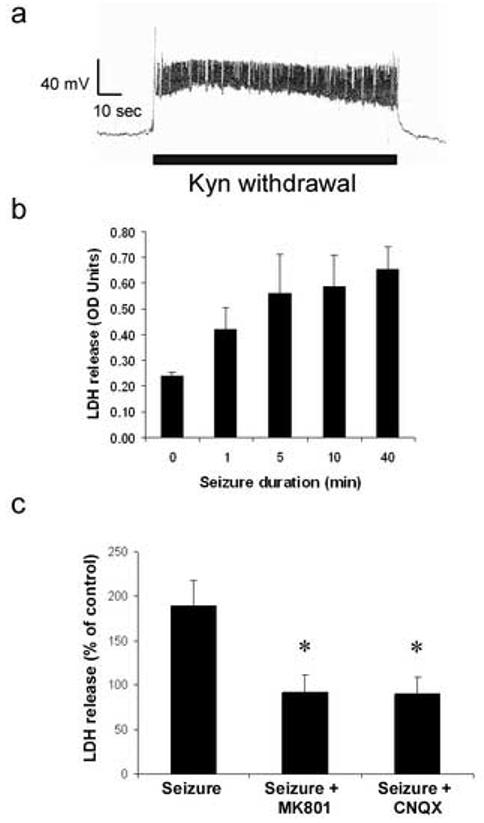
Kynurenic acid withdrawal-induced seizure elicits time-dependent cell death. a) Withdrawal of kynurenic acid from cultures chronically treated with kynurenic acid and Mg2+ results in seizure-like activity. b) Increasing seizure duration increases the amount of LDH released from neurons. Note seizure duration greater that 5 min results in maximal cell death. Media samples were taken 24 hours following seizure for LDH assay. c) Seizure induced cell death is reduced by the NMDA antagonist MK801 (10 μM) and the AMPA antagonist CNQX (20 μM). Cells were incubated with drug during 40 min seizure period. Data shown are mean ± sem (n= 5), * denotes P<0.01 vs. effect of seizure (post hoc Dunnet's test).
In our previous study we subjected cells to a permanent withdrawal of kynurenic acid (Meller, et al. 2003). In order to match our in vivo seizure models we investigated the effect of changing the seizure to a physiologically plausible duration. In preliminary experiments, we discovered that shortening the withdrawal period to 40 min also induced a similar increase in LDH release from cells, when measured 24 hours later (Fig. 1b). Using this 40 min seizure duration we investigated the temporal profile of LDH release from hippocampal cells following seizure. A near maximal increase in LDH release was observed within 8 hours of the seizure, suggesting that mechanisms that regulate cell death are activated within this time frame (<8 hours) (not shown).
To investigate the relative contribution of ionotropic glutamate receptors to this injury cells were incubated with MK801 (NMDA antagonist) and CNQX (AMPA antagonist) and injury following seizure was evaluated. MK801 (10 μM) blocked cell death 24 hours following 40 min seizures (Fig. 1c). CNQX (20 μM) also blocked cell death following 40 min seizures. (Fig 1c). Taken together these data suggest that withdrawal of kynurenic acid for 40 min induces cell death, which is mediated by activation of both NMDA and AMPA/ kainate glutamate receptors.
3.2. Increased DNA damage following seizures
We used a TUNEL assay to detect DNA strand cleavage following seizure. In control (non-seizure-treated cells) TUNEL staining was not evident. We observed an increase in TUNEL positive cells following seizures (70 %), and many of these cells exhibited morphologies typical of apoptosis, i.e. nuclear blebbing and condensation (see Fig. 2).
Figure 2.
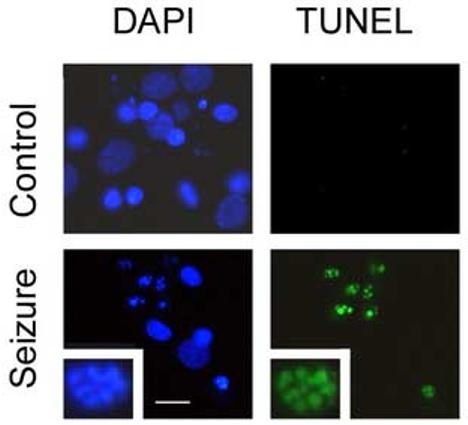
Increased DNA damage following seizure. a) Control or seizure-treated cells were recovered for 24 hours and DNA damage assessed by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL). Cells were counterstained with DAPI to show nuclear morphology. Note the clumped nuclear morphology in seizure treated cells, inset 40 × magnified view of nucleus of seizure treated cell. Scale bar = 20 μm.
3.3. Caspase 8 and Caspase 12 cleavage precedes Caspase 9 cleavage following seizures
In order to determine the mechanisms involved in seizure-induced cell death we investigated the cleavage (and activation) of caspase enzymes. In preliminary experiments using whole cell lysates we detected caspase 3, 8, 9 and 12 cleavage following seizures, but not cleavage of caspase 1, 2, 6, 7 and 10 (not shown). In order to understand more about these caspase's cellular location following activation, we performed immunoblotting on cell fractions purified by differential centrifugation, (Henshall, et al. 2004, Schindler, et al. 2004, Shinoda, et al. 2004b). Fig. 3a shows a panel verifying the quality of fractionation.
Figure 3.
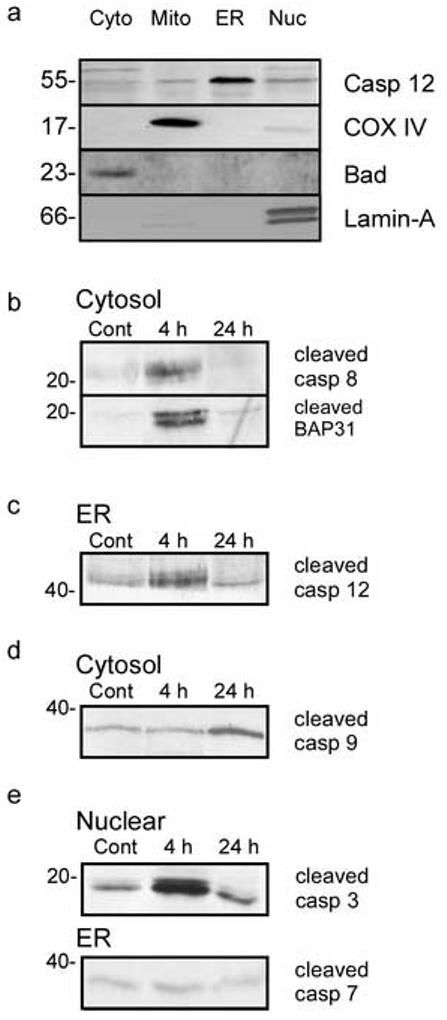
Caspase cleavage following seizures. a) Assessment of fraction purity. Fractions were blotted for caspase 12, cytochrome oxidase IV, Bad and lamin-A as specific markers for microsomal (ER), mitochondrial (mito) cytoplasmic (cyto) and nuclear (nuc) fractions. b) Increased cleavage of caspase 8 and its substrate BAP31 4 hours following seizure. Cytosol fractions were prepared and immunoblotted for cleaved caspase 8 and BAP31. c) Increased cleavage of caspase 12 following seizure. Microsomal fractions were prepared and immunoblotted for cleaved 12. d) Caspase 9 cleavage increase 24 hours following seizure. Cytosol fractions were prepared and immunoblotted for cleaved caspase 9. e) Increased nuclear caspase 3 cleavage following seizures. Nuclear fractions were prepared and immunoblotted for cleaved caspase 3. Caspase 7 cleavage in microsomal fractions did not increase following seizure. Images shown are representative blots of duplicate experiments which gave similar results. Numbers at the side of the blots represent molecular weight in kDa.
Caspase 8 is cleaved when the death receptor-mediated extrinsic cell death pathway is activated (Schneider and Tschopp 2000). There was an increase in cytoplasmic caspase 8 cleavage 4 hours following seizures (Fig. 3b). Interestingly levels of the 57 kDa parent band were also increased in the microsomal fraction at both 4 and 24 hours (not shown). Additionally, we examined the cleavage of a caspase 8 substrate, BAP31 following seizure (Breckenridge, et al. 2002, Ng and Shore 1998). In agreement with the cleavage of caspase 8, we observe cleavage of BAP31 to the 20 kDa fragment at 4 hours (Fig. 3b). This suggests that caspase 8 is active 4 hours following seizure.
Caspase 12 is an initiator caspase which is activated by endoplasmic reticulum stress and the death receptor scaffolding protein TRAF2 (Yoneda, et al. 2001). We observed an increase in a 42 kDa cleavage fragment of caspase 12 in the microsomal/ ER fraction at 4 hours post-seizure (Fig. 3c). We also observed a small increase in TRAF-2 levels in microsomal/ ER fractions 4 hours following seizure (not shown).
As a marker of the intrinsic cell death pathway, we investigated cytosolic caspase 9 cleavage. Caspase 9 levels and cleavage in the cytosol were increased at 24 hours, but not 4 hours following seizure (Fig. 3d). This suggests that on a temporal basis either caspase 8 or caspase 12 are more likely candidates for initiating caspase 3 cleavage and cell death pathway initiation following seizures, than caspase 9.
Consistent with our whole cell lysates data we observed an increase in nuclear caspase 3 cleavage at 4 hours, which was slightly reduced 24 hours following seizure (Fig. 3e). This suggests that the initiating pathway for cell death following seizures is activated within 4 hours. Finally, we did not observe an increase in caspase 7 cleavage following seizure (Fig. 3e), suggesting other executioner caspases are not involved in seizure induced cell death in vitro, which is consistent with our rat in vivo studies (Henshall, et al. 2002).
3.4. Affinity labeling of activated caspases following seizure
Having established evidence for caspase 8 cleavage and activation following seizure, we used biotin-zVAD-fmk to label activated caspases following seizures, as shown by others (Tu, et al. 2006). VAD is a “common substrate” for caspases (Sohn, et al. 2005). Cells were incubated with biotin-zVAD-fmk and subject to seizure and recovered for 4 hours. Standard immunoprecipitation techniques were used to separate activated caspases. Incubation of hippocampal cells with biotin-zVAD-fmk for 4 hours following seizure resulted in the precipitation of caspase 8 and to a lesser extent caspase 3 (Fig. 4). In contrast we did not observe pull down of caspase 2, 9 or 12 (Fig. 4). Taken together these data suggest an apical role of the extrinsic cell death pathway-associated caspase 8 following seizures, but does not rule out the later activation of other caspases.
Figure 4.
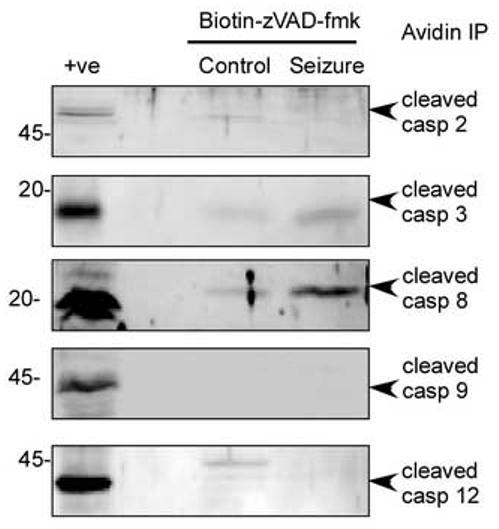
Immunoprecipitation of active caspases reveals caspase 8 is activated following seizure. Cells were incubated with biotin-zVAD-fmk (1 μM) and subject to seizures and 4 h recovery. Bound caspases were precipitated with avidin-agarose and immunoblotted for caspase 2, 3,8 9 and 12. As a positive control cortical brain lysate from 100 min MCAO treated rats was used (see (Meller, et al. 2005)). Note lack of cleaved caspase 2 9 and 12, and the increase in cleaved caspase 8, 4 hours following seizure. Caspase 3 was also cleaved following seizure. Images shown are representative blots from 3 separate experiments with identical results. Numbers at the side of the blots represent molecular weight in kDa.
3.5. Blockade of Caspase 8 but not Caspase 9 reduces seizure-induced cell death
Using peptide inhibitors of the caspase enzymes, we investigated the effect of caspase blockade of cell death following seizures. Blocking caspase 8 with Ac-IETD-CHO (10 μM) caused a significant decrease in seizure–induced cell death (Fig. 5). In contrast, blocking caspase 9 with Ac-LEHD-CHO (10 μM) caused a non-significant decrease in LDH release from hippocampal cultures following seizure (Fig. 5). Basal levels of LDH release were slightly reduced in cells treated with both Ac-IETD-CHO and Ac-LEHD-CHO, but this was not significant (Fig. 5).
Figure 5.
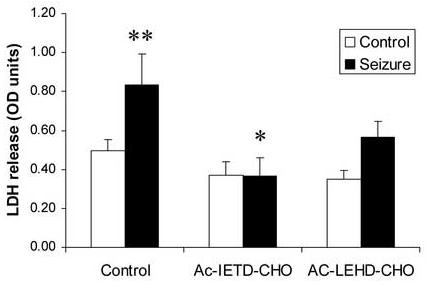
Blockade of caspase 8 reduces seizure-induced cell death. Cells were incubated with the caspase 8 inhibitor Ac-IETD-CHO or caspase 9 inhibitor Ac-LEHD-CHO (both 10 μM) for 10 min prior to seizure, and then for 24 hours following 40 min seizure. Cell death was assessed by LDH release assay. Data shown are mean ± sem (n= 4), * denotes P<0.01 vs. effect of seizure (Dunnet's post hoc test), ** denotes P<0.01 vs. non-seized control.
4. Discussion
Our studies confirm that removal of chronic glutamate receptor blockade from hippocampal cultures results in seizure-like activity that leads to time-dependent cell death mediated by NMDA and AMPA glutamate receptors. The seizure-like activity results in DNA damage and the appearance of cells with morphology consistent with that observed in programmed cell death.
The seizures activate caspase enzymes, with caspase 3, 8 and 12 showing activation prior to caspase 9. Finally, blocking caspase 8 rather than caspase 9 was more effective at preventing cell death following seizures. Taken together our data suggest that following seizures, extrinsic cell death pathway activation occurs prior to intrinsic cell death pathway activation, leading to DNA damage and cell death.
Our in vitro model of seizure-induced injury is based on the model of Furshpan and Potter (Furshpan 1989, Furshpan 1991, Furshpan and Potter 1989). Our study suggests that hippocampal cells are quite sensitive to seizure with as little as 5 min worth of seizure causing an increase in cell death (Fig. 1a). Further since we observe a delay between seizure cessation and cell death, this suggests that cells die by active cell death mechanisms.
Cell death following seizures was blocked by both MK801 and CNQX, which suggest that seizure-induced cell death is mediated by NMDA and AMPA receptors respectively, in agreement with studies on spontaneous seizures in vitro (McBain, et al. 1988). Interestingly, when kynurenic acid was permanently withdrawn, cell death was not blocked by CNQX (Meller, et al. 2003). MK801 and CNQX both block the increase in caspase 3 cleavage observed in whole cell lysates following seizures (not shown), which suggests that following withdrawal of kynurenic acid, activation of multiple glutamate receptors leads to seizures in the culture, which result in caspase activation and cell death. As a caveat it is worth noting that while CNQX has been shown to block NMDA as well as kainate receptors (Lester, et al. 1989, Long, et al. 1990, McBain, et al. 1988); the electrophysiological profile of the hippocampal neurons undergoing seizures in the presence of CNQX would suggest a predominant effect of blocking fast-AMPA/kainate receptors (Meller, et al. 2003). We did not study the role of the metabotropic glutamate receptors, however this area is worthy of further investigation given the sometimes-contradictory role of different metabotropic glutamate receptors subtypes in protection and neuronal cell death, for example (Maiese, et al. 2000, Poli, et al. 2003).
On the basis of our experiments, we suggest that following seizures the extrinsic cell death pathway may play an important role in initiating seizure-induced cell damage. Specifically, we observe increased caspase 8 cleavage prior to caspase 9 cleavage, and that caspase 8 inhibitors were more effective than caspase 9 inhibitor at blocking cell death following seizures (Figs 2, 3 and 4). While counter to the dogma suggesting that glutamate excitotoxicity results in mitochondrial dysfunction and intrinsic cell death pathway activation, our data are consistent with our previous in vitro and in vivo studies. In our previous in vitro study, we did not show any protection by blocking Bad-mediated cell death with FK506, and suggested a redundant role for this mechanism (Meller, et al. 2003); We interpret this with the caveat that the kynurenic acid withdrawal was permanent. In our previous in vivo studies we have consistently observed activation of the extrinsic cell death pathway caspases prior to intrinsic cell death caspases (Henshall, et al. 2001a, Henshall, et al. 2001b). Specifically we observed activation and cleavage of caspase 8 immediately following 40 min seizure, where as caspase 9 cleavage largely emerged 4 hours following seizure. While the timing may be slightly different between our in vivo and in vitro models, we still observe a primacy of caspase 8 activation.
While Ac-IETD-CHO and Ac-LEHD-CHO are deemed inhibitors of caspase 8 and 9, respectively (www.biomol.com), it has recently been shown that these substrates are not as specific as perhaps first considered (Sohn, et al. 2005). To further confirm our data we used affinity labeling, which showed activation of caspase 8 occurs prior to activation of caspase 9. Hence taken together, our data are consistent with the view that activation of caspase 8, and perhaps the extrinsic pathway, occurs soon after seizures.
In light of our present observations we are left with an intriguing question; what is the initiating factor following seizures which activates caspase 8 and yet leaves caspase 9 inactive? Typically caspases are cleaved in a cascade, hence an upstream initiator caspase would be a likely candidate. While the initiator caspase 2 is rapidly cleaved following seizures in vivo, the selective caspase 2 inhibitor VDVAD is an inefficient inhibitor of seizure-induced hippocampal damage (Henshall, et al. 2001c). In our model we did not observe caspase 2 cleavage, nor was caspase 2 pulled down in our affinity assay, hence making it unlikely to be the apical caspase in the cell death cascade. Another candidate caspase would be caspase 12 since we observe it to be cleaved rapidly following seizures (Fig. 3). However, caspase 8 is not a known caspase 12 substrate. Furthermore, caspase 12 was not pulled down in our affinity assay, which suggests it is not a primary caspase in the cascade following seizures. The therapeutic potential of caspase 12 may also be limited due to the premature termination of the expression of the protein due to the inclusion of a stop codon in its sequence, except in a subgroup of humans (Fischer, et al. 2002). However the intriguing link between calcium influx following seizure and the fact that both caspase 8 and caspase 12 were cleaved in the endoplasmic reticulum (ER) (which is a calcium store), make the role of caspase 12 and the ER in regulating seizure-induced cell damage worthy of further investigation.
The calpain family of proteases may regulate caspase activation as well as mediating excitotoxic damage to neurons (Blomgren, et al. 2001, Higuchi, et al. 2005, Lankiewicz, et al. 2000, Nakagawa and Yuan 2000, Takano, et al. 2005). Indeed raised intracellular calcium levels observed following seizures could activate calpains, which could process and activate caspases. Caspase 12 is a calpain substrate, but this yields a cleavage fragment of 35 kDa (Nakagawa and Yuan 2000), rather than the 42 kDa fragment we observed following seizure (Fig. 3). Calpain-mediated cleavage of caspase 9 yields fragments of 35, 25, and 15 kDa (Chua, et al. 2000) whereas we observe a 37-39 kDa fragment. As such given the weight of the cleaved caspase product and the lack of additional bands, our results are more consistent of a role for either APAF-1-mediated or caspase −3 mediated late cleavage of caspase 9 (Fujita, et al. 2001), than a mechanism involving calpains. Calpains can also cleave and inactivate caspase-7 (Chua, et al. 2000), and we did not observe caspase-7 cleavage in our model, despite detecting its expression (Fig. 3e). Taken together, our data suggests that broad calpain activation probably does not account for the present observations. However, given the role of calcium in seizure-induced cell death, that calpains are activated upstream of caspases following other apoptosis inducing stimuli (Waterhouse, et al. 1998) and that calpains can cleave caspase 8 (Chua, et al. 2000), further study of calpains in our system is under current investigation.
An alternative explanation would be to suggest that death receptor-mediated activation of caspase 8 initiates seizure-induced cell death. While not yet investigated in our model, this would fit with our in vivo evidence that describes the assembly of the death receptor signaling scaffold complex following seizures (Shinoda, et al. 2003). Furthermore it would allow cleavage and activation of caspase 8 without prior activation of other caspases, as observed in our data. We have shown that seizure-induced cell death was blocked by a TNFα (although not a Fas) neutralizing antibody (Shinoda, et al. 2003), however the role of TNF in seizure-induced cell death is currently complicated by the fact that TNFR1 knockout mice show greater vulnerability to neurotoxic insults than wildtype controls (Gary, et al. 1998), implying a pleiotropic effect of TNF. Whether the TNF receptor is activated following seizures in our model is currently under investigation.
Our studies have focused on understanding the mechanisms by which a period of harmful seizures leads to neuronal damage, hence the utility of this model is to assist with our studies to investigate salient features of seizure-induced brain damage. In vivo models of epilepsy are complex, involving whole animals, multiple brain circuits and necessitate detailed endpoint quantification of seizure-induced damage to the brain. As such the simplicity of the in vitro system to specifically test hypotheses prior to such involved in vivo studies is appealing. While the withdrawal of kynurenic acid is an artificial paradigm by which to induce seizures (although by this caveat many experimental models of seizure must also be considered artificial), it must however be pointed that our approach to studying seizure-induced cell damage typically utilizes in vitro, in vivo and clinical samples for example (Shinoda, et al. 2004b), rather than relying on one model alone.
One potential area where our model may have advantages over other in vitro models is in the study of injurious vs. non-injurious seizures. It has been shown that not all seizures result in cell death and in our animal models only seizures characterized as (even brief) high amplitude poly-spike transients result in damage to the hippocampus, where as prolonged periods of low amplitude epileptiform trains do not, even though the behavioral correlates of these two seizure types are indistinguishable (Shinoda, et al. 2004a). In our in vitro model, seizure-like electrical activity induced by kynurenic acid withdrawal is maintained in the presence of MK801, but the cell death-promoting feature of our seizure-like activity is not (Meller, et al. 2003). As such this may represent a correlate to the in vivo model and is currently being investigated.
In summary our data support caspase 8 as the critical modulator of seizure-induced cell death in vitro. Brief or repetitive seizures, and SE, cause significant neuronal loss by a programmed/active cell death mechanism (Bengzon, et al. 1997, Henshall, et al. 2000b, Henshall, et al. 2002, Kotloski, et al. 2002, Narkilahti, et al. 2003, Zhang, et al. 1998). Inhibition of the cell death pathways using pharmacological or molecular approaches confers substantial (≥50%) protection against seizure-induced neuronal death in both in vivo and in vitro models (Culmsee, et al. 2001, Henshall, et al. 2001b, Roy, et al. 2002, Viswanath, et al. 2000), although optimal protection may require both anticonvulsant and neuroprotective treatment. Clearly, further studies are required to determine effective therapeutic regimes to reduce activation of these pathways following seizure.
Acknowledgement
We wish to thank Gordon Shore (McGill University, Canada) for the kind gift of the BAP31 antibody.
Footnotes
Grant information
Funded by NIH grants NS41935 (DCH), NS39016 (RPS and RM) and Science Foundation Ireland Grant B466 (DCH).
References
- Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 1998;281:1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- Bengzon J, Kokaia Z, Elmer E, Nanobashvili A, Kokaia M, Lindvall O. Apoptosis and proliferation of dentate gyrus neurons after single and intermittent limbic seizures. Proc. Natl. Acad. Sci. U. S. A. 1997;94:10432–10437. doi: 10.1073/pnas.94.19.10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomgren K, Zhu C, Wang X, Karlsson JO, Leverin AL, Bahr BA, Mallard C, Hagberg H. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia-ischemia: a mechanism of “pathological apoptosis”? J. Biol. Chem. 2001;276:10191–10198. doi: 10.1074/jbc.M007807200. [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Nguyen M, Kuppig S, Reth M, Shore GC. The procaspase-8 isoform, procaspase-8L, recruited to the BAP31 complex at the endoplasmic reticulum. Proc. Natl. Acad. Sci. U. S. A. 2002;99:4331–4336. doi: 10.1073/pnas.072088099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao G, Minami M, Pei W, Yan C, Chen D, O'Horo C, Graham SH, Chen J. Intracellular bax translocation after transient cerebral ischemia: implications for a role of the mitochondrial apoptotic signaling pathway in ischemic neuronal death. J. Cereb. Blood Flow Metab. 2001;21:321–333. doi: 10.1097/00004647-200104000-00001. [DOI] [PubMed] [Google Scholar]
- Chua BT, Guo K, Li P. Direct cleavage by the calcium-activated protease calpain can lead to inactivation of caspases. J. Biol. Chem. 2000;275:5131–5135. doi: 10.1074/jbc.275.7.5131. [DOI] [PubMed] [Google Scholar]
- Coulter DA, DeLorenzo RJ. Basic mechanisms of status epilepticus. Adv. Neurol. 1999;79:725–733. [PubMed] [Google Scholar]
- Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, Greig NH, Mattson MP. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid beta-peptide. J. Neurochem. 2001;77:220–228. doi: 10.1046/j.1471-4159.2001.t01-1-00220.x. [DOI] [PubMed] [Google Scholar]
- Eldadah BA, Faden AI. Caspase pathways, neuronal apoptosis, and CNS injury. J. Neurotrauma. 2000;17:811–829. doi: 10.1089/neu.2000.17.811. [DOI] [PubMed] [Google Scholar]
- Fischer H, Koenig U, Eckhart L, Tschachler E. Human caspase 12 has acquired deleterious mutations. Biochem. Biophys. Res. Commun. 2002;293:722–726. doi: 10.1016/S0006-291X(02)00289-9. [DOI] [PubMed] [Google Scholar]
- Fujikawa DG, Ke X, Trinidad RB, Shinmei SS, Wu A. Caspase-3 is not activated in seizure-induced neuronal necrosis with internucleosomal DNA cleavage. J. Neurochem. 2002;83:229–240. doi: 10.1046/j.1471-4159.2002.01152.x. [DOI] [PubMed] [Google Scholar]
- Fujita E, Egashira J, Urase K, Kuida K, Momoi T. Caspase-9 processing by caspase-3 via a feedback amplification loop in vivo. Cell Death Differ. 2001;8:335–344. doi: 10.1038/sj.cdd.4400824. [DOI] [PubMed] [Google Scholar]
- Furshpan EJ. A cell-culture approach to the study of seizure activity. Q. J. Exp. Physiol. 1989;74:975–985. doi: 10.1113/expphysiol.1989.sp003373. [DOI] [PubMed] [Google Scholar]
- Furshpan EJ. Seizure-like activity in cell culture. Epilepsy Res. 1991;10:24–32. doi: 10.1016/0920-1211(91)90091-s. [DOI] [PubMed] [Google Scholar]
- Furshpan EJ, Potter DD. Seizure-like activity and cellular damage in rat hippocampal neurons in cell culture. Neuron. 1989;3:199–207. doi: 10.1016/0896-6273(89)90033-0. [DOI] [PubMed] [Google Scholar]
- Gary DS, Bruce-Keller AJ, Kindy MS, Mattson MP. Ischemic and excitotoxic brain injury is enhanced in mice lacking the p55 tumor necrosis factor receptor. J. Cereb. Blood Flow Metab. 1998;18:1283–1287. doi: 10.1097/00004647-199812000-00001. [DOI] [PubMed] [Google Scholar]
- Griffiths T, Evans MC, Meldrum BS. Status epilepticus: the reversibility of calcium loading and acute neuronal pathological changes in the rat hippocampus. Neuroscience. 1984;12:557–567. doi: 10.1016/0306-4522(84)90073-3. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Araki T, Schindler CK, Shinoda S, Lan JQ, Simon RP. Expression of death-associated protein kinase and recruitment to the tumor necrosis factor signaling pathway following brief seizures. J. Neurochem. 2003;86:1260–1270. doi: 10.1046/j.1471-4159.2003.01934.x. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Bonislawski DP, Skradski SL, Araki T, Lan J-Q, Schindler CK, Meller R, Simon RP. Formation of the Apaf-1/cytochrome c complex precedes activation of caspase-9 during seizure-induced neuronal death. Cell Death Differ. 2001a;8:1169–1181. doi: 10.1038/sj.cdd.4400921. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Bonislawski DP, Skradski SL, Meller R, Lan J-Q, Simon RP. Cleavage of Bid may amplify caspase-8-induced neuronal death following focally evoked limbic seizures. Neurobiol. Dis. 2001b;8:568–580. doi: 10.1006/nbdi.2001.0415. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Chen J, Simon RP. Involvement of caspase-3-like protease in the mechanism of cell death following focally evoked limbic seizures. J. Neurochem. 2000a;74:1215–1223. doi: 10.1046/j.1471-4159.2000.741215.x. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Schindler CK, So NK, Lan JQ, Meller R, Simon RP. Death-associated protein kinase expression in human temporal lobe epilepsy. Ann. Neurol. 2004;55:485–494. doi: 10.1002/ana.20001. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Sinclair J, Simon RP. Spatio-temporal profile of DNA fragmentation and its relationship to patterns of epileptiform activity following focally evoked limbic seizures. Brain Res. 2000b;858:290–302. doi: 10.1016/s0006-8993(99)02452-x. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Skradski SL, Bonislawski DP, Lan JQ, Simon RP. Caspase-2 activation is redundant during seizure-induced neuronal death. J. Neurochem. 2001c;77:886–895. doi: 10.1046/j.1471-4159.2001.00291.x. [DOI] [PubMed] [Google Scholar]
- Henshall DC, Skradski SL, Meller R, Araki T, Minami M, Schindler CK, Lan JQ, Bonislawski DP, Simon RP. Expression and differential processing of caspases 6 and 7 in relation to specific epileptiform EEG patterns following limbic seizures. Neurobiol. Dis. 2002;10:71–87. doi: 10.1006/nbdi.2002.0505. [DOI] [PubMed] [Google Scholar]
- Hermann BP, Seidenberg M, Bell B. The neurodevelopmental impact of childhood onset temporal lobe epilepsy on brain structure and function and the risk of progressive cognitive effects. Prog. Brain Res. 2002;135:429–438. doi: 10.1016/S0079-6123(02)35040-4. [DOI] [PubMed] [Google Scholar]
- Hesdorffer DC, Verity CM. Risk factors. In: Engel JJ, Pedley TA, editors. Epilepsy: A comprehensive textbook. Lippincott-Raven; Philadelphia: 1997. pp. 59–67. [Google Scholar]
- Higuchi M, Tomioka M, Takano J, Shirotani K, Iwata N, Masumoto H, Maki M, Itohara S, Saido TC. Distinct mechanistic roles of calpain and caspase activation in neurodegeneration as revealed in mice overexpressing their specific inhibitors. J. Biol. Chem. 2005;280:15229–15237. doi: 10.1074/jbc.M500939200. [DOI] [PubMed] [Google Scholar]
- Kotloski R, Lynch M, Lauersdorf S, Sutula T. Repeated brief seizures induce progressive hippocampal neuron loss and memory deficits. Prog. Brain Res. 2002;135:95–110. doi: 10.1016/S0079-6123(02)35010-6. [DOI] [PubMed] [Google Scholar]
- Lankiewicz S, Marc Luetjens C, Truc Bui N, Krohn AJ, Poppe M, Cole GM, Saido TC, Prehn JH. Activation of calpain I converts excitotoxic neuron death into a caspase-independent cell death. J. Biol. Chem. 2000;275:17064–17071. doi: 10.1074/jbc.275.22.17064. [DOI] [PubMed] [Google Scholar]
- Lester RA, Quarum ML, Parker JD, Weber E, Jahr CE. Interaction of 6-cyano-7-nitroquinoxaline-2,3-dione with the N-methyl-D-aspartate receptor-associated glycine binding site. Mol. Pharmacol. 1989;35:565–570. [PubMed] [Google Scholar]
- Liou AKF, Clark RS, Henshall DC, Yin XM, Chen J. To die or not to die for neurons in ischemia, traumatic brain injury and epilepsy: a review on the stress-activated signaling pathways and apoptotic pathways. Prog. Neurobiol. 2003;69:103–142. doi: 10.1016/s0301-0082(03)00005-4. [DOI] [PubMed] [Google Scholar]
- Long SK, Smith DA, Siarey RJ, Evans RH. Effect of 6-cyano-2,3-dihydroxy-7-nitro-quinoxaline (CNQX) on dorsal root-, NMDA-, kainate- and quisqualate-mediated depolarization of rat motoneurones in vitro. Br. J. Pharmacol. 1990;100:850–854. doi: 10.1111/j.1476-5381.1990.tb14103.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiese K, Vincent A, Lin SH, Shaw T. Group I and group III metabotropic glutamate receptor subtypes provide enhanced neuroprotection. J. Neurosci. Res. 2000;62:257–272. doi: 10.1002/1097-4547(20001015)62:2<257::AID-JNR10>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- McBain CJ, Boden P, Hill RG. The kainate/quisqualate receptor antagonist, CNQX, blocks the fast component of spontaneous epileptiform activity in organotypic cultures of rat hippocampus. Neurosci. Lett. 1988;93:341–345. doi: 10.1016/0304-3940(88)90106-1. [DOI] [PubMed] [Google Scholar]
- Meller R, Minami M, Cameron JA, Impey S, Chen D, Lan JQ, Henshall DC, Simon RP. CREB-mediated Bcl-2 protein expression after ischemic preconditioning. J. Cereb. Blood Flow Metab. 2005;25:234–246. doi: 10.1038/sj.jcbfm.9600024. [DOI] [PubMed] [Google Scholar]
- Meller R, Schindler CK, Chu XP, Xiong ZG, Cameron JA, Simon RP, Henshall DC. Seizure-like activity leads to the release of BAD from 14-3-3 protein and cell death in hippocampal neurons in vitro. Cell Death Differ. 2003;10:539–547. doi: 10.1038/sj.cdd.4401206. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Yuan J. Cross-talk between two cysteine protease families. Activation of caspase-12 by calpain in apoptosis. J. Cell Biol. 2000;150:887–894. doi: 10.1083/jcb.150.4.887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narkilahti S, Nissinen J, Pitkanen A. Administration of caspase 3 inhibitor during and after status epilepticus in rat: effect on neuronal damage and epileptogenesis. Neuropharmacology. 2003;44:1068–1088. doi: 10.1016/s0028-3908(03)00115-1. [DOI] [PubMed] [Google Scholar]
- Ng FW, Shore GC. Bcl-XL cooperatively associates with the Bap31 complex in the endoplasmic reticulum, dependent on procaspase-8 and Ced-4 adaptor. J. Biol. Chem. 1998;273:3140–3143. doi: 10.1074/jbc.273.6.3140. [DOI] [PubMed] [Google Scholar]
- Poli A, Beraudi A, Villani L, Storto M, Battaglia G, Di Giorgi Gerevini V, Cappuccio I, Caricasole A, D'Onofrio M, Nicoletti F. Group II metabotropic glutamate receptors regulate the vulnerability to hypoxic brain damage. J. Neurosci. 2003;23:6023–6029. doi: 10.1523/JNEUROSCI.23-14-06023.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puig B, Ferrer I. Caspase-3-associated apoptotic cell death in excitotoxic necrosis of the entorhinal cortex following intraperitoneal injection of kainic acid in the rat. Neurosci. Lett. 2002;321:182–186. doi: 10.1016/s0304-3940(01)02518-6. [DOI] [PubMed] [Google Scholar]
- Reed JC. Bcl-2 family proteins. Oncogene. 1998;17:3225–3236. doi: 10.1038/sj.onc.1202591. [DOI] [PubMed] [Google Scholar]
- Roy M, Hom JJ, Sapolsky RM. HSV-mediated delivery of virally derived anti-apoptotic genes protects the rat hippocampus from damage following excitotoxicity, but not metabolic disruption. Gene Ther. 2002;9:214–219. doi: 10.1038/sj.gt.3301642. [DOI] [PubMed] [Google Scholar]
- Schindler CK, Shinoda S, Simon RP, Henshall DC. Subcellular distribution of Bcl-2 family proteins and 14-3-3 within the hippocampus during seizure-induced neuronal death in the rat. Neurosci. Lett. 2004;356:163–166. doi: 10.1016/j.neulet.2003.11.048. [DOI] [PubMed] [Google Scholar]
- Schneider P, Tschopp J. Apoptosis induced by death receptors. Pharm. Acta Helv. 2000;74:281–286. doi: 10.1016/s0031-6865(99)00038-2. [DOI] [PubMed] [Google Scholar]
- Shinoda S, Araki T, Lan JQ, Schindler CK, Simon RP, Taki W, Henshall DC. Development of a model of seizure-induced hippocampal injury with features of programmed cell death in the BALB/c mouse. J. Neurosci. Res. 2004a;76:121–128. doi: 10.1002/jnr.20064. [DOI] [PubMed] [Google Scholar]
- Shinoda S, Schindler CK, Meller R, So NK, Araki T, Yamamoto A, Lan JQ, Taki W, Simon RP, Henshall DC. Bim regulation may determine hippocampal vulnerability after injurious seizures and in temporal lobe epilepsy. J. Clin. Invest. 2004b;113:1059–1068. doi: 10.1172/JCI19971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda S, Skradski SL, Araki T, Schindler CK, Meller R, Lan J-Q, Taki W, Simon RP, Henshall DC. Formation of a tumour necrosis factor receptor 1 molecular scaffolding complex and activation of apoptosis signal-regulating kinase 1 during seizure-induced neuronal death. Eur. J. Neurosci. 2003;17:2065–2076. doi: 10.1046/j.1460-9568.2003.02655.x. [DOI] [PubMed] [Google Scholar]
- Sohn D, Schulze-Osthoff K, Janicke RU. Caspase-8 can be activated by interchain proteolysis without receptor-triggered dimerization during drug-induced apoptosis. J. Biol. Chem. 2005;280:5267–5273. doi: 10.1074/jbc.M408585200. [DOI] [PubMed] [Google Scholar]
- Sztriha L, Joo F, Szerdahelyi P. Accumulation of calcium in the rat hippocampus during kainic acid seizures. Brain Res. 1985;360:51–57. doi: 10.1016/0006-8993(85)91219-3. [DOI] [PubMed] [Google Scholar]
- Takano J, Tomioka M, Tsubuki S, Higuchi M, Iwata N, Itohara S, Maki M, zSaido TC. Calpain mediates excitotoxic DNA fragmentation via mitochondrial pathways in adult brains: evidence from calpastatin mutant mice. J. Biol. Chem. 2005;280:16175–16184. doi: 10.1074/jbc.M414552200. [DOI] [PubMed] [Google Scholar]
- Tu S, McStay GP, Boucher LM, Mak T, Beere HM, Green DR. In situ trapping of activated initiator caspases reveals a role for caspase-2 in heat shock-induced apoptosis. Nat Cell Biol. 2006;8:72–77. doi: 10.1038/ncb1340. [DOI] [PubMed] [Google Scholar]
- Viswanath V, Wu Z, Fonck C, Wei Q, Boonplueang R, Andersen JK. Transgenic mice neuronally expressing baculoviral p35 are resistant to diverse types of induced apoptosis, including seizure-associated neurodegeneration. Proc. Natl. Acad. Sci. U. S. A. 2000;97:2270–2275. doi: 10.1073/pnas.030365297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasterlain CG, Fujikawa DG, Penix L, Sankar R. Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia. 1993;34:S37–53. doi: 10.1111/j.1528-1157.1993.tb05905.x. [DOI] [PubMed] [Google Scholar]
- Waterhouse NJ, Finucane DM, Green DR, Elce JS, Kumar S, Alnemri ES, Litwack G, Khanna K, Lavin MF, Watters DJ. Calpain activation is upstream of caspases in radiation-induced apoptosis. Cell Death Differ. 1998;5:1051–1061. doi: 10.1038/sj.cdd.4400425. [DOI] [PubMed] [Google Scholar]
- Xiong Z, Lu W, MacDonald JF. Extracellular calcium sensed by a novel cation channel in hippocampal neurons. Proc. Natl. Acad. Sci. U. S. A. 1997;94:7012–7017. doi: 10.1073/pnas.94.13.7012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan W, Samson M, Jegou B, Toppari J. Bcl-w forms complexes with Bax and Bak, and elevated ratios of Bax/Bcl-w and Bak/Bcl-w correspond to spermatogonial and spermatocyte apoptosis in the testis. Mol. Endocrinol. 2000;14:682–699. doi: 10.1210/mend.14.5.0443. [DOI] [PubMed] [Google Scholar]
- Yin XM, Luo Y, Cao G, Bai L, Pei W, Kuharsky DK, Chen J. Bid-mediated mitochondrial pathway is critical to ischemic neuronal apoptosis and focal cerebral ischemia. J. Biol. Chem. 2002;277:42074–42081. doi: 10.1074/jbc.M204991200. [DOI] [PubMed] [Google Scholar]
- Yoneda T, Imaizumi K, Oono K, Yui D, Gomi F, Katayama T, Tohyama M. Activation of caspase-12, an endoplastic reticulum (ER) resident caspase, through tumor necrosis factor receptor-associated factor 2- dependent mechanism in response to the ER stress. J. Biol. Chem. 2001;276:13935–13940. doi: 10.1074/jbc.M010677200. [DOI] [PubMed] [Google Scholar]
- Yuan J, Yankner BA. Apoptosis in the nervous system. Nature. 2000;407:802–809. doi: 10.1038/35037739. [DOI] [PubMed] [Google Scholar]
- Zhang LX, Smith MA, Li XL, Weiss SR, Post RM. Apoptosis of hippocampal neurons after amygdala kindled seizures. Brain Res. Mol. Brain Res. 1998;55:198–208. doi: 10.1016/s0169-328x(97)00316-1. [DOI] [PubMed] [Google Scholar]