

Abstract
Rubredoxin from the hyperthermophile Pyrococcus furiosus is the most thermostable protein characterized to date with an estimated global unfolding rate of 10−6 s−1 at 100°C. In marked contrast to these slow global dynamics, hydrogen exchange experiments here demonstrate that conformational opening for solvent access occurs in the ≈millisecond time frame or faster at 28°C for all amide positions. Under these conditions all backbone amides with exchange protection factors between 104 and 106, for which EX2 exchange kinetics were directly verified, have exchange activation energy values within 2–3 kcal/mol of that observed for unstructured peptides. The conformational flexibility of this protein is thus sufficient for water and base catalyst access to the exchanging amide with quite limited structural disruption. The common hypothesis that enhanced conformational rigidity in the folded native state underlies the increased thermal stability of hyperthermophile proteins is not supported by these data.
The remarkable thermal stability of proteins from so-called hyperthermophiles, microorganisms that grow at temperatures above 90°C, is commonly thought to be the result of enhanced conformational rigidity in the folded native state. Quantitative comparisons between thermodynamic global stability and local conformational dynamics have been complicated by the irreversible denaturation behavior of most hyperthermophile proteins studied. However, indirect support for such a correlation comes from the observation that the unfolding rates of hyperthermophile proteins appear to be substantially slower than those of the homologous mesophile proteins. For example, rubredoxin from the mesophile Clostridium pasteurianum unfolds 102-103 faster than the structurally homologous hyperthermophile Pyrococcus furiosus rubredoxin under the same conditions (1). In a potentially related phenomenon, a number of studies on homologous series of mesophile, thermophile, and hyperthermophile enzymes have found that their maximal catalytic rates are similar at the optimal growth temperature of the organisms from which they are obtained even though these growth temperatures differ by 70°C or more (2–5). The most commonly invoked explanation of this surprising constancy in maximal catalytic rates is based on a direct extension of the aforementioned hypothesized coupling of local conformational dynamics and thermodynamic global stability. It is argued that for each member of an homologous enzyme series a certain degree of conformational fluctuations is required for optimal catalysis. The presumably more rigid hyperthermophile enzymes require a much higher temperature to achieve the requisite conformational dynamics. Because increased conformational fluctuations are expected to lead to decreased global stability, thermodynamic stability and maximal catalytic activity are argued to be fundamentally linked (3, 6, 7). From the practical point of view, this model predicts that increased thermal stabilization and enhanced catalytic activity cannot be simultaneously achieved. Both theoretical (8) and experimental (7, 9) arguments have been offered against the universality of this proposed coupling of rigidity, stability, and catalysis. However, to date no other explanation of the observed constancy of maximal catalytic rates for mesophile and hyperthermophile enzymes has gained comparable acceptance.
Amide hydrogen exchange measurements can provide a useful monitor of both global thermodynamic stability and local conformational kinetics. For conformationally protected amide hydrogens, exchange with solvent occurs according to the general scheme:
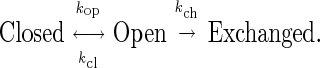 |
1 |
Under all conditions the observed exchange rate, kex, must be slower than the conformational opening rate, kop. Under most experimental conditions used in protein exchange studies, the rate of conformational closing, kcl, is much greater than the chemical exchange rate of the exposed amide kch, leading to pre-exchange equilibration of the open-closed conformational transition. In this so-called EX2 condition, kex = (kop/kcl)⋅kch, where the ratio kch/kex is referred to as the amide protection factor. For the neutral and basic pH conditions considered herein, kch is in turn proportional to the hydroxide ion concentration [OH−]. kch values estimated from peptide model studies have been widely used to predict a conformational equilibrium value ΔGconf [= −RT ln(kop/kcl)] from amide exchange data. At conditions well removed from the denaturation boundaries, the activation energy values for protein amide exchange are often of similar magnitude to those observed for the exchange reaction in freely accessible peptides (i.e., 17 kcal/mol) (10). Activation energies in this regime provide a primary operational definition for amide exchange by “local” conformational fluctuations. Under conditions nearer to denaturation, a number of amides generally exhibit much higher activation energies that are similar in magnitude to those deduced from calorimetric denaturation studies. For a number of proteins, the ΔGconf values derived from exchange data of the most slowly exchanging amides have been shown to agree with the calorimetric free energy measurements, indicating hydrogen exchange via global unfolding (11–13).
Amide exchange experiments have been used far less frequently to monitor the kinetics of the conformational opening step. The transfer rate of 1H spins migrating between the 1H2O and 1HN frequencies can be directly measured for rapidly exchanging amides (kex ≈0.5 s−1 to 100 s−1). Slower exchange yields no observable transfer whereas faster exchange broadens the amide resonance beyond detection. Magnetization transfer experiments have been used to monitor protein hydrogen exchange for solvent-exposed positions that exchange in this time regime near neutral pH (14–16). However, this technique has not been applied to proteins stable at high pH where the intrinsic chemical exchange rates are 105-106 faster. The rubredoxin from P. furiosus is ideally suited for such studies. In addition to a denaturation temperature at neutral pH estimated to be near 200°C, the CD signal of the Zn2+ form of the protein is essentially unchanged from 2°C at pH 6.5 to 94°C at pH 11.6 (17).
Amide exchange measurements were carried out on the Zn2+ form of 15N-labeled P. furiosus rubredoxin at pH values from 7.17 to 12.51 (200 mM sodium chloride and 50 mM sodium phosphate) for 3°C, 28°C, and 53°C. The maximal thermodynamic stability of this protein is estimated to occur in the range of 30°C to 60°C (17). In Fig. 1 (Right) representative fast heteronuclear single quantum correlation (18) spectra indicate the 1H-15N amide cross peaks for which the initial 1H magnetization starts at the amide resonance and remains there during the pulse sequence. In contrast, the CLEANEX-PM (19) spectra (Fig. 1 Left) indicate magnetization that initially resides in the 1H2O resonance and subsequently transfers to the amide resonance during the CLEANEX-PM mixing sequence. The intrinsic chemical exchange rate kch for a solvent exposed amide differs by 50- to 100-fold between the conditions of the top and middle as well as for the middle and bottom panels of Fig. 1. As illustrated in Fig. 1 Left, the set of amide hydrogens exhibiting magnetization transfer from 1H2O are largely distinct for the three given pH and temperature conditions. Furthermore, nearly all of the amide positions in the protein exhibit magnetization transfer from 1H2O in one of these three spectra.
Figure 1.

Clean chemical exchange-phase modulated (CLEANEX-PM) (Left) and fast heteronuclear single quantum correlation (FHSQC) (Right) spectra of 15N-labeled P. furiosus rubredoxin. The CLEANEX-PM (19) spectra indicate 1H signals that initiate in the 1H2O resonance and then transfer to the 1H amide resonance during the mixing period of the pulse sequence, whereas the FHSQC (18) spectra indicate 1H signals that remain at the amide resonance throughout the pulse sequence. The 15N-labeled sample expressed in Escherichia coli possesses an added N-terminal methionine residue. The thermal stabilities of the native and methionine-added forms are essentially equivalent (21). Initial rate values were obtained by using eight CLEANEX-PM mixing periods ranging from 5.19 ms to 41.52 ms in even increments. 1H2O T1 relaxation corrections of 10%, 25%, and 25% at 3°C (2-s delay), 28°C (2-s delay), and 53°C (4-s delay), respectively, were measured (19). pH values are reported at 25°C. Amide exchange activation energies are corrected for the 3,500-cal/mol ionization enthalpy for the third pK of sodium phosphate (37). In the 3°C pH 7.17 CLEANEX-PM spectra, the C-terminal Asp-53 yielded the largest negative cross peak because of incomplete cancellation between the rotating-frame and longitudinal nuclear Overhauser effects. This finding is indicative of significant motion faster than the slow tumbling limit. This cross peak yields an apparent exchange rate of −0.14 s−1 with a resultant negligible effect on the estimated kex values.
The bottom pair of spectra demonstrate that all of the slowest exchanging amides in the protein (Fig. 1 Right, fast heteronuclear single quantum correlation spectrum) exhibit magnetization transfer from the 1H2O resonance (Fig. 1 Left, CLEANEX-PM spectrum). Hence, the remarkable conclusion is that the native hydrogen bonding is disrupted for all amide hydrogens of this protein, and water plus the hydroxide catalyst gain direct access in less than a sec. Both EX2 kinetics and low activation energies of amide exchange could be explicitly verified for the majority of the protein amides. The proportionality between the observed exchange rate kex and [OH−] is illustrated in Fig. 2 for several of the amides that exhibited observable magnetization transfer across much of the higher pH range studied. As summarized in Table 1, these CLEANEX-PM experiments yield hydrogen exchange rates for every amide and indole position in P. furiosus rubredoxin, where kOH− = kex/[OH−].
Figure 2.

pH dependence of amide exchange for residues from P. furiosus rubredoxin at 28°C. Gly-9 (■), Gly-17 (●), Thr-27, (▴), and Glu-47 (⧫) illustrate residues distributed throughout the protein structure that give observable magnetization transfer cross peaks across a 1.5–2.0 pH unit span in the high pH range. EX2 behavior is observed up to pH 12.51. Best-fit lines of unit slope indicate proportionality between the exchange rate and [OH−].
Table 1.
Amide exchange rates kOH− (M−1·s−1) for P. furiosus rubredoxin at 28°C
| Position | Rate | Position | Rate |
|---|---|---|---|
| Ala-1 | 2.2 × 108 | Glu-30 | 6.4 × 105 |
| Lys-2 | 2.9 × 106 | Glu-31 | 850 |
| Trp-3 | 90 | Leu-32 | 350 |
| Val-4 | 110 | Asp-34 | 2.0 × 108 |
| Cys-5 | 80 | Asp-35 | 2.6 × 107 |
| Lys-6 | 470 | Trp-36 | 2,300 |
| Ile-7 | 40 | Val-37 | 120 |
| Cys-8 | 20 | Cys-38 | 40 |
| Gly-9 | 850 | Ile-40 | ∼20 |
| Tyr-10 | 40 | Cys-41 | ∼20 |
| Ile-11 | 370 | Gly-42 | 220 |
| Tyr-12 | 90 | Ala-43 | 90 |
| Asp-13 | 5,900 | Lys-45 | 2.5 × 104 |
| Glu-14 | 1.5 × 105 | Ser-46 | 1.3 × 107 |
| Asp-15 | 7,200 | Glu-47 | 1,100 |
| Ala-16 | 6,000 | Phe-48 | 210 |
| Gly-17 | 8,200 | Glu-49 | 230 |
| Asp-18 | 8,000 | Lys-50 | 2.1 × 105 |
| Asp-20 | 4.3 × 107 | Leu-51 | 1.6 × 104 |
| Asn-21 | 3.1 × 104 | Glu-52 | 1.6 × 107 |
| Gly-22 | 7.8 × 106 | Asp-53 | 3.8 × 106 |
| Ile-23 | 4,600 | ||
| Ser-24 | 5.0 × 107 | Side chain | |
| Gly-26 | 1.6 × 104 | Trp-3 ring | 3.5 × 104 |
| Thr-27 | 5400 | Trp-36 ring | 2,800 |
| Lys-28 | 4.1 × 104 | Asn-21 Nδ1 | 1.0 × 105 |
| Phe-29 | 8,400 | Asn-21 Nδ2 | 2.6 × 105 |
The amide 1HN exchange values for Lys-28, Phe-29, and Trp-36 as well as for indole 1HN of Trp-3 and Trp-36 were fitted for kex data below pH 11. All positions having kOH− values between 150 s−1 and 5 × 105 s−1 predict an optimal slope for log kex vs. pH between 0.85 and 1.2. The pH (7.17, 10.38, 10.95, 11.49, 12.02, and 12.51) and temperature (3°C, 28°C, and 53°C) data observed for each amide then were fitted to a slope of log kex vs. pH set to 1.0 and an activation energy of 17 kcal/mol. The pH and the temperature-dependence analyses were satisfactorily fitted with a 25% standard error in the observed rates. The rates for all other amides (kOH− > 5 × 105 s−1 or kOH− < 150 s−1) were estimated assuming EX2 kinetics and a 17-kcal/mol activation energy.
Deviations from linearity in the log kex vs. pH plot for these data should be detectable at kex rates approximately 10-fold below the kop rate. Amides with kOH− > 1,000 s−1 in Table 1 have been directly observed to have EX2 behavior up to kex values of ≈100 s−1 at the limit of the CLEANEX-PM experiment. Hence in these cases kop rates ≥ 1,000 s−1 can be concluded. EX2 behavior appears to extend to at least pH 12.51 for the slower exchanging amide positions (i.e., kOH− < 1,000 s−1). Therefore, for these amides the numerical values of kOH− listed in Table 1 should approximate the lower limits for kop. As a result, it can be concluded that conformational fluctuations on the magnitude scale of hydrogen bond rupture and access to water and base catalyst occur in the ≈ millisecond time frame or faster throughout the entire protein.
The main-chain amides of Lys-28, Phe-29, and Trp-36 as well as the two indole HN positions exhibit a marked decrease in the slope of the log kex vs. pH data above pH 11. This behavior suggests transition into the kinetic regime in which the conformational opening rate has become limiting for kex. In particular, kex data for both the main-chain HN of Phe-29 and the indole HN of Trp-3 appear to extrapolate to an apparent conformational opening rate of 70 s−1 at 28°C. These two groups form a tertiary hydrogen bonding interaction with the Oɛ1 of the Glu-14 side chain, which is a characteristic interaction of the thermophile rubredoxin not seen in rubredoxins from mesophilic organisms (20, 21). These results suggest that disruption of this tertiary hydrogen bond interaction occurs more slowly than that for most, if not all, binary hydrogen bonding interactions in the protein. It should be noted that the increased occurrence of ion pairs in hyperthermophile proteins has been directly implicated in decreased rates of denaturation in this protein (1) as well as others (22) in addition to their more commonly proposed role in global stability (20, 23–25).
Conformational opening rates of ≈103 s−1 demonstrated herein contrast sharply with estimates of kop based on indirect analysis of 2H2O exchange experiments. Recent studies have yielded conformational opening rate estimates of 10−2 to 10−3 s−1 (26) and 10−5 to 10−6 s−1 (27) for the slowest exchanging amides of proteins measured under low activation energy conditions. On the other hand, the present data are consistent with the familiar result of protein amide exchange occurring on the scale of years. The exchange rate of 2.5 × 10−7 min−1 (i.e., 0.13 yr−1) for Tyr-23 of bovine pancreatic trypsin inhibitor at 36°C and pH 4.6 (28) corresponds to a kOH− value of ≈10 s−1. Likewise, in nearly all cases the apparent ΔGconf derived from Table 1 agree reasonably well with the values for this protein derived from 2H2O exchange experiments in low denaturant concentrations carried out at much lower pH (17). The two noteworthy exceptions are Cys-5 and Cys-38, which have amide exchange processes that appear to reflect larger-scale fluctuations (i.e., global and “subglobal”) under the conditions of the previous study.
Activation energies of exchange for the backbone amides were determined over the temperature range of 3°C to 53°C. Sufficient data were available for all amides exhibiting protection factors of 104 to 106 [i.e., kOH− of 3 × 104 to 3 × 102 at 28°C given high salt (10) and isotope effect corrections (29)]. The precision of the activation energy determinations is estimated to be 2–3 kcal/mol. The activation energies were found to be statistically indistinguishable from 17 kcal/mol in every case except for Leu-51. In Fig. 3A the exchange data of this residue is plotted for 3°C (●), 28°C (▴), and 53°C (▪) in which the low and high temperature data have been scaled to that for 28°C assuming a 17-kcal/mol activation energy. For comparison, the corresponding temperature-dependence data for Glu-47 is presented in Fig. 3B. Optimal scaling of the Leu-51 exchange data occurs with an apparent activation energy of 13.5 kcal/mol. The amide of Leu-51 is hydrogen-bonded to the main-chain carbonyl of Lys-2 (21) with no obvious structural justification for its reduced apparent activation energy.
Figure 3.

Activation energy analysis of amide exchange for residues from P. furiosus rubredoxin. (A) The exchange rate data for Leu-51 at 28°C (▴). The corresponding data at 3°C (●) and 53°C (■) have been scaled to 28°C assuming a 17-kcal/mol activation energy. Optimal alignment of the data from the three temperatures is obtained with an apparent activation energy of 13.5 kcal/mol. (B) The far more typical case of Glu-47 in which a 17-kcal/mol activation energy factor adequately scales the data from the three different temperatures as in A.
In the high pH range considered herein, only hydrogen bonded amides gave sufficient exchange data for the activation energy analysis. These data indicate a maximum enthalpy contribution of 2–3 kcal/mol to the activated exchange state provided by the protein structural changes, which represents only 3–5% of the activation enthalpy commonly observed for protein unfolding (30). Although the hydrogen bond of the exchanging amide surely must be disrupted, setting a limit on the scale of other structural disruptions is less definitive. Estimation of the enthalpy of disruption of a protein hydrogen bond vary from ≈10 kcal/mol for unhydrated amides (31) to ≈1 kcal/mol when the amide is hydrated (32). Given the involvement of other enthalpic contributions as well, the very low and uniform activation energies observed herein appear consistent with having only the hydrogen bond of the exchanging amide disrupted in the activated state of exchange.
Amide exchange by local fluctuations has been proposed to be an effective monitor of the thermodynamic microstability of proteins (33). The distribution of amide protection factors for P. furiosus rubredoxin are typical of those observed for a large number of mesophile proteins. For protection factors up to at least 106, activation energies for exchange near the feasible lower limit are observed throughout this protein under conditions of global thermodynamic stability. The conformational flexibility is sufficient for exchange to occur with minimal enthalpic perturbations beyond disruption of the exchanging hydrogen bond. Hence by these criteria the microstability of this highly thermostable protein is indistinguishable from that of typical mesophile proteins. When combined with ≈103 s−1 local fluctuation opening rates at 28°C contrasted to a global unfolding rate of 10−6 s−1 at 100°C for this protein (1), the present data suggest that conformational fluctuations required for amide exchange monitored herein vs. those required for global unfolding must be very weakly coupled.
As noted, amide protection factors take the form of equilibrium constants, and they have commonly been interpreted in terms of conformational equilibria. On the other hand, on occasion amide protection factors also have been invoked as measures of conformational dynamics. In particular, IR monitoring of hydrogen-deuterium exchange for homologous mesophile and thermophile enzymes has shown increased amide protection factors for the thermophile enzymes when measured under similar conditions (6, 34). Although no direct information on the conformational opening or closing rates is provided by the amide protection factors, those authors have interpreted the increased amide protection factors of the thermophile enzymes as indicative of decreased conformational flexibility, which in turn was used to explain the decreased enzymatic activity of the thermophile enzymes under these conditions. If as assumed in these earlier studies, amide exchange is monitoring conformational motions that in some sense overlap with the conformational motions involved in enzyme catalysis, then the conformational dynamics of amide exchange that occur in the time frame of maximal turnover rates should be suppressed in the thermophile enzymes. The present study demonstrates that, on the contrary, moderate scale conformational dynamics in the millisecond time frame and faster are ubiquitous throughout the structure of the most thermostable protein characterized to date. This time frame corresponds to the maximal turnover rates for a great proportion of enzymes. Hence, these amide exchange measurements do not provide evidence for a systematic rigidification of local fluctuations on a molecule-wide scale, which might explain the constancy of maximal catalytic rates at physiological temperatures observed in various comparisons of homologous mesophile and thermophile enzymes.
These results do not imply that modulation of conformational fluctuations in the active site region is irrelevant to catalysis. A large number of mesophile enzymes have been proposed to have greater conformational flexibility in the active site as compared with the remainder of the protein. Mutagenesis studies of phage T4 lysozyme have directly demonstrated that active site residues are not optimized for stability (35). Furthermore, evolutionary changes localized to the active site region of marine lactate dehydrogenases appear to provide kinetic adaptation to at least modest physiological temperature differences (36). To mutually satisfy requirements for both catalytic activity and structural stability imposed by their higher physiological temperature, hyperthermophile enzymes may partition conformational fluctuations between the active site region and the rest of the protein differently from what occurs in mesophile enzymes. Mutations that modulate conformational flexibility in the active site region of hyperthermophile enzymes potentially might enhance catalytic reactivity with comparatively modest effects on global stability.
Acknowledgments
This research was performed under the auspices of the Laboratory Directed Research and Development Program at Los Alamos National Laboratory. Additional support was provided by American Chemical Society Grant PF-4254 (to M.W.W.A.) and National Science Foundation Grant MCB 9809060 (to M.W.W.A.).
Abbreviation
- CLEANEX-PM
clean chemical exchange-phase modulated
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.040569697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.040569697
References
- 1.Cavagnero S, Debe D A, Zhou Z H, Adams M W W, Chan S I. Biochemistry. 1998;37:3369–3376. doi: 10.1021/bi9721795. [DOI] [PubMed] [Google Scholar]
- 2.Graves J E, Somero G N. Evolution. 1982;36:97–106. doi: 10.1111/j.1558-5646.1982.tb05014.x. [DOI] [PubMed] [Google Scholar]
- 3.Varley P G, Pain R H. J Mol Biol. 1991;220:531–538. doi: 10.1016/0022-2836(91)90028-5. [DOI] [PubMed] [Google Scholar]
- 4.Robb F T, Park J B, Adams M W W. Biochim Biophys Acta. 1992;1120:267–272. doi: 10.1016/0167-4838(92)90247-b. [DOI] [PubMed] [Google Scholar]
- 5.Brown S H, Sjoholm C, Kelley R M. Biotechol Bioeng. 1993;41:878–886. doi: 10.1002/bit.260410907. [DOI] [PubMed] [Google Scholar]
- 6.Wrba A, Schweiger A, Schultes V, Jaenicke R, Zavodszky P. Biochemistry. 1990;29:7584–7592. doi: 10.1021/bi00485a007. [DOI] [PubMed] [Google Scholar]
- 7.Giver L, Gershenson A, Freskgard P O, Arnold F H. Proc Natl Acad Sci USA. 1998;95:12809–12813. doi: 10.1073/pnas.95.22.12809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lazaridis T, Lee I, Karplus M. Protein Sci. 1997;6:2589–2605. doi: 10.1002/pro.5560061211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vandenberg B, Vriend G, Veltman O R, Venema G, Eijsink V G H. Proc Natl Acad Sci USA. 1998;95:2056–2060. doi: 10.1073/pnas.95.5.2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bai Y, Milne J S, Mayne L, Englander S W. Proteins Struct Funct Genet. 1993;17:75–86. doi: 10.1002/prot.340170110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kim K S, Fuchs J A, Woodward C K. Biochemistry. 1993;32:9600–9608. doi: 10.1021/bi00088a012. [DOI] [PubMed] [Google Scholar]
- 12.Orban J, Alexander P, Bryan P. Biochemistry. 1994;33:5702–5710. doi: 10.1021/bi00185a006. [DOI] [PubMed] [Google Scholar]
- 13.Bai Y W, Milne J S, Mayne L, Englander S W. Proteins Struct Funct Genet. 1994;20:4–14. doi: 10.1002/prot.340200103. [DOI] [PubMed] [Google Scholar]
- 14.Gemmecker G, Jahnke W, Kessler H. J Am Chem Soc. 1993;115:11620–11621. [Google Scholar]
- 15.Grzesiek S, Bax A. J Biomol NMR. 1993;3:627–638. doi: 10.1007/BF00198368. [DOI] [PubMed] [Google Scholar]
- 16.Mori S, Johnson M O, Berg J M, van Zijl P C M. J Am Chem Soc. 1994;116:11982–11984. [Google Scholar]
- 17.Hiller R, Zhou Z H, Adams M W W, Englander S W. Proc Natl Acad Sci USA. 1997;94:11329–11332. doi: 10.1073/pnas.94.21.11329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mori S, Abeygunawardana C, Johnson M O, van Zijl P C M. J Magn Reson B. 1995;108:94–98. doi: 10.1006/jmrb.1995.1109. [DOI] [PubMed] [Google Scholar]
- 19.Hwang T L, van Zijl P C M, Mori S. J Biomol NMR. 1998;11:221–226. doi: 10.1023/a:1008276004875. [DOI] [PubMed] [Google Scholar]
- 20.Day M W, Hsu B T, Joshua-Tor L, Park J B, Zhou Z H, Adams M W W, Rees D C. Protein Sci. 1992;1:1494–1507. doi: 10.1002/pro.5560011111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bau R, Rees D C, Kurtz D M, Scott R A, Huang H S, Adams M W W, Eidsness M K. J Biol Inorg Chem. 1998;3:484–493. [Google Scholar]
- 22.Pappenberger G, Schurig H, Jaenicke R. J Mol Biol. 1997;274:676–683. doi: 10.1006/jmbi.1997.1421. [DOI] [PubMed] [Google Scholar]
- 23.Teplyakov A V, Kuranova I P, Harutyunyan E H, Vainshtein B, Frommel C, Hohne W E, Wilson K S. J Mol Biol. 1990;214:261–279. doi: 10.1016/0022-2836(90)90160-n. [DOI] [PubMed] [Google Scholar]
- 24.Jaenicke R, Schurig H, Beaucamp N, Ostendorp R. Adv Protein Chem. 1996;48:181–269. doi: 10.1016/s0065-3233(08)60363-0. [DOI] [PubMed] [Google Scholar]
- 25.Elcock A H. J Mol Biol. 1998;284:489–502. doi: 10.1006/jmbi.1998.2159. [DOI] [PubMed] [Google Scholar]
- 26.Arrington C B, Robertson A D. Biochemistry. 1997;36:8686–8691. doi: 10.1021/bi970872m. [DOI] [PubMed] [Google Scholar]
- 27.Kragelund B B, Heinemann B, Knudsen J, Poulsen F M. Protein Sci. 1998;7:2237–2248. doi: 10.1002/pro.5560071101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wüthrich K, Wagner G. J Mol Biol. 1979;130:1–18. doi: 10.1016/0022-2836(79)90548-5. [DOI] [PubMed] [Google Scholar]
- 29.Connelly G P, Bai Y, Jeng M F, Englander S W. Proteins. 1993;17:87–92. doi: 10.1002/prot.340170111. [DOI] [PubMed] [Google Scholar]
- 30.Privalov P L. Adv Protein Chem. 1979;33:167–241. doi: 10.1016/s0065-3233(08)60460-x. [DOI] [PubMed] [Google Scholar]
- 31.Makhatadze G I, Privalov P L. J Mol Biol. 1993;232:639–659. doi: 10.1006/jmbi.1993.1416. [DOI] [PubMed] [Google Scholar]
- 32.Scholtz J M, Marqusee S, Baldwin R L, York E J, Stewart J M, Santoro M, Bolen S W. Proc Natl Acad Sci USA. 1991;88:2854–2858. doi: 10.1073/pnas.88.7.2854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Privalov P L, Tsalkova T N. Nature (London) 1979;280:693–696. doi: 10.1038/280693a0. [DOI] [PubMed] [Google Scholar]
- 34.Zavodszky P, Kardos J, Svingor A, Petsko G A. Proc Natl Acad Sci USA. 1998;95:7406–7411. doi: 10.1073/pnas.95.13.7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shoichet B K, Baase W A, Kuroki R, Matthews B W. Proc Natl Acad Sci USA. 1995;92:452–456. doi: 10.1073/pnas.92.2.452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fields P A, Somero G N. Proc Natl Acad Sci USA. 1998;95:11476–11481. doi: 10.1073/pnas.95.19.11476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pitzer K S. J Am Chem Soc. 1937;59:2365–2371. [Google Scholar]