

Abstract
The development and progression of microvascular complications have been extensively documented in a cohort of type 1 diabetic subjects enrolled in the Diabetes Control and Complications Trial (DCCT) and followed in the Epidemiology of Diabetes Interventions and Complications (EDIC) study. We describe the association of genetic variation in the ACE gene in 1,365 DCCT/EDIC subjects with incident persistent microalbuminuria (n = 312) and severe nephropathy (n = 115). We studied three markers (rs1800764, insertion/deletion, and rs9896208) in the ACE gene that allowed us to capture genetic variation in the common haplotypes occurring at frequencies of >5% in Caucasians. Compared with the more frequent genotype (D/I) for the insertion/deletion polymorphism, in multivariate models, the I/I genotype conferred a lower risk for persistent microalbuminuria (hazard ratio [HR] 0.62 [95% CI 0.43–0.89], P = 0.009) and severe nephropathy (0.56 [0.32–0.96], P = 0.033). Variation at the two other markers, rs1800764 and rs9896208, were also associated with these renal outcomes. In addition, homozygosity for the common haplotype TIC (which corresponded to the T, insertion, and C alleles at the three markers, rs1800764, insertion/deletion, and rs9896208, respectively) versus the CDT/TIC haplotype pair was associated with lower risk for development of persistent microalbuminuria (HR 0.49 [0.32–0.75], P = 0.0009) and severe nephropathy (0.41 [0.22–0.78], P = 0.006). Our findings in the DCCT/EDIC cohort provide strong evidence that genetic variation at the ACE gene is associated with the development of nephropathy in patients with type 1 diabetes.
Microvascular and neurologic complications of type 1 diabetes result in significant morbidity and mortality. The Diabetes Control and Complications Trial (DCCT) demonstrated that intensive therapy aimed at reducing glycemic exposure reduced the development and progression of long-term complications by as much as 76% compared with conventional therapy (1). In addition to the importance of intensive diabetes management, it is clear that there are other factors, including genetic ones, that contribute to the development of these complications. Epidemiologic studies (2,3) conducted before the use of intensive therapy suggested that ~30% of patients with type 1 diabetes developed diabetic nephropathy after 20 years of diabetes; however, modern diabetes care has markedly reduced the incidence of diabetic nephropathy to 13–16% (4 – 6) after 20 years’ diabetes duration. A number of studies have documented familial clustering of diabetic nephropathy (7–9). In the DCCT, there was an increased risk of microalbuminuria (albumin excretion rate [AER] >28 μg/min) in diabetic relatives of microalbuminuria-positive versus -negative DCCT subjects in the secondary intervention cohort (odds ratio 5.4 [95% CI 2.2–13.7], P < 0.001) after adjustment for covariates (10). Given the evidence for familial clustering of diabetic nephropathy, we have begun a systematic study of candidate genes associated with the development of nephropathy in the DCCT subjects.
ACE plays an important role in the renin-angiotensin-aldosterone pathway and has been studied extensively as a putative mediator of diabetic nephropathy. This enzyme cleaves the COOH-terminal dipeptide of angiotensin I to produce angiotensin II and inactivates bradykinin by removal of COOH-terminal peptides. Increased ACE activity increases intraglomerular pressure (11) and can lead to glomerulosclerosis (12). Plasma ACE activity, a highly heritable trait, is encoded by one of the three known isozymes transcribed from the ACE gene, which is composed of 26 exons located on chromosome 17q23 (13). There are many biallelic single nucleotide polymorphisms (SNPs) within and flanking this gene (14) that are associated with ACE activity; however, the exact etiologic variant(s) is/are as yet undetermined, as these polymorphisms are in strong linkage disequilibrium with each other (15). The most extensively studied polymorphism is the insertion/deletion of a 287-bp Alu repeat in intron 16 (16).
Since the initial report of the protective effect of the I/I insertion/deletion genotype in the development of diabetic nephropathy in type 1 diabetes (17), there have been many other studies of the association of this polymorphism with elevated urinary excretion of albumin and/or nephropathy in patients with type 1 and type 2 diabetes. Meta-analyses (18 –20) have suggested that discrepant results among studies may be partially attributed to differences in study design, diabetes phenotype (type 1 diabetes versus type 2 diabetes), ethnic composition, methods of quantitation, and estimation of AER (e.g., single measures of AER versus sustained elevations in AER) and definitions of diabetic nephropathy (microalbuminuria versus macroalbuminuria).
The DCCT and its long-term follow-up, the Epidemiology of Diabetes Interventions and Complications (EDIC) study, have measured renal function, albuminuria, and the development of nephropathy for up to 17 years using standardized methods. We have analyzed the association of the insertion/deletion polymorphism of the ACE gene with the time to development of persistent elevation of microalbuminuria and severe nephropathy in 1,365 Caucasian subjects in the DCCT/EDIC study. To capture the genetic variation in the most common Caucasian haplotypes at the ACE locus occurring at frequencies >5% (15), we also genotyped two SNPs (http://www.ncbi.nlm.nih.gov/SNP/), rs1800764 and rs9896208, flanking the insertion/deletion polymorphism in the 5′and 3′ untranslated region and analyzed the association of variation at these three loci with nephropathy outcomes using Cox proportional hazards models of complication-free survival. To reduce population heterogeneity, we restrict the analysis to self-identified “white” subjects only, which represent 96% of the original cohort.
RESEARCH DESIGN AND METHODS
The DCCT study design (21) is described in the online appendix (available at http://diabetes.diabetesjournals.org).
Phenotypic characterization of nephropathy
Two nephropathy outcomes, persistent microalbuminuria and severe nephropathy, were defined using repeated 4-h AER measurements obtained yearly during the DCCT and every 2nd year during EDIC. Persistent microalbuminuria was defined as an AER ≥20.8 μg/min on two consecutive assessments. The definition of severe nephropathy is also based on repeated measures of AER and represents the progression from persistent microalbuminuria to an AER ≥208 μg/min or renal replacement therapy (dialysis or transplant). Time to outcome development or censoring was determined as number of visit years from DCCT baseline up to and including the 8th year of EDIC follow-up (2001); mean (±SD) follow-up from DCCT entry was 13.5 ± 2.6 years. Subjects meeting the criteria for persistent microalbuminuria at DCCT baseline and DCCT year 1 (n = 66) were excluded from subsequent analyses of that outcome. Due to the DCCT exclusion criteria, no subjects had severe nephropathy at baseline.
Determination of ACE genotypes
Genetic variation at the ACE gene has been extensively studied, and haplotypes have been determined (15,22). Fifteen haplotypes were identified by measured haplotype analysis in families, but because of extensive linkage disequilibrium between markers, only four common haplotypes had frequencies >5% (15). In addition to the insertion/deletion polymorphism, we genotyped two SNPs that allowed us to capture the four most common haplotypes occurring with frequency >5% in Caucasian populations. The intermarker distance between the 5′ SNP, rs1800764 (T7715C) (15), and the insertion/deletion is 15.4 kb and between the insertion/deletion and the 3′ SNP, rs9896208 (T33569C) (15), is 10.4 kb (see online appendix for genotyping protocol).
Statistical analysis
The primary analysis was to evaluate the association between each of the ACE markers and the risk of complications, taking into account the DCCT/EDIC study design and potential confounding factors, including all physiological traits known to be associated with variation in risk of renal complications. The relative risks associated with different marker genotypes were estimated by the hazard ratio (HR) in the Cox proportional hazards model, with and without including explanatory covariates in the model (see online appendix for further description of analysis).
RESULTS
Examination of the distribution of baseline covariates among genotype groups (Table 1) revealed a significant association between BMI and two of the ACE variants, as well as a weak association between triglyceride level and the insertion/deletion genotype. A significant association (P = 0.03) was also present between use of ACE inhibitors during EDIC and the genotype at rs1800764 (Table 1). Due to the clinical correlation of the development of microalbuminuria and the subsequent institution of an ACE inhibitor, we repeated this analysis after exclusion of the individuals who received an ACE inhibitor after the development of persistent microalbuminuria. There was no significant association of ACE inhibitor use with the rs1800764 genotype in subjects with persistent microalbuminuria (P = 0.30) or severe nephropathy (P = 0.07). There was also no association of non-ACE inhibitor antihypertensive medications with any of the markers (P = 0.59). As expected, many of these baseline covariates were significantly associated with persistent microalbuminuria and severe nephropathy (online appendix Tables A1 and A2).
TABLE 1.
Association of DCCT baseline variables with ACE polymorphisms
| ACE polymorphisms
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| n | rs1800764
|
Insertion/deletion
|
rs9896208
|
||||||||||
| Variable Genotypes | CC | CT | TT | P | II | ID | DD | P | CC | CT | TT | P | |
| Total cohort | 1,365 | 258 | 698 | 409 | 316 | 680 | 369 | 560 | 612 | 193 | |||
| Sex | |||||||||||||
| Male | 717 | 126 | 374 | 217 | 168 | 364 | 185 | 296 | 315 | 106 | |||
| Female | 648 | 132 | 324 | 192 | 0.41 | 148 | 316 | 184 | 0.56 | 264 | 297 | 87 | 0.69 |
| DCCT treatment | |||||||||||||
| Intensive | 671 | 124 | 332 | 215 | 163 | 337 | 171 | 294 | 286 | 91 | |||
| Conventional | 694 | 134 | 366 | 194 | 0.25 | 153 | 343 | 198 | 0.38 | 266 | 326 | 102 | 0.12 |
| Cohort | |||||||||||||
| Primary | 686 | 134 | 347 | 205 | 151 | 351 | 184 | 279 | 312 | 95 | |||
| Secondary | 679 | 124 | 351 | 204 | 0.82 | 165 | 329 | 185 | 0.52 | 281 | 300 | 98 | 0.88 |
| Use of ACE inhibitors during EDIC | |||||||||||||
| Yes | 408 | 68 | 230 | 110 | 90 | 208 | 110 | 167 | 185 | 56 | |||
| No | 885 | 176 | 429 | 280 | 0.030 | 210 | 435 | 240 | 0.77 | 364 | 391 | 130 | 0.87 |
| Age at diagnosis (years) | 1,365 | 20.8 ± 7.9 | 21.5 ± 8.1 | 21.0 ± 8.2 | 0.41 | 20.9 ± 8.3 | 21.5 ± 8.1 | 21.0 ± 7.9 | 0.43 | 21.1 ± 8.1 | 21.3 ± 8.1 | 21.3 ± 7.7 | 0.66 |
| Age at baseline (years) | 1,365 | 26.3 ± 7.0 | 27.1 ± 7.0 | 26.7 ± 7.3 | 0.33 | 26.7 ± 7.3 | 26.9 ± 7.1 | 26.7 ± 6.9 | 0.92 | 26.8 ± 7.2 | 26.8 ± 7.0 | 27.0 ± 7.1 | 0.86 |
| Type 1 diabetes duration (years) | 1,365 | 5.5 ± 4.1 | 5.6 ± 4.2 | 5.7 ± 4.2 | 0.80 | 5.9 ± 4.2 | 5.5 ± 4.1 | 5.7 ± 4.2 | 0.28 | 5.8 ± 4.2 | 5.4 ± 4.2 | 5.8 ± 4.1 | 0.20 |
| HbA1c at eligibility (%) | 1,365 | 9.0 ± 1.6 | 9.0 ± 1.6 | 9.1 ± 1.6 | 0.46 | 9.1 ± 1.6 | 9.0 ± 1.6 | 9.0 ± 1.6 | 0.69 | 9.1 ± 1.5 | 9.0 ± 1.7 | 9.0 ± 1.6 | 0.50 |
| BMI (kg/m2) | 1,365 | 22.9 ± 2.7 | 23.5 ± 2.8 | 23.5 ± 2.8 | 0.01 | 23.5 ± 2.9 | 23.5 ± 2.8 | 23.1 ± 2.7 | 0.02 | 23.4 ± 2.9 | 23.4 ± 2.7 | 23.2 ± 2.8 | 0.77 |
| Mean arterial pressure (mmHg) | 1,365 | 86 ± 9 | 87 ± 9 | 86 ± 8 | 0.70 | 87 ± 8 | 86 ± 9 | 86 ± 9 | 0.95 | 87 ± 9 | 86 ± 9 | 87 ± 9 | 0.44 |
| Total cholesterol (mg/dl) | 1,365 | 178 ± 34 | 176 ± 33 | 176 ± 34 | 0.90 | 175 ± 34 | 177 ± 33 | 176 ± 33 | 0.67 | 177 ± 33 | 175 ± 33 | 178 ± 34 | 0.84 |
| HDL cholesterol (mg/dl) | 1,365 | 52 ± 13 | 50 ± 12 | 49 ± 11 | 0.12 | 49 ± 11 | 50 ± 12 | 51 ± 13 | 0.15 | 50 ± 12 | 50 ± 12 | 51 ± 12 | 0.45 |
| Triglycerides (mg/dl) | 1,365 | 79 ± 40 | 80 ± 42 | 87 ± 61 | 0.16 | 89 ± 67 | 78 ± 36 | 82 ± 48 | 0.06 | 84 ± 56 | 80 ± 38 | 80 ± 50 | 0.27 |
| AER (μn) | 1,365 | 10 ± 10 | 10 ± 12 | 12 ± 15 | 0.78 | 12 ± 15 | 10 ± 12 | 10 ± 10 | 0.05 | 12 ± 14 | 10 ± 10 | 10 ± 17 | 0.30 |
Data are means ± SD. χ2 P values are reported for categorical traits; other values are Kruskal-Wallis P values. The following variables were Ln transformed to normalize the data for statistical analysis: eligibility HbA1c, total cholesterol, HDL cholesterol, triglycerides, and AER.
During follow-up between DCCT entry and EDIC year 8, 312 subjects developed persistent microalbuminuria (of which 66 had events at DCCT baseline and were excluded from analyses). Of these individuals, 115 also developed a more severe renal phenotype, including clinical grade proteinuria with an AER >208 μg/min (n = 114) and/or a renal end point (n = 12 patients with either dialysis or transplant).
The quality characteristics of the genotype data were assessed by replicate Centre d’Etude du Polymorphisme Humain (CEPH) control samples, and of 498 replicate genotypes at the three markers, only two genotyping errors occurred corresponding to an observed genotyping error rate of 0.4%. The minor allele frequencies for rs1800764, insertion/deletion, and rs9896208 were 0.45, 0.47, and 0.37, respectively. No significant deviations from Hardy-Weinberg equilibrium were observed.
Proportional hazards analysis of the ACE polymorphisms adjusted for DCCT year of entry, cohort, and treatment strata indicate that genotype variation at each of the markers was significantly associated with the risk of developing persistent microalbuminuria (Table 2). Specifically, as depicted in Fig. 1, individuals with either the T/T, I/I, or C/C genotypes at rs1800764, insertion/deletion, and rs9896208, respectively, had a lower risk of progression to persistent microalbuminuria during a maximum of 17 years of follow-up from DCCT baseline. Individuals with the C/C genotype at rs9896208 had a significantly lower risk of development of persistent microalbuminuria (HR 0.70 [95% CI 0.53–0.93], P = 0.015) (Fig. 1 and Table 2) and severe nephropathy (0.60 [0.39 – 0.92], P = 0.020) (online appendix Fig. A1 and Table A2). Genotypes at rs1800764 and insertion/deletion, however, were not significantly (P > 0.05) associated with severe nephropathy in analyses that did not take any of the DCCT baseline physiological trait variables into account (Table 2).
TABLE 2.
Univariate proportional hazards analysis of ACE genotype and haplotype with renal outcomes
| Grades of diabetic nephropathy
|
|||||||
|---|---|---|---|---|---|---|---|
| ACE genotype
|
Persistent microalbuminuria (n = 246)
|
Severe nephropathy (n = 115)
|
|||||
| Single locus | Genotype comparisons | HR | 95% CI | Wald P value | HR | 95% CI | Wald P value |
| rs1800764 | CC vs. CT | 0.94 | 0.68–1.32 | 0.74 | 0.67 | 0.39–1.15 | 0.15 |
| TT vs. CT | 0.64 | 0.47–0.88 | 0.006* | 0.70 | 0.45–1.10 | 0.12 | |
| Insertion/deletion | DD vs. DI | 1.02 | 0.76–1.37 | 0.87 | 0.96 | 0.62–1.48 | 0.86 |
| II vs. DI | 0.67 | 0.47–0.95 | 0.026* | 0.69 | 0.41–1.15 | 0.15 | |
| rs9896208 | TT vs. CT | 1.09 | 0.76–1.56 | 0.63 | 0.96 | 0.57–1.61 | 0.88 |
| CC vs. CT | 0.70 | 0.53–0.93 | 0.015* | 0.60 | 0.39–0.92 | 0.020* | |
| Haplotype rs1800764, insertion/deletion, rs9896208 | |||||||
| CDT/CDT | 0.94 | 0.60–1.48 | 0.78 | 0.90 | 0.47–1.73 | 0.75 | |
| TIC/TIC | 0.56 | 0.37–0.85 | 0.007* | 0.53 | 0.29–0.97 | 0.04* | |
| CDC/CDT | 0.83 | 0.47–1.45 | 0.51 | 0.56 | 0.22–1.43 | 0.22 | |
| CDC/TIC | 0.78 | 0.51–1.19 | 0.25 | 0.69 | 0.36–1.33 | 0.27 | |
| CDT/other | 1.06 | 0.69–1.62 | 0.79 | 1.04 | 0.57–1.88 | 0.90 | |
| TIC/other | 0.66 | 0.42–1.06 | 0.09 | 0.77 | 0.40–1.49 | 0.44 | |
| Other/other | 0.71 | 0.34–1.49 | 0.36 | 0.54 | 0.16–1.76 | 0.30 | |
Each model was adjusted for study design (treatment, cohort, cohort by treatment interaction, and stratified by year of entry into DCCT). Indicated haplotypes are compared with the most frequent haplotype pair (CDT/TIC).
P < 0.05.
FIG. 1.
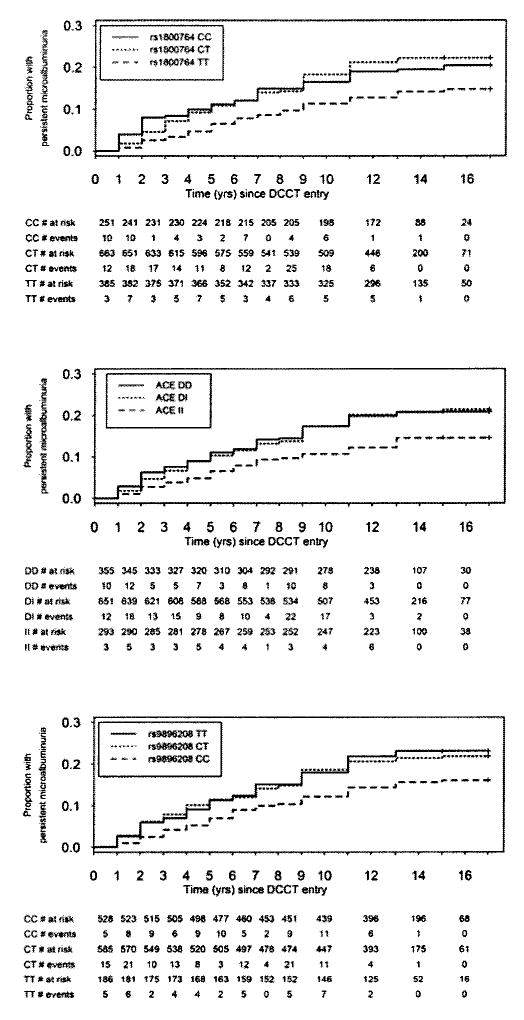
Cumulative incidence of persistent microalbuminuria according to ACE genotype. The time was calculated from DCCT baseline to the first consecutive timed AER collection >20.8 μg/min in the DCCT or EDIC follow-up periods in individuals genotyped for three ACE polymorphisms: rs1800764, insertion/deletion, and rs9896208. For univariate analysis, see Table 2.
Three-locus haplotypes were estimated using a Bayesian reconstruction algorithm implemented in the PHASE (version 2.1) software (see online appendix for haplotype construction). In the DCCT/EDIC cohort, 92% of chromosomes had one of the four common haplotypes previously reported. In univariate analysis, individuals homozygous for the TIC haplotype (which corresponded to the T, insertion, and C alleles at the three markers, rs1800764, insertion/deletion, and rs9896208, respectively) had a lower risk of persistent microalbuminuria and severe nephropathy when compared with individuals with the most common CDT/TIC haplotype pair (Table 2).
When we adjusted for the additional covariates (Table 3 legend), the association of each of the three ACE variants with the development of persistent microalbuminuria remained significant (Table 3). Values of R2 indicating the proportion of explained variability in survival times to persistent microalbuminuria were calculated as 0.8, 0.6, and 0.8% for rs1800764, insertion/deletion, and rs9896208 markers, respectively. For severe nephropathy, adjustment for covariates in the multivariate model strengthened the genotype associations (Table 3). At rs1800764, individuals with the T/T genotype had a lower risk than C/T heterozygotes. Individuals with the I/I genotype at the insertion/deletion also had lower risk for severe nephropathy than the D/I heterozygotes, and similarly at rs9896208, individuals with the C/C genotype had a lower risk than those with the C/T genotype. As measured by the R2 statistic, genotypes at rs1800764, insertion/deletion, and rs9896208 accounted for 0.4, 0.4, and 0.6%, respectively, of explained variability in risk for severe nephropathy.
TABLE 3.
Multivariate proportional hazards analysis of ACE genotype and renal outcomes
| Grades of diabetic nephropathy
|
|||||||
|---|---|---|---|---|---|---|---|
| ACE genotype
|
Persistent microalbuminuria (n = 246)
|
Severe nephropathy (n = 115)
|
|||||
| Single locus | Genotype comparisons | HR | 95% CI | Wald P value | HR | 95% CI | Wald P value |
| rs1800764 | CC vs. CT | 0.98 | 0.69–1.38 | 0.90 | 0.69 | 0.38–1.23 | 0.20 |
| TT vs. CT | 0.61 | 0.44–0.84 | 0.003* | 0.62 | 0.39–0.98 | 0.043* | |
| Insertion/deletion | DD vs. DI | 0.98 | 0.72–1.33 | 0.91 | 0.89 | 0.56–1.39 | 0.60 |
| II vs. DI | 0.62 | 0.43–0.89 | 0.009* | 0.56 | 0.32–0.96 | 0.033* | |
| rs9896208 | TT vs. CT | 1.12 | 0.77–1.62 | 0.56 | 0.95 | 0.54–1.65 | 0.85 |
| CC vs. CT | 0.65 | 0.49–0.88 | 0.005* | 0.54 | 0.34–0.85 | 0.008* | |
| Haplotype rs1800764, insertion/deletion, rs9896208 | |||||||
| CDT/CDT | 0.94 | 0.58–1.51 | 0.79 | 0.79 | 0.38–1.64 | 0.53 | |
| TIC/TIC | 0.49 | 0.32–0.75 | 0.0009* | 0.41 | 0.22–0.78 | 0.006* | |
| CDC/CDT | 0.70 | 0.39–1.25 | 0.22 | 0.52 | 0.19–1.39 | 0.19 | |
| CDC/TIC | 0.68 | 0.43–1.06 | 0.09 | 0.59 | 0.29–1.18 | 0.14 | |
| CDT/other | 0.89 | 0.57–1.38 | 0.60 | 0.89 | 0.48–1.65 | 0.71 | |
| TIC/other | 0.56 | 0.35–0.92 | 0.02* | 0.58 | 0.29–1.16 | 0.12 | |
| Other/other | 0.76 | 0.36–1.63 | 0.48 | 0.51 | 0.15–1.71 | 0.27 | |
HRs and Wald tests for ACE genotype association with the time (years) from DCCT entry to the development of persistent microalbuminuria (246 events and 1,053 censored; 66 subjects had AER >20.8 μg/min at DCCT baseline and were therefore excluded from the CoxPH analysis) and severe nephropathy (115 events and 1,250 censored) in multivariate Cox proportional hazards analyses stratified by year of entry into the DCCT. Each locus model was adjusted for cohort, treatment, cohort by treatment interaction, and covariate effects, including linear and quadratic terms for age at diagnosis of type 1 diabetes, duration of type 1 diabetes prior to DCCT entry, sex, HbA1c at DCCT eligibility, and DCCT baseline measures for BMI, mean blood pressure, triglyceride, total cholesterol, HDL cholesterol, and the time-dependent updated HbA1c weighted mean. Indicated haplotypes are compared with the most frequent haplotype pair (CDT/TIC).
P < 0.05.
In multivariate proportional hazards analysis, individuals homozygous for the TIC haplotype compared with individuals with CDT/TIC had a HR of 0.49 (95% CI 0.32–0.75, P = 0.0009) and 0.41 (0.22–0.78, P = 0.006) for development of persistent microalbuminuria and severe nephropathy, respectively (Table 3). These three marker haplotypes explained 1.2% of variability in risk for persistent microalbuminuria and 0.8% for severe nephropathy. It appears that homozygosity for the C allele at the last marker, rs9896208, alone does not confer the substantially lower risk associated with the TIC/TIC haplotype, as the haplotype pair CDC/TIC, which is also homozygous for the C allele at rs9896208, is not distinguishable from CDT/TIC in these models (P = 0.09). The significantly lower risk of the TIC haplotype therefore appears to reside primarily in the homozygosity of the T allele and the insertion allele of rs1800764 and insertion/deletion or other unmeasured variation on that haplotype.
DISCUSSION
Given the previous contradictory or weakly positive cross-sectional studies of the ACE gene polymorphisms, we examined their role in the development of microalbumin excretion and severe nephropathy in the DCCT/EDIC cohort. Because of the extensive linkage disequilibrium within this genomic region, there are relatively few common haplotypes in Caucasians. Using only the insertion/deletion and two SNPs, we are able to capture the genetic variation that is present in the four most common haplotypes occurring at frequencies >5% in Caucasian subjects. This approach is superior to measurement of variation at only a single marker locus (such as the insertion/deletion, on which many of the previous cross-sectional studies are solely based) because it captures the common variation spanning ~25 kb of genomic sequence encompassing the ACE gene. Using this approach, we are less likely to miss an association between polymorphic variation at a gene and nephropathy outcomes if it exists. At the same time, we are spared the task of genotyping all polymorphic variants in each haplotype to capture this variation.
When the ACE polymorphisms were examined against our nephropathy outcomes during a period of up to 17 years of observation, we determined that subjects with certain ACE genotypes had a significantly lower risk in progression to persistent microalbuminuria as well as severe nephropathy, which included subjects with AER >208 μg/min, dialysis, or transplant. The effect of the ACE genotypes persisted even when controlling in multivariate models for known covariates that are associated with microalbumin excretion in the DCCT/EDIC cohort, which specifically included sex, age at diagnosis, diabetes duration, DCCT baseline BMI, mean blood pressure, triglycerides, total cholesterol, HDL cholesterol, time-dependent updated HbA1c weighted mean, cohort, and treatment.
A major quantitative trait loci for circulating ACE activity lies within a 16-kb region between intron 5 and downstream of exon 26 of the ACE gene (15). We speculate that genetic variation at the markers that we have analyzed, or at other loci in strong linkage disequilibrium with these markers, affects ACE enzyme activity, which in turn contributes to the development of diabetic nephropathy. Supporting this hypothesis are data from studies of transgenic diabetic mice engineered to have three copies of the Ace gene with resultant ACE enzyme activity 153% relative to two copy diabetic mice (23). The three copy mice had increased blood pressure and overt proteinuria compared with the two copy mice after 12 weeks of streptozotocin-induced diabetes, supporting the hypothesis that a genetic increase in ACE activity was sufficient to accelerate diabetic nephropathy in animal models (23).
The current study provides more definitive support for the role of ACE polymorphisms in diabetic nephropathy than the many previous cross-sectional studies (18,20). Based on our multivariate models adjusted for covariate effects, the three-marker ACE haplotypes explain 0.8% of variability in persistent microalbuminuria and 0.5% of variability in severe nephropathy. The previous studies may therefore lack statistical power to detect small effects of the ACE insertion/deletion polymorphism on renal outcomes. The pooled odds ratios for elevated urinary albumin excretion for I/I versus I/D and D/D genotypes in a meta-analysis (including different ethnic groups) in type 1 diabetes was 0.72 (95% CI 0.51–1.01) with a borderline significant P value of 0.06 (20). Three type 1 diabetes studies (17,24,25) in that meta-analysis reported odds ratios with 95% CIs excluding unity that indicated a protective effect of the I/I genotype on nephropathy.
The DCCT clinical trial was not initially designed as a genetic study, and to guard against population stratification, a potential source of bias, analyses were limited to subjects self-reported as “white.” In addition, the modest number of severe renal end points limits our power to detect small genetic effects. Despite this, there are many strengths of this study. The DCCT/EDIC cohort represents one of the largest collections of genetic and clinical information on subjects with type 1 diabetes. Biochemical measures were performed in one central laboratory, and the cohort is phenotypically well characterized, with longitudinal prospective clinical data on complication status for up to 17 years. The attrition from the EDIC study has been very low, and clinical data continues to be acquired on >90% of those DCCT participants who took part in EDIC. Finally, our genotyping strategy is superior to the many previous cross-sectional studies that only genotyped one polymorphic variant (i.e., insertion/deletion) and subsequently were unable to capture the common variation spanning the entire ACE gene.
Our findings in the DCCT/EDIC cohort therefore offer definitive evidence that in Caucasians, genetic variation at the ACE gene is associated not only with the onset of microalbuminuria but also with severe diabetic nephropathy in subjects with type 1 diabetes.
Supplementary Material
Acknowledgments
A.P.B. is supported by a Banting and Best Diabetes Center new investigator award. A.D.P. holds a Canada Research Chair in Genetics of Complex Diseases and receives support from Genome Canada and the Premier Research Excellence Award. S.B.B. holds a Senior Investigator Award (Canadian Institutes of Health Research) and receives support from the Network of Centres of Excellence (Canada) in Mathematics (MITACS). This work is supported by National Institutes of Health subcontract (N01-6-2204) and by contracts with the Division of Diabetes, Endocrinology and Metabolic Diseases of the National Institute of Diabetes and Digestive and Kidney Diseases; by the General Clinical Research Centers Program, National Center for Research Resources; and by Genentech through a Cooperative Research and Development Agreement with the National Institute of Diabetes and Digestive and Kidney Diseases.
Footnotes
Additional information for this article can be found in an online appendix at http://diabetes.diabetesjournals.org.
AER, albumin excretion rate; DCCT, Diabetes Control and Complications Trial; EDIC, Epidemiology of Diabetes Interventions and Complications; SNP, single nucleotide polymorphism.
References
- 1.The Diabetes Control and Complications Trial Research Group. The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med. 1993;329:977–986. doi: 10.1056/NEJM199309303291401. [DOI] [PubMed] [Google Scholar]
- 2.Andersen AR, Christiansen JS, Andersen JK, Kreiner S, Deckert T. Diabetic nephropathy in type 1 (insulin-dependent) diabetes: an epidemiological study. Diabetologia. 1983;25:496–501. doi: 10.1007/BF00284458. [DOI] [PubMed] [Google Scholar]
- 3.Krolewski AS, Warram JH, Christlieb AR, Busick EJ, Kahn CR. The changing natural history of nephropathy in type I diabetes. Am J Med. 1985;78:785–794. doi: 10.1016/0002-9343(85)90284-0. [DOI] [PubMed] [Google Scholar]
- 4.Hovind P, Tarnow L, Rossing K, Rossing P, Eising S, Larsen N, Binder C, Parving HH. Decreasing incidence of severe diabetic microangiopathy in type 1 diabetes. Diabetes Care. 2003;26:1258–1264. doi: 10.2337/diacare.26.4.1258. [DOI] [PubMed] [Google Scholar]
- 5.Nordwall M, Bojestig M, Arnqvist HJ, Ludvigsson J. Declining incidence of severe retinopathy and persisting decrease of nephropathy in an unselected population of type 1 diabetes: the Linkoping Diabetes Complications Study. Diabetologia. 2004;47:1266–1272. doi: 10.1007/s00125-004-1431-6. [DOI] [PubMed] [Google Scholar]
- 6.Pambianco G, Zgibor J, Orchard T. Temporal trends in type 1 diabetes (T1D): coronary artery disease (CAD), proliferative retinopathy (PR) and overt nephropathy (ON) (Abstract) Diabetes. 2003;52 (Suppl 1):A40. [Google Scholar]
- 7.Quinn M, Angelico MC, Warram JH, Krolewski AS. Familial factors determine the development of diabetic nephropathy in patients with IDDM. Diabetologia. 1996;39:940–945. doi: 10.1007/BF00403913. [DOI] [PubMed] [Google Scholar]
- 8.Seaquist ER, Goetz FC, Rich S, Barbosa J. Familial clustering of diabetic kidney disease: evidence for genetic susceptibility to diabetic nephropathy. N Engl J Med. 1989;320:1161–1165. doi: 10.1056/NEJM198905043201801. [DOI] [PubMed] [Google Scholar]
- 9.Borch-Johnsen K, Norgaard K, Hommel E, Mathiesen ER, Jensen JS, Deckert T, Parving HH. Is diabetic nephropathy an inherited complication? Kidney Int. 1992;41:719–722. doi: 10.1038/ki.1992.112. [DOI] [PubMed] [Google Scholar]
- 10.The Diabetes Control and Complications Trial Research Group. Clustering of long-term complications in families with diabetes in the Diabetes Control and Complications Trial. Diabetes. 1997;46:1829–1839. [PubMed] [Google Scholar]
- 11.Alhenc-Gelas F, Baussant T, Hubert C, Soubrier F, Corvol P. The angiotensin converting enzyme in the kidney. J Hypertens. 1989;(Suppl 7):S9–S13. doi: 10.1097/00004872-198909007-00003. (discussion S14) [DOI] [PubMed] [Google Scholar]
- 12.Hall JE, Guyton AC, Jackson TE, Coleman TG, Lohmeier TE, Trippodo NC. Control of glomerular filtration rate by renin-angiotensin system. Am J Physiol. 1977;233:F366–F372. doi: 10.1152/ajprenal.1977.233.5.F366. [DOI] [PubMed] [Google Scholar]
- 13.Cambien F, Alhenc-Gelas F, Herbeth B, Andre JL, Rakotovao R, Gonzales MF, Allegrini J, Bloch C. Familial resemblance of plasma angiotensin-converting enzyme level: the Nancy Study. Am J Hum Genet. 1988;43:774–780. [PMC free article] [PubMed] [Google Scholar]
- 14.Doria A, Warram JH, Rich SS, Krolewski AS. Angiotensin I-converting enzyme (ACE): estimation of DNA haplotypes in unrelated individuals using denaturing gradient gel blots. Hum Genet. 1994;94:117–123. doi: 10.1007/BF00202855. [DOI] [PubMed] [Google Scholar]
- 15.Soubrier F, Martin S, Alonso A, Visvikis S, Tiret L, Matsuda F, Lathrop GM, Farrall M. High-resolution genetic mapping of the ACE-linked QTL influencing circulating ACE activity. Eur J Hum Genet. 2002;10:553–561. doi: 10.1038/sj.ejhg.5200847. [DOI] [PubMed] [Google Scholar]
- 16.Rigat B, Hubert C, Alhenc-Gelas F, Cambien F, Corvol P, Soubrier F. An insertion/deletion polymorphism in the angiotensin I-converting enzyme gene accounting for half the variance of serum enzyme levels. J Clin Invest. 1990;86:1343–1346. doi: 10.1172/JCI114844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marre M, Bernadet P, Gallois Y, Savagner F, Guyene TT, Hallab M, Cambien F, Passa P, Alhenc-Gelas F. Relationships between angiotensin I converting enzyme gene polymorphism, plasma levels, and diabetic retinal and renal complications. Diabetes. 1994;43:384–388. doi: 10.2337/diab.43.3.384. [DOI] [PubMed] [Google Scholar]
- 18.Fujisawa T, Ikegami H, Kawaguchi Y, Hamada Y, Ueda H, Shintani M, Fukuda M, Ogihara T. Meta-analysis of association of insertion/deletion polymorphism of angiotensin I-converting enzyme gene with diabetic nephropathy and retinopathy. Diabetologia. 1998;41:47–53. doi: 10.1007/s001250050865. [DOI] [PubMed] [Google Scholar]
- 19.Staessen JA, Wang JG, Ginocchio G, Petrov V, Saavedra AP, Soubrier F, Vlietinck R, Fagard R. The deletion/insertion polymorphism of the angiotensin converting enzyme gene and cardiovascular-renal risk. J Hypertens. 1997;15:1579–1592. doi: 10.1097/00004872-199715120-00059. [DOI] [PubMed] [Google Scholar]
- 20.Tarnow L, Gluud C, Parving HH. Diabetic nephropathy and the insertion/deletion polymorphism of the angiotensin-converting enzyme gene. Nephrol Dial Transplant. 1998;13:1125–1130. doi: 10.1093/ndt/13.5.1125. [DOI] [PubMed] [Google Scholar]
- 21.The DCCT Research Group: The Diabetes Control and Complications Trial (DCCT): design and methodologic considerations for the feasibility phase. Diabetes. 1986;35:530–545. [PubMed] [Google Scholar]
- 22.Rieder MJ, Taylor SL, Clark AG, Nickerson DA. Sequence variation in the human angiotensin converting enzyme. Nat Genet. 1999;22:59–62. doi: 10.1038/8760. [DOI] [PubMed] [Google Scholar]
- 23.Huang W, Gallois Y, Bouby N, Bruneval P, Heudes D, Belair MF, Krege JH, Meneton P, Marre M, Smithies O, Alhenc-Gelas F. Genetically increased angiotensin I-converting enzyme level and renal complications in the diabetic mouse. Proc Natl Acad Sci U S A. 2001;98:13330–13334. doi: 10.1073/pnas.231476798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marre M, Jeunemaitre X, Gallois Y, Rodier M, Chatellier G, Sert C, Dusselier L, Kahal Z, Chaillous L, Halimi S, Muller A, Sackmann H, Bauduceau B, Bled F, Passa P, Alhenc-Gelas F. Contribution of genetic polymorphism in the renin-angiotensin system to the development of renal complications in insulin-dependent diabetes: Genetique de la Nephropathie Diabetique (GENEDIAB) study group. J Clin Invest. 1997;99:1585–1595. doi: 10.1172/JCI119321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Demurov LM, Chistiakov DA, Chugunova LA, Shamkhalova MSh, Shestakova MV, Anokhin EE, Kondrat’ev IaIu, Dedov II, Nosikov VV. Polymorphism of the insertion/deletion type in the angiotensin-converting enzyme gene in normal subjects and among patients with vascular complications. Mol Biol (Mosk) 1997;31:59–62. (article in Russian) [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.