

Abstract
Combinatorial regulation of transcription involves binding of transcription factors to DNA as well as protein–protein interactions between them. In this paper, we demonstrate the existence of a mutual transcriptional antagonism between the thyroid hormone receptor (TR) and the cyclic AMP response element binding protein (CREB), which involves a direct association of both transcription factors. TR inhibits transcriptional activity of CREB and represses activation of cAMP response element (CRE)-containing promoters. TR does not bind to the CRE in vitro, but in vivo the liganded receptor is tethered to the promoter through protein–protein interactions. In turn, expression of CREB reduces TR-dependent transcriptional responses. The association of TR with CREB inhibits the ability of protein kinase A to phosphorylate CREB at Ser133, and leads to a reduction in the ligand-dependent recruitment of the p160 coactivators by TR. These results indicate the existence of a transcriptional cross-talk between CREB and TR signalling pathways, which can have important functional consequences.
Keywords: CREB/CREB phosphorylation/thyroid hormone receptor/transcriptional repression
Introduction
The actions of the thyroid hormone triiodothyronine (T3) are mediated by nuclear receptors (TRs), which normally act by binding to DNA response elements (TREs) located in regulatory regions of target genes (Zhang and Lazar, 2000). The nuclear receptors have a modular structure with several regions (A/B, C, D and E). The C region contains the DNA binding domain (DBD), composed of two zinc fingers, and the E domain is responsible for ligand binding and dimerization. Transcriptional regulation by the receptors is achieved through autonomous activation functions (AFs): a constitutive N-terminal AF-1 in the A/B region, and a ligand-dependent AF-2 located in the C-terminal E region of the ligand binding domain (LBD) (Aranda and Pascual, 2001). TRs bind corepressors, found within multicomponent complexes, that have histone deacetylase activity (Guenther et al., 2000; Li et al., 2000). Upon ligand binding the receptors undergo a conformational change, which allows the recruitment of multiple coactivator complexes in an AF-2-dependent manner (Glass and Rosenfeld, 2001; McKenna and O’Malley, 2002). Some of these proteins are chromatin remodelling factors, some (such as CBP/p300 and the p160 coactivators) possess histone acetylase activity, and others may interact directly with the basic transcriptional machinery (Ito and Roeder, 2001; Rachez and Freedman, 2001).
Nuclear receptors can also modulate gene expression by mechanisms that are independent of DNA binding and that instead appear to involve protein–protein interactions. Thus, they can alter the expression of genes that do not contain a hormone response element through positive or negative interference with the activity of other transcription factors, a mechanism generally referred to as transcriptional cross-talk. For example, some nuclear receptors can negatively regulate target gene promoters that carry AP-1 or NF-κB binding sites, without binding to these DNA elements themselves (Desbois et al., 1991; Zhang et al., 1991; Pfhal, 1993; Saatcioglu et al., 1997; Gottlicher et al., 1998; Karin and Chang, 2001). It was originally proposed that the receptors contact the basic leucine zipper region of c-Jun or the rel homology domain of the p65 subunit of NF-κB directly, and that this interaction inhibits binding to their corresponding cognate sites (Yang-Yen et al., 1990; McKay and Cidlowsy, 1998). However, using chromatin immunoprecipitation (ChIP) assays it has been recently reported that these protein– protein interactions allow recruitment of receptors to the promoter without altering c-Jun or NF-κB occupancy (Nissen and Yamamoto, 2000; Rogatsky et al., 2001). It has also been reported recently that corepressor complexes inhibit Jun-N-terminal kinase (JNK) activation through the associated GPS2 subunit and thus could potentially provide a mechanism for hormone-mediated antagonism of AP-1 function (Zhang et al., 2002).
We have demonstrated previously that T3 represses transcription of the pituitary-specific transcription factor GHF-1/Pit-1, which does not contain a TRE, by a mechanism that involves transcriptional interference with two cyclic AMP response elements (CREs) present in its promoter. The hormone reduces basal levels of GHF-1/Pit-1 promoter activity and antagonizes its response to cyclic AMP and the tumor promoter TPA (12-O-tetradecanoylphorbol-13-acetate) (Sánchez-Pacheco et al., 1995). Repression of the pituitary thyroid-stimulating hormone α (TSHα) gene expression by T3 represents a feedback mechanism for controlling thyroid hormone levels. In this case, two CREs present in the proximal promoter are also critical regulatory elements for repression by T3 (Tagami et al., 1999). Therefore, a transcriptional cross-talk between TRs and transcription factors that bind to the CREs appear to be important for the inhibitory effect of T3 on the expression of genes that play a key role in pituitary function.
CREB (cyclic AMP response element binding protein) is the main transcriptional regulator which binds to the CREs (Montminy and Bilezitjian, 1987; Mayr and Montminy, 2001). CREB belongs to the family of transcription factors characterized by a C-terminal basic leucine-zipper (b-Zip) domain responsible for dimerization and binding to DNA. The N-terminus contains a kinase-inducible domain (KID) and a glutamine-rich domain termed Q2, and both are necessary for full CREB activation in response to external stimuli. Cyclic AMP is thought to transactivate the CREs via phosphorylation of CREB at Ser133 in the KID domain by protein kinase A (PKA) (González and Montminy, 1989). The same residue can be phosphorylated in response to multiple kinases, which are activated by different signalling pathways (Shaywitz and Greenberg, 1999). Phosphorylated CREB then binds to its coactivator CBP (CREB-binding protein), which facilitates unwinding of DNA and interacts with the basal transcriptional machinery (Chrivia et al., 1993; Arias et al., 1994). The competition for limiting concentrations of CBP in cells could be involved in the antagonism between nuclear receptors and CRE-mediated transcription, where the limiting amount of CBP factors would be partitioned between the liganded receptors and phosphorylated CREB. A similar mechanism could, at least in part, be responsible for the antagonistic actions of nuclear receptor ligands on AP-1 activity, based on the requirement of CBP for both classes of transcription factors (Kamei et al., 1996).
In this work we have examined the mechanisms by which T3 induces repression of CRE-containing promoters. We have found that T3 represses CREB phosphorylation at Ser133, as well as the transcriptional activity of CREB. A direct protein–protein interaction between TR and CREB appears to be involved in the transcriptional antagonism observed. TR associates with CREB, and both the leucine zipper of CREB as well as the DNA binding domain (DBD) of TR are required for this interaction. The association of TR with CREB leads to a reduced ability of PKA to phosphorylate CREB in vitro, which could be responsible for the reduced cellular phosphorylation of CREB. In addition, interaction with CREB leads to a reduction in the ligand-dependent recruitment of the p160 coactivator ACTR by TR. Finally, overexpression of CREB reduces ligand-dependent transcriptional activation of a TRE-containing promoter, demonstrating the existence of a mutual antagonism between the CREB and TR signalling pathways that could have important implications in transcriptional responses of cells expressing both transcription factors.
Results
T3 antagonizes CRE-mediated transcription in pituitary cells by a mechanism that does not involve titration of CBP
To examine the effect of T3 on CRE-mediated transcriptional responses, transient transfection assays were carried out in pituitary GH4C1 cells, which express high endogenous levels of TRs (Figure 1A). The GHF-1–CAT construct contains two CREs at positions –197 and –153 (McCormick et al., 1990), the ICER–CAT plasmid possesses four CREB-binding motifs (Molina et al., 1993), and the SOM–CAT construct contains a somatostatin promoter fragment with one CRE motif (Montminy et al., 1986). Activity of these promoters was stimulated by forskolin, and to a lesser extent by TPA. Our previous observations with the GHF-1 promoter showed that the CREs mediate this activation, as deletion of these motifs abolished the response to both stimulators (Sánchez-Pacheco et al., 1995). Additionally, a maximal induction was found with the combination of forskolin and TPA that acted synergistically to induce CRE-mediated transcription. Figure 1A shows that treatment with T3 was able to markedly reduce the response of the different CRE-containing promoters, even under conditions of maximal stimulation.

Fig. 1. T3 represses CRE-mediated transactivation, irrespective of cellular CBP levels. (A) GH4C1 cells were used for transient transfection with the indicated CRE-containing constructs. Assays were carried out in cells incubated for 48 h with T3, and with TPA or forskolin (FK) during the last 4 h. CAT activity was quantitated and is expressed as the fold induction over the values obtained in the corresponding untreated cells. (B) The GHF-1 promoter construct (10 µg) was cotransfected into GH4C1 cells, with 10 µg of an expression vector for CBP or with the same amount of a non-coding vector. CAT activity was determined in cells treated as in (A).
Expression of CRE-mediated transcription by T3 could be a consequence of competition for limiting amounts of CBP, a common coactivator for TRs and CREB. If this hypothesis were true, overexpression of CBP should reverse transrepression by T3. However, as shown in Figure 1B, this is not observed in GH4C1 cells, in which transfection with an expression vector for CBP did not further induce GHF-1 promoter activity and had little, if any, influence on the inhibitory effect of T3.
T3 inhibits CREB phosphorylation
To analyse whether a reduction in CREB phosphorylation might underlie the T3-dependent repression of the CRE-containing promoters, the amount of phosphorylated CREB (P-CREB) in GH4C1 cells was assessed by western blot using an antibody specific for the Ser133-phosphorylated form of CREB. As shown in Figure 2A, incubation with forskolin for 15 min caused a marked increase in the levels of P-CREB, and T3 was able to reduce P-CREB levels significantly. This effect was already observed after 2 h of incubation with the hormone, and was maintained after 24 h of treatment. T3 also reduced the low basal levels of P-CREB observed in the untreated cells. In contrast, the amount of total CREB remained unaltered upon treatment with either T3 or forskolin, as analysed with an antibody recognizing total CREB.

Fig. 2. T3 inhibits CREB phosphorylation. (A) Western blot analysis of GH4C1 cell extracts with phospho-CREB (P-CREB) and total CREB antisera after treatment with T3 for the indicated time periods and forskolin (FK) for the last 15 min. The upper panel shows quantitation of the P-CREB blot. (B) The cells were incubated with T3 for 24 h and/or FK during the last 15 min, fixed, and analysed by indirect immunofluorescence with the P-CREB antibody. Background (Bkg) immunofluorescence obtained in the absence of the antibody is also shown. (C) Western blot analysis with P-CREB and total CREB antisera in GH4C1 cells treated with FK for 15 min, or with TPA for the indicated time periods. In (D) the cells were incubated for T3 for 24 h and with TPA for 15 min. Immunofluorescence assays with the P-CREB antibody under the same conditions are shown in (E).
Immunofluorescence assays were used to determine whether changes in nuclear P-CREB levels were detectable in GH4C1 cells treated with T3 (Figure 2B). P-CREB staining was seen in the nuclei of the untreated pituitary cells, and this staining was strongly reduced after incubation with T3. On the other hand, incubation with forskolin caused a strong nuclear labelling, and T3 was able to reduce P-CREB staining in the nuclei. Interestingly, some cytoplasmic staining could be observed in hormone-treated cells. By comparison, immunohistochemistry performed with the antibody recognizing total CREB did not show changes after the different treatments (data not shown).
Since TPA increased CRE-mediated transcription, we assessed the possibility that it could also induce CREB phosphorylation in GH4C1 cells. As shown in Figure 2C, TPA induced a strong and sustained increase of P-CREB levels in pituitary cells. In fact, as examined after 15 min of incubation, P-CREB levels were similar in cells treated with forskolin and TPA, despite the lower transcriptional activity of the tumor promoter (Figure 1). Figure 2D illustrates that T3 was also able to inhibit the levels of P-CREB significantly in cells incubated with TPA. That TPA increases nuclear P-CREB levels and T3 reduces this response was also observed using immunofluorescence with the anti-P-CREB antibody (Figure 2E). In addition, incubation with ligands of tyrosine kinase receptors such as IGF-1, EGF or bFGF induced CREB phosphorylation in GH4C1 cells, and incubation with T3 also reduced the response to these growth factors (not shown).
T3 represses CREB transcriptional activity
To detect the direct effects of T3 on CREB-mediated transcription, a GAL–CREB expression plasmid was cotransfected into GH4C1 cells with a reporter plasmid containing binding motifs for GAL4. GAL–CREB consists of the GAL4-DBD fused to full-length CREB. As expected, treatment with forskolin increased GAL–CREB-dependent gene expression (Figure 3A). TPA also stimulated GAL–CREB activity, albeit with a reduced potency, and a maximal induction was again found in the presence of both compounds together. In addition, T3 was able to strongly reduce the response to forskolin, TPA and the combination of the two, thus indicating that T3 directly reduces CREB-mediated transcription in GH4C1 cells. Similar experiments were performed with a mutant GAL–CREB hybrid in which Ser133 was replaced by an alanine. This mutation almost totally abolished induction by the stimuli tested here. Furthermore, activity of a plasmid expressing only the GAL-DBD was not significantly affected by forskolin, TPA or T3.

Fig. 3. The thyroid hormone receptor inhibits CREB transcriptional activity. In (A), GH4C1 cells were transfected with 5 µg Gal-DBD alone (Gal), or fused to either native CREB (Gal-CREB) or CREB mutated in Ser133 (Gal-CREBAla133). These constructs were cotransfected with 10 µg of the reporter plasmid 4xUAS-LUC, and luciferase activity was determined in cells treated with T3 for 48 h and/or TPA and forskolin (FK) during the last 4 h. Reporter activity is expressed as the fold induction over the values obtained in the corresponding untreated cells. (B) HeLa cells were cotransfected with 3 µg of 4xUAS-LUC and 1.5 µg of Gal-CREB, either alone or in combination with an expression vector for TR (1 µg). The Gal-DBD plasmid was also used as a control. Luciferase activity was determined as in (A), after incubation with T3 and/or FK and TPA.
In order to analyse whether the antagonistic effect of TR on CREB-mediated transcription was also observed in other cell types, the GAL–CREB fusion construct was transfected into HeLa cells (Figure 3B). TPA was a better stimulator of the transcriptional activity of CREB than forskolin, which was a very poor stimulator in these cells, although it increased the response to the phorbol ester. These cells express low endogenous levels of TRs and, accordingly, incubation with T3 had no effect on CREB transcriptional activity. In contrast, in cells transfected with TR, T3 caused a profound reduction of CREB-mediated transcription. Interestingly, even in the absence of ligand, expression of TR produced a decrease in the response to TPA or forskolin plus TPA. On the other hand, TR did not significantly alter the activity of the GAL-DBD plasmid (data not shown). Therefore, TRs appear to be strong transrepressors of CREB transcriptional activity, irrespective of cell type.
The inhibitory effect of T3 is sensitive to TSA
Repression of AP-1-mediated activation by TR is blocked by trichostatin A (TSA) (Nissen and Yamamoto, 2000), a general inhibitor of histone deacetylases (HDACs). In contrast, glucocorticoid receptor (GR) regulation of AP-1 activity, at the same response element, is resistant to the inhibitor (Rogatsky et al., 2001). To analyse whether changes in acetylation of histones or other protein targets can modulate the antagonism of CREB-mediated transcription by TR, the inhibitory effect of T3 on GHF-1 promoter activity in GH4C1 cells was also tested in the presence of TSA. As shown in Figure 4A, TR-mediated inhibition appears to be sensitive to TSA, since T3 was unable to repress promoter stimulation by forskolin and TPA in the presence of the inhibitor. Furthermore, as shown in Figure 4B, repression of CREB transcriptional activity by T3, analysed using the GAL–CREB chimera, was also significantly reversed in cells incubated with TSA.

Fig. 4. Trichostatin A (TSA) modulates the inhibitory effect of T3 on CREB-dependent transcription. In (A), GH4C1 cells were transfected with the GHF-1–CAT construct and incubated for 48 h with T3 in the presence and absence of 200 nM TSA. When indicated, the cells were also treated with TPA and forskolin (FK) during the last 4 h. CAT activity is expressed as the fold induction over the values obtained in the untreated cells. In (B), GH4C1 cells were transfected with Gal-DBD alone (Gal) or with Gal–CREB. Luciferase activity was determined as in (A), after incubation with T3, TSA and/or FK and TPA.
CREB interacts with TR
Transrepression by nuclear receptors can involve protein– protein interactions with other transcription factors. To address whether inhibition of CREB-mediated transcription by TR could result from the association of both factors, pull-down assays with [35S]CREB and the fusion protein glutathione S-transferase (GST)–TR were carried out. As shown in Figure 5A, [35S]CREB bound specifically to GST–TR with similar strength in the presence and absence of T3. A series of mutants of TR were used to delineate the protein domains involved in the interaction with CREB. Figure 5B shows that deletion of the A/B and C domains of the receptor (residues 1–120) abolished this interaction. The involvement of the C domain, which contains the receptor DBD, was proved by using different DBD mutants. When Cys51, a conserved residue in the first zinc finger that participates in the coordination of the zinc ion, was substituted by an arginine, the association between TR and CREB was strongly reduced. Mutation of lysines 72 and 112 at the base of the first and second zinc fingers, respectively, also reduced interaction. In contrast, mutation of a single lysine at position 101 at the tip of the first zinc finger did not affect the association with CREB (data not shown).

Fig. 5. In vitro interaction of TR with CREB. (A) Pull-down assay performed with 1 µg GST–0 or GST–TR and 10 µl of 35S-labelled CREB in the presence and absence of 1 µM T3. Input represents 20% of the protein used. (B) Schematic representation of TR showing the different functional domains. The wild-type receptor (wt) and the indicated TR mutants were used in pull-down assays with [35S]CREB. (C) Representation of CREB domains that were used in the interaction assay with [35S]TR in the presence and absence of T3. (D) In vivo interaction of TR with CREB. GH4C1 cells were transfected with HA-TR and CREB, and were treated with T3 for 24 h and forskolin for the last 15 min. Immunoprecipitates (1.2 mg) with 1 µg anti-HA antiserum (α-HA) or with a control serum were subjected to western blot analysis together with 10% of the cell extract (input). A non-specific band (n.s.) appearing in the immunoprecipitates is labelled with an asterisk.
The domain of CREB involved in association with TR was also mapped using [35S]TR and different CREB fragments fused to GST (Nakajima et al., 1997) (Figure 5C). Neither the KID domain, nor the glutamine-rich transcriptional activation domain interacted with the receptor. However, the leucine zipper of CREB was sufficient for a significant interaction with TR, which was again observed both in the presence and absence of T3.
To prove that CREB and TR can form a complex in mammalian cells, co-immunoprecipitation assays were also performed. In Figure 5D, GH4C1 cells were transfected with expression vectors for CREB and a hemagglutanin (HA)-tagged receptor. CREB was detected by western blot in immunoprecipitates obtained with an anti-HA antibody, demonstrating that TR and CREB can interact in the cells. Interaction was weak or absent in the untreated cells, whereas a clear signal was obtained in cells incubated with T3 and/or forskolin. This suggests that, in contrast with the results obtained in vitro, interaction with CREB in vivo is increased in the presence of T3.
TR inhibits in vitro phosphorylation of CREB by PKA
The interaction with TR might induce a conformational change in CREB that could alter the accessibility of Ser133 to kinases. To test this possibility, in vitro phosphorylation of recombinant purified CREB by PKA was performed with excess amounts of enzyme and [32P]ATP in the presence and absence of GST–TR. Figure 6A shows that CREB is phosphorylated by the enzyme (lane 5), and that 32P incorporation was not affected in the presence of GST alone (lane 2). TR was also a target of PKA (lane 1), which is known to phosphorylate the receptor at several residues (Goldberg et al., 1988; Leitman et al., 1996; Tzagarakis-Foster and Privalsky, 1998). Interestingly, when both proteins were incubated together, phosphorylation of GST–TR was not affected but P32 incorporation into CREB was strongly reduced. This occurred both in the presence and absence of T3 (Figure 6A, lanes 3 and 4). That the interaction of TR with CREB is required for inhibition of CREB phosphorylation is illustrated in Figure 6B, in which the potency of native TR with that of the DBD mutants was compared. Whereas TR reduced CREB phosphorylation significantly, the TR mutants C51G and K72,112R, which did not interact with CREB, did not alter phosphorylation. In contrast, the DBD mutant K101R was as potent as the wild-type receptor in reducing 32P incorporation. This mutant also interacts normally with CREB in in vitro pull-down assays.

Fig. 6. Association with TR inhibits CREB phosphorylation by PKA. (A) Recombinant CREB (50 ng) was incubated with [γ-32P]ATP in the presence of PKA. Phosphorylation assays were performed in the presence and absence of GST–TRα (100 ng) or the same amount of GST alone. Labelled proteins are shown using arrows. In (B), the wild-type receptor (wt) and the indicated TR mutants were used in the phosphorylation assays.
T3 causes TR recruitment to the CRE-containing promoter in vivo, but does not alter occupancy by total CREB
To analyse binding of CREB to the CRE sites in vivo, we carried out ChIP assays in GH4C1 cells (Figure 7). Primers amplifying a GHF-1 promoter fragment containing two CRE motifs were used. Promoter occupancy by CREB was constitutive and was not altered by treatment with T3 or forskolin (Figure 6A); therefore, association with TR does not appear to reduce binding of total CREB to its cognate site. It has been shown that nuclear receptors can repress AP-1-activated transcription by tethering to the bound AP-1 factor (Rogatsky et al., 2001). Likewise, although TR is unable to bind the CRE motifs directly (data not shown), it could be targeted to the GHF-1 promoter by tethering to the bound CREB by protein–protein interactions. As a direct test of in vivo recruitment of TR to the promoter, ChIP assays with anti-TR antibodies were also performed. As shown in Figure 7A, TR was recruited to the GHF-1 promoter in the presence of T3, whereas forskolin did not induce occupancy. An antibody against tubulin, used as a negative control, did not precipitate the GHF-1 promoter under the same conditions. Figure 7B shows the results obtained after quantification by real-time PCR of ChIP assays performed after different time periods of incubation with T3. Normalized to control tubulin antibody, the TR and CREB antibodies yielded a 9- and 60-fold enrichment, respectively, of the GHF-1-containing fragment in control cells. Incubation with T3 caused a significant and sustained increase in the amount of TR bound to the promoter (>10-fold between 4 and 24 h). This increase was significantly reversed after 36 h of hormone treatment. In contrast with results obtained with the TR antibody, incubation with T3 or forskolin caused only minimal changes in promoter occupancy by CREB.

Fig. 7. In vivo occupancy of the GHF-1 promoter by TR and CREB. (A) Soluble chromatin was extracted from GH4C1 cells treated with T3 for 24 h or with forskolin for the last 15 min, and immunoprecipitated with antibodies against TR, CREB or tubulin. DNA was amplified using primers that cover the GHF-1 promoter. A representative PCR at 23 cycles of amplification is shown. (B) ChIP assays from the chromatin of cells incubated with T3 for the indicated time periods and with forskolin for 15 min were quantified using real-time PCR. PCR signals obtained in the corresponding untreated cells were arbitrarily set to 1 and values were calculated compared with this. Each data point corresponds to two to four independent values.
Interaction with TR is required for repression of CREB transcriptional activity by T3
To test whether the association of the receptor with CREB was required for T3-mediated transrepression, transcriptional activity of CREB was determined in HeLa cells expressing either the wild-type receptor or the C51G mutant. Whereas, in agreement with the data presented in Figure 3B, T3 strongly reduced basal and induced transcriptional activity of the Gal–CREB construct in cells expressing the native receptor, the hormone had little effect in cells expressing the TR mutant, which does not interact with CREB (Figure 8). These results show that the association of both factors appears to be important for transrepression by T3.
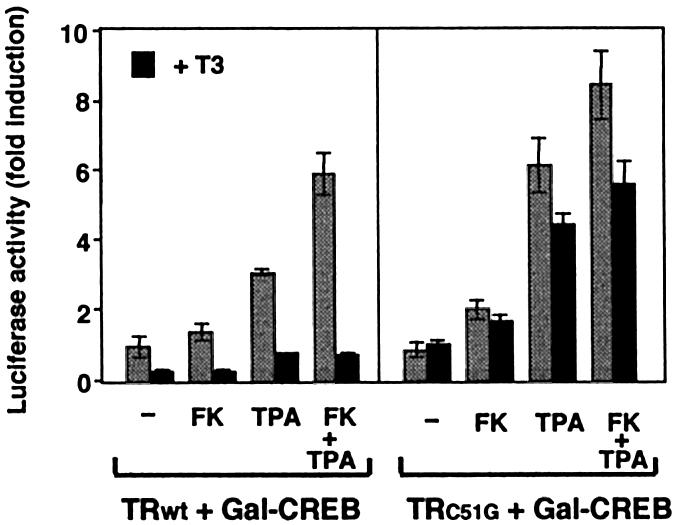
Fig. 8. Mutant TRC51G that does not interact with CREB does not repress CREB transcriptional activity. Reporter activity was determined in HeLa cells transfected with Gal–CREB in combination with an expression vector for native TR or the C51G mutant. Luciferase activity was determined as in Figure 3, after incubation with T3 and/or FK and TPA.
CREB represses TR-mediated transactivation
In order to examine whether the transcriptional antagonism between CREB and TR is mutual, we also analysed the effect of CREB on the activity of a reporter plasmid containing a consensus palindromic TRE. As shown in Figure 9A, T3 stimulated the activity of this construct in GH4C1 cells, and transfection of CREB significantly decreased T3-mediated transactivation. Interestingly, transfection of the transcription factor ICER, which is essentially composed of a b-Zip, had a similar effect. This suggests that the equivalent domain of CREB, which is responsible for interaction with the receptor, is sufficient for the observed antagonism. That the repressive effect of CREB also occurs in HeLa cells is shown in Figure 8B (right panel). In these cells, which express low receptor levels, T3 did not increase reporter activity. However, activation of the TRE-containing plasmid was found after transfection with TR, and coexpression of CREB strongly reduced this response. To analyse whether non-phosphorylatable CREB inhibited TR-dependent transcription, the receptor was also cotransfected with the CREAla133 mutant. It can be observed that mutant CREB was as potent as the native transcription factor in repressing the response to T3.

Fig. 9. CREB reduces coactivator recruitment by T3 and antagonizes T3-mediated transactivation. (A) In the left panel, GH4C1 cells were transfected with 2 µg of the reporter plasmid MMTV-TREpal-LUC, and 2 µg of vectors for CREB or ICERγI. In the right panel HeLa cells were transfected with the reporter plasmid and 2 µg of TRα, in the presence and absence of 2 µg of vectors coding either native CREB or the CREBAla133 mutant. Cells were treated with T3 for 24 h and reporter activity was determined. (B) Pull-down assays were performed with in vitro translated TR (5 µl) and GST–ACTR or GST–SMRT (500 ng) in the presence and absence of recombinant CREB, and with and without T3 (1 µM). Quantitation of the radioactivity in each band is shown in the lower panel.
Interaction with CREB reduces coactivator recruitment by TR
Association of TR with CREB could trigger a change in receptor conformation that could affect recruitment of coregulators. This possibility was explored by pull-down assays performed with [35S]TR and GST fusions of the corepressor SMRT or the coactivator ACTR, in the presence and absence of purified CREB. As shown in Figure 8B, no significant binding of GST alone to 35S-labelled TR was observed. In addition, we did not observe any interaction between [35S]CREB and GST–ACTR (data not shown). However, [35S]TR was specifically retained by GST–ACTR immobilized in glutathione agarose beads, and this binding was increased significantly by the addition of T3. The addition of CREB caused a significant reduction in the amount of ACTR bound to the receptor. As expected, the corepressor SMRT bound strongly to TR in the absence of T3, and was released from the receptor upon hormone incubation. In contrast to the observed decrease in coactivator binding, CREB did not alter the association of TR with the corepressor.
Discussion
Nuclear receptors can repress gene transcription in a ligand-dependent manner by interfering with the activity of other transcription factors. In this work we have demonstrated that TR can inhibit CRE-dependent gene expression without binding to this DNA motif. We have observed that T3 represses the activity of different CRE-containing promoters, which in pituitary cells are stimulated in a synergistic manner by cAMP and phorbol esters. This repression is not restricted to this cell type, since it also occurs in other TR-expressing cell types. Furthermore, in cells that do not express the receptor transfection of TR can repress CRE-mediated transcription, even in the absence of ligand (M.Méndez-Pertuz and A.Aranda, unpublished observations).
It has been proposed that competition for common transcriptional mediators could be involved in the transcriptional antagonism between nuclear receptors and the AP-1 or NF-κB factors. This was suggested by the finding that the inhibitory effects of retinoic acid and glucocorticoid receptors on AP-1-dependent activation could be significantly reversed by expression of CBP or p300 (Kamei et al., 1996). However, overexpression of CBP did not abolish the repressive effect of T3 on CRE sites, showing that competition for limiting amounts of CBP does not appear to be responsible for repression. In agreement with these findings, glucocorticoids have recently been demonstrated to maintain their repressive capacity at NF-κB and AP-1 sites, irrespective of the coactivator levels in the cells, suggesting the existence of alternative mechanisms for this transcriptional antagonism (De Bosscher et al., 2000, 2001). In contrast, and similar to the results obtained with AP-1–TR antagonism (Nissen and Yamamoto, 2000), the inhibitory effect of T3 was significantly blocked by TSA, an HDAC inhibitor, showing that changes in histone acetylation are involved in TR–CREB antagonism. Interestingly, it has very recently been shown that HDAC1 associates with, and blocks, CREB phosphorylation via a stable interaction with the PP1 phosphatase (Canettieri et al., 2003).
Our results show that in pituitary cells, incubation with T3 represses CREB transcriptional activity as measured in a GAL–CREB system. As in other cell types (Brindle et al., 1995; Seternes et al., 1999), phorbol esters were less efficient than cAMP in stimulating CREB activity, but both compounds cooperated to produce a strong stimulation that was markedly reduced by T3. A difference between the results obtained in GH4C1 cells and in HeLa cells was that in HeLa cells, forskolin was only effective at inducing CREB transactivation when combined with the tumour promoter, and in these cells, which express low amounts of TRs, T3 was unable to reduce CREB transcriptional activity. However, expression of TR in HeLa cells reduced the response to both compounds even in the absence of ligand, and T3 caused a drastic decrease in both basal and stimulated CREB transcriptional activity.
As activation of CREB is caused by phosphorylation at Ser133, it was not surprising that T3 inhibited CREB phosphorylation in GH4C1 cells. Not only forskolin, but also TPA and several growth factors elicited Ser133 phosphorylation, and T3 was able to inhibit the response to the different stimuli. It is interesting that even though the phorbol ester and growth factors induced comparable levels of CREB phosphorylation, forskolin was much more potent in activating CREB-mediated transcription. This is compatible with results obtained in other cell types (Mayr et al., 2001).
In the case of the transcriptional repression of AP-1 and NF-κB-dependent gene expression by nuclear receptors, the binding motifs for those factors can act as ‘tethering’ response elements at which the transcription factor binds to the element, and the receptor associates with the transcription factor through protein–protein interactions (Gottlicher et al., 1998). Our work shows that this appears to be the mechanism by which TR can repress transcription of genes containing CREs, as the receptor does not bind directly to the CRE but can interact with CREB, which binds to this site. Indeed, with ChIP assays, we have found that T3 induces recruitment of TR to the GHF-1 promoter that contains two CREs, but does not contain a TRE. ChIP assays have also shown that in vivo, CREB occupancy of the CRE sites is not altered in T3-treated cells. These results are similar to those obtained in other models of transcriptional cross-talk, in which the interaction of receptors with c-Jun or the p65 subunit of NF-κB does not affect in vivo promoter occupancy by these transcription factors (Nissen and Yamamoto, 2000; Rogatsky et al., 2001).
We have also found that TR associates with CREB both in vivo and in vitro, and that this interaction in cells is increased in the presence of T3. Unexpectedly, the hormone did not influence the in vitro binding of TR to CREB. It is possible that the concentrations of TR and CREB used in the in vitro assays are sufficient to detect interactions in the absence of T3. It has been also suggested that the receptors used in these assays could be in an active conformation, even in the absence of ligand (Hong et al., 1996). Additionally, it should be noted that the unoccupied receptor can repress CREB-induced transcription when overexpressed in cells. It is then likely that, at physiological levels, the interaction between TR and CREB is labile in vivo in the absence of hormone, and that additional factors are needed to enhance this interaction and to provoke the T3-dependent repression of CREB transcriptional activity.
The b-Zip of CREB and the DBD of TR are involved in the association between both factors, as the leucine zipper of CREB was sufficient to interact with the receptor, and mutations in TR that alter the structure of the DBD dramatically reduced its binding to CREB. The b-Zip is a powerful dimerization domain, highly conserved in a large family of transcription factors, and the DBD is also the most conserved domain in the nuclear receptor superfamily. This suggests that the interaction of nuclear receptors with b-Zip domains could be a rather general phenomenon. In agreement with this hypothesis, c-Jun (Schule et al., 1990; Zhou et al., 1999; Lin et al., 2002), the hematopoietic protein p45/NF-E2 (Cheng et al., 1997), the Epstein–Barr virus-encoded BZLF-1 protein (Pfitzner et al., 1995) or GT198, a recently identified coactivator (Ko et al., 2002), have been shown to contact different nuclear receptors through this domain.
Since PKA plays a key role in cAMP-mediated transcriptional responses, we analysed the possibility that TR could affect phosphorylation of CREB by this kinase. The results obtained demonstrated that the receptor did indeed strongly repress PKA-mediated CREB phosphorylation. In contrast, phosphorylation of TR, which is also a direct target of this kinase (Tzagarakis-Foster and Privalsky, 1998) was not affected in the presence of CREB. A protein–protein contact of TR with CREB was also required for inhibition of in vitro CREB phosphorylation, since mutant receptors that show a markedly reduced ability to interact with this factor failed to reduce CREB phosphorylation by PKA in vitro. These results suggest that association with TR induces a conformational change in CREB, which makes the KID domain inaccessible to the kinase and can result in the inhibition of CREB phosphorylation observed in the cells. This structural change could also affect accessibility of the transcription factor to other kinases, as suggested by the finding that T3 also reduces the response to TPA or growth factors, which elicit CREB phosphorylation through a different signalling pathway. A TR mutant that did not interact with CREB was unable to repress the transcriptional activity of the latter, showing that protein–protein interactions between TR and CREB appear to be required for the transcriptional antagonism.
We have also demonstrated that coactivator recruitment by TR is decreased in the presence of CREB. Although the receptor DBD is responsible for the interaction with CREB, this domain does not participate directly in binding to coactivators, which is mediated by the AF-2 region in the LBD. This suggests that association of the receptor DBD with CREB could also cause a conformational change in the receptor, which is propagated to the LBD and reduces coactivator binding. This reduction is most likely related to the decreased TR-mediated transcriptional activity found in cells transfected with CREB. CREB belongs to a family of cAMP-responsive transcription factors that includes CREB, ATF1, CREM and ICER (De Cesare and Sassone-Corsi, 2000). Antagonism of TR-mediated responses is not restricted to CREB, and more likely extends to other members of the CREB family since expression of ICER also reduces the transcriptional response to T3. Furthermore, the CREB mutant S133A also inhibits TR-dependent transcription, indicating that protein–protein interactions, rather than phosphorylation of the transcription factor, appear to be important for the antagonism observed.
Transcription factors of the CREB family are the key regulators in many important physiological events, including development, endocrine system function and brain function (Mayr and Montminy, 2001), which are also profoundly influenced by the thyroid hormones. We show here the ability of TR to interact with b-Zip proteins and to mutually regulate their transcriptional activity. It can be envisioned that thyroid hormones could repress transcription of genes that do not contain a TRE, but that do contain a CRE, by antagonizing their response to different stimuli that phosphorylate transcription factors of the CREB family. In turn, transcriptional responses of TRE-containing genes would be attenuated in cells expressing high levels of b-Zip factors. It is, however, not unlikely that T3 and different cellular stimuli could cooperate to activate transcription of the subset of genes having both TRE and CRE motifs in their promoters. In summary, our results show the existence of a complex cross-talk between the thyroid hormone receptors and membrane signal transduction pathways, which ultimately activate transcription factors of the CREB family. Coordination of their functions on complex promoters can impart selectivity and flexibility to target gene activation and have important functional consequences.
Materials and methods
Expression vectors and transfections
CRE-containing promoters fused to CAT (McCormick et al., 1990; Molina et al., 1993; Montminy and Bilezikjian, 1987), a construct containing a palindromic TRE (Umesono and Evans, 1989) and expression vectors for TRα (Sap et al., 1986), CREB (Montminy and Bilezikjian, 1987), ICER Iγ (Molina et al., 1993) or CBP (Chakravarti et al., 1996), have been described previously. The GAL–CREB chimeras were a gift from Silvio Gutkind (National Institutes of Health, Bethesda, MD). GH4C1 cells were transfected by electroporation and HeLa cells were transfected by calcium phosphate as described previously (Tolón et al., 2000). The amount of DNA in each transfection was kept constant by the addition of appropriate non-coding vectors. Unless otherwise stated, cells were incubated for 48 h in the presence or absence of 5 nM T3 (GH4C1) or 100 nM T3 (HeLa cells), and with 100 nM TPA or 10 µM forskolin for the last 4 h in Dulbecco’s modified Eagle’s medium supplemented with 10% AG1-X8 resin and charcoal-stripped newborn calf serum. All data shown are means ± standard deviations, obtained from at least four independent transfections, and the experiments were repeated at least twice with similar relative differences in regulated expression.
Western blotting
GH4C1 cells were collected in lysis buffer (0.5% SDS, 50 mM Tris–HCl pH 7.5, 150 mM NaCl, and 1.25 µg/µl of pepstatin, leupeptin and aprotinin). Forty micrograms were run on a 12% acrylamide gel and transferred to permeabilized nylon membranes (Immobilon P). Blots were incubated overnight at 4°C with a 1:1000 dilution of antibodies recognizing specifically either CREB phosphorylated in Ser133 (P-CREB) (catalogue no. 9191S; New England Biolabs) or total CREB (Santa Cruz Biotechnology), followed by a second incubation with a peroxidase-conjugated secondary antibody. Blots were revealed by enhanced chemiluminiscence (ECL; Amersham Pharmacia Biotech).
Immunofluorescence
For immunofluorescence, GH4C1 cells were fixed with 4% formaldehyde for 30 min at 4°C, and washed with phosphate-buffered saline contaning 0.05% Triton X-100 as described previously (Sánchez-Pacheco et al., 1995). The cells were incubated with 5% non-immune goat serum for 20 min to avoid non-specific binding, and overnight with the CREB or P-CREB antibodies at a 1:1000 dilution. A second antibody conjugated with rhodamine at a 1:250 dilution was used as a marker.
CREB phosphorylation assays
Recombinant CREB was expressed in Escherichia Coli BL-21 and purified, after induction with IPTG, by heating to 95°C for 20 min. The protein (50 ng) was incubated with [γ-32P]ATP (1 µCi) in the presence of an excess (4 µg) of the catalytic subunit of PKA (Sigma) in phosphorylation buffer (25 mM Tris–HCl pH 7.5, 10 mM MgCl2, 1 mM dithiothreitol, 50 mM KCl) for 1 h at 37°C. Phosphorylation assays were performed in the presence and absence of GST–TRα fusion proteins (100 ng), or the same amount of GST alone. Labelled proteins were analysed by SDS–PAGE and autoradiography.
GST pull-down assays
GST–TRα cloned into the pGEX 2TK-P vector was used to generate point mutations in residues C51, K72,112 and K102 with the QuikChange™ site-directed mutagenesis kit (Stratagene). GST–ACTR (Chen et al., 1997) and GST–SMRT (Chen and Evans, 1995), as well as GST fusion constructs containing different CREB domains (Nakajima et al., 1997), were also used. Recombinant proteins were synthesized, purified on glutathione–Sepharose resin and analysed by SDS–PAGE. 35S-labelled CREB and TRα were generated with the TNT T7 Quick coupled in vitro transcription and translation kit and used in pull-down assays with 1 µg of GST or GST-fused proteins as described previously (Tolón et al., 2000).
Immunoprecipitation
For immunoprecipitation, GH4C1 cells were cotransfected with 20 µg of expression vectors for CREB and full-length TRα with a HA-tag. The cells were harvested in 200 µl lysis buffer and ∼1500 µg of cell extracts were incubated with anti-HA antibody at 4°C overnight and for 1 h with protein A–Sepharose™ 4B (catalogue no. 17-0780-01; Amersham Biosciences). The cells were either untreated or treated with T3 5 nM for 24 h, and 10 µM forskolin for 15 min. The immunocomplexes were resolved by SDS–PAGE and analysed by western blot with the CREB antibodies.
Chromatin immunoprecipitation
GH4C1 cells grown in 100 cm2 dishes were treated with T3 for different time periods, or with forskolin for 15 min. Cells were cross-linked with formaldehyde (1% final concentration) and sonicated. The average DNA fragment size of 100–600 bp was assessed by agarose gel electrophoresis. For chromatin immunoprecipitations, 200 µl chromatin samples were diluted 2-fold in IP buffer (1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 8.0, 150 mM NaCl, protease inhibitors) and precleared with 80 µl of a 50% protein A–Sepharose slurry containing 10 µg sonicated salmon sperm DNA and 1 mg/ml bovine serum albumin, for 1 h with agitation at 4°C. Twenty microlitres of each sample were saved as ‘total genomic input’, and the rest of the lysate was used for immunoprecipitation with CREB or TR antibodies (1 µg/sample). An equivalent amount of anti-tubulin antibody was used as a negative control. After an overnight incubation at 4°C, 50 µl of a 50% protein A–Sepharose slurry were added and immunocomplexes were recovered. The beads were washed five times: once with buffer containing 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl pH 8.0, 150 mM NaCl and protease inhibitors, again with the same buffer at high-salt concentration (500 mM), once with a buffer containing 0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris–HCl pH 8.1, and twice with TE (10 mM Tris, 1 mM EDTA). The immunocomplexes were eluted twice from the beads by adding 500 µl 1% SDS in 0.1 M NaHCO3 at room temperature for 30 min. For the reversal of cross-links, 20 µl of 5 M NaCl was added and the samples were incubated at 65°C for 4 h. The DNA was extracted with phenol–chloroform, ethanol-precipitated in the presence of 20 µg of glycogen at –20°C overnight and resuspended in 20 µl of TE buffer. The precipitated DNA was quantified by real-time PCR using SYBR Green. A –264/–69 fragment of the rat GHF-1/Pit-1 gene was amplified using a primer pair (5′-GCCACACCAGTAGTTTGAG-3′ and 5′-CTCCGTG GTCCTCCCAGCTCC-3′). PCR signals were always corrected for the PCR signal obtained with the total DNA IP input. The ratio obtained with the corresponding antibody in untreated cells was arbitrarily set as 1.
Acknowledgments
Acknowledgements
We are grateful to Drs R.Evans, M.Montminy, S.Gutkind, J.R.Naranjo and M.Vallejo for reagents. We also thank M.Vallejo and J.Martin for helpful discussions. This work was supported by grants BMC2001-2275 from the Ministerio de Ciencia y Tecnología, 08.1/0047.1/2001 from the Comunidad de Madrid and 02-101 from the AICR.
References
- Aranda A. and Pascual,A. (2001) Nuclear hormone receptors and gene expression. Physiol. Rev., 81, 1269–1304. [DOI] [PubMed] [Google Scholar]
- Arias J., Alberts,A.S., Brindle,P., Claret,F.X., Smeal,T., Karin,M., Feramisco,J. and Montminy,M. (1994) Activation of cAMP and mitogen responsive genes relies on a common nuclear factor. Nature, 370, 226–229. [DOI] [PubMed] [Google Scholar]
- Brindle P., Nakajima,T. and Montminy,M. (1995) Multiple protein kinase A-regulated events are required for transcriptional induction by cAMP. Proc. Natl Acad. Sci. USA, 92, 10521–10525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canettieri G., Morantte,I., Guzman,E., Asahara,H., Herzig,S., Anderson,S.D., Yates,J.R. and Montminy,M. (2003) Attenuation of a phosphorylation-dependent activator by an HDAC-PP1 complex. Nat. Struct. Biol., 10, 175–181. [DOI] [PubMed] [Google Scholar]
- Chakravarti D., LaMorte,V.J., Nelson,M.C., Nakajima,T., Schulman,I.G., Juguilon,H., Montminy,M. and Evans,R.M. (1996) Role of CBP/p300 in nuclear receptor signaling. Nature, 383, 99–102. [DOI] [PubMed] [Google Scholar]
- Chen J.D. and Evans,R.M. (1995) A transcriptional co-repressor that interacts with nuclear hormone receptors. Nature, 377, 454–457. [DOI] [PubMed] [Google Scholar]
- Chen H., Lin,R.J., Schiltz,R.L., Chakravarti,D., Nash,A., Nagy,L., Privalsky,M.L., Nakatani,Y. and Evans,R.M. (1997) Nuclear receptor coactivator ACTR is a novel histone acetyltransferase and forms a multimeric activation complex with p/CAF and CBP/p300. Cell, 90, 569–580. [DOI] [PubMed] [Google Scholar]
- Cheng X., Reginato,M.J., Andrews,N.C. and Lazar,M.A. (1997) The transcriptional integrator CREB-binding protein mediates positive cross talk between nuclear hormone receptors and the hematopoietic bZip protein p45/NF-E2. Mol. Cell. Biol., 17, 1407–1416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chrivia J.C., Kwok,R.P., Lamb,N., Hagiwara,M., Montminy,M.R. and Goodman,R.H. (1993) Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature, 365, 855–859. [DOI] [PubMed] [Google Scholar]
- De Bosscher K., Vanden Berghe,W., Vermeulen,L., Plaisance,S., Boone,E. and Haegeman,G. (2000) Glucocorticoids repress NF-κB-driven genes by disturbing the interaction of p65 with the basal transcription machinery, irrespective of coactivators levels in the cell. Proc. Natl Acad. Sci. USA, 97, 3919–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bosscher K., Vanden Berghe,W. and Haegeman,G. (2001) Glucocorticoid repression of AP-1 is not mediated by competition for nuclear coactivators. Mol. Endocrinol., 15, 219–227. [DOI] [PubMed] [Google Scholar]
- De Cesare D. and Sassone-Corsi,P. (2000) Transcriptional regulation by cyclic AMP-responsive factors. Prog. Nucleic Acid Res. Mol. Biol., 64, 343–369. [DOI] [PubMed] [Google Scholar]
- Desbois C., Aubert,D., Legrand,C., Pain,B. and Samarut,J. (1991) A novel mechanism of action for v-ErbA: abrogation of the inactivation of transcription factor AP-1 by retinoic acid and thyroid hormone receptors. Cell, 67, 731–740. [DOI] [PubMed] [Google Scholar]
- Glass C.K. and Rosenfeld,M.G. (2001) Coregulator codes of transcriptional regulation by nuclear receptors. J. Biol. Chem., 276, 36865–36868. [DOI] [PubMed] [Google Scholar]
- Goldberg Y., Glineur,C., Gesquiere,J.D., Riscouart,A., Sap,J., Vennstrom,B. and Ghysdael,J. (1988) Activation of protein kinase C or cAMP-dependent protein kinase increases phosphorylation of the c-erbA-encoded thyroid hormone receptor and of the v-erbA-encoded protein. EMBO J., 7, 2425–2433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- González G.A. and Montminy,M.R. (1989) Cyclic AMP stimulates somatostatin gene transcription by phosphorylation of CREB at serine 133. Cell, 59, 675–680. [DOI] [PubMed] [Google Scholar]
- Gottlicher M., Heck,S. and Herrlich,P. (1998) Transcriptional cross-talk, the second mode of steroid hormone action. J. Mol. Med., 76, 480–489. [DOI] [PubMed] [Google Scholar]
- Guenther M.G., Lane,W.S., Fischle,W., Verdin,E., Lazar,M.A. and Shiekhattar,R. (2000) A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev., 14, 1048–1057. [PMC free article] [PubMed] [Google Scholar]
- Hong H., Kohli,K., Triveri,A., Johnson,D.L. and Stallcup,M.R. (1996) GRIP1, a novel mouse protein that serves as a transcriptional coactivator in yeast for the hormone binding domains of steroid receptors. Proc. Natl Acad. Sci. USA, 93, 4948–4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito M. and Roeder,R.G. (2001) The TRAP/SMCC/Mediator complex and thyroid hormone receptor function. Trends Endocrinol. Metab., 12, 127–134. [DOI] [PubMed] [Google Scholar]
- Kamei Y. et al. (1996) A CBP integrator complex mediates transcriptional activation and AP-1 inhibition by nuclear receptors. Cell, 85, 403–414. [DOI] [PubMed] [Google Scholar]
- Karin M. and Chang,L. (2001) AP-1-glucocorticoid receptor crosstalk taken to a higher level. J. Endocrinol., 169, 447–451. [DOI] [PubMed] [Google Scholar]
- Ko L., Cardona,G.R., Henrion-Caude,A. and Chin,W.W. (2002) Identification and characterization of a tissue-specific coactivator, GT198, that interacts with the DNA-binding domains of nuclear receptors. Mol. Cell. Biol., 22, 357–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leitman D.C., Costa,C.H., Graf,H., Baxter,J.D. and Ribeiro,R.C. (1996) Thyroid hormone activation of transcription is potentiated by activators of cAMP-dependent protein kinase. J. Biol. Chem., 271, 21950–21955. [DOI] [PubMed] [Google Scholar]
- Li J., Wang,J., Wang,J., Nawaz,Z., Liu,J.M., Qin,J. and Wong,J. (2000) Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J., 19, 4342–4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin F., Kolluri,S.K., Chen,G.Q. and Zhang,X.K. (2002) Regulation of retinoic acid-induced inhibition of AP-1 activity by orphan receptor chicken ovalbumin upstream promoter-transcription factor. J. Biol. Chem., 277, 21414–21422. [DOI] [PubMed] [Google Scholar]
- Mayr B. and Montminy,M.R. (2001) Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell. Biol., 2, 599–609. [DOI] [PubMed] [Google Scholar]
- Mayr B., Canettieri,G. and Montminy,M.R. (2001) Distinct effects of cAMP and mitogenic signals on CREB-binding protein recruitment impart specificity to target gene activation via CREB. Proc. Natl Acad. Sci. USA, 98, 10936–10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick A., Brady,H., Theill,L.E. and Karin,M. (1990) Regulation of the pituitary-specific homeobox gene GHF1 by cell-autonomous and environmental cues. Nature, 345, 829–832. [DOI] [PubMed] [Google Scholar]
- McKay L.I. and Cidlowski,J.A. (1998) Cross-talk between nuclear factor-κB and the steroid hormone receptors: mechanisms of mutual antagonism. Mol. Endocrinol., 12, 45–56. [DOI] [PubMed] [Google Scholar]
- McKenna N.J. and O’Malley,B.W. (2002) Combinatorial control of gene expression by nuclear receptors and coregulators. Cell, 108, 465–474. [DOI] [PubMed] [Google Scholar]
- Molina C.A., Foulkes,N.S. and Sassone-Corsi,P. (1993) Inductibility and negative autoregulation of CREM: an alternative promoter directs the expression of ICER, an early response repressor. Cell, 76, 875–886. [DOI] [PubMed] [Google Scholar]
- Montminy M.R. and Bilezikjian,L.M. (1987) Binding of a nuclear protein to the cyclic-AMP response element of the somatostatin gene. Nature, 328, 175–178. [DOI] [PubMed] [Google Scholar]
- Montminy M.R., Sevarino,K.A., Wagner,J.A., Mandel,G. and Goodman,R.H. (1986) Identification of a cyclic-AMP-responsive element within the rat somatostatin gene. Proc. Natl Acad. Sci. USA, 83, 6682–6686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima T., Uchida,C. anderson,S.F., Parvin,J.D. and Montminy,M. (1997) Analysis of a cAMP-responsive activator reveals a two-component mechanism for transcriptional induction via signal-dependent factors. Genes Dev., 11, 738–747. [DOI] [PubMed] [Google Scholar]
- Nissen R.M. and Yamamoto,K.R. (2000) The glucocorticoid receptor inhibits NFκB by interfering with serine-2 phosphorylation of the RNA polymerase II carboxy-terminal domain. Genes Dev., 14, 2314–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfhal M. (1993) Nuclear receptor/AP-1 interaction. Endocrinol. Rev., 14, 651–658. [Google Scholar]
- Pfitzner E., Becker,P., Rolke,A. and Schule,R. (1995) Functional antagonism between the retinoic acid receptor and the viral transactivator BZLF1 is mediated by protein–protein interactions. Proc. Natl Acad. Sci. USA, 92, 12265–12269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachez C. and Freedman,L.P. (2001) Mediator complexes and transcription. Curr. Opin. Cell Biol., 13, 274–280. [DOI] [PubMed] [Google Scholar]
- Rogatsky I., Zarember,K.O. and Yamamoto,K.R. (2001) Factor recruitment and TIF2/GRIP1 corepressor activity at a collagenase-3 response element that mediates regulation by phorbol esters and hormones. EMBO J., 20, 6071–6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saatcioglu F. et al. (1997) Mutations in the conserved C-terminal sequence in thyroid hormone receptor dissociate hormone-dependent activation from interference with AP-1 activity. Mol. Cell. Biol., 17, 4687–4695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez-Pacheco A., Palomino,T. and Aranda,A. (1995) Negative regulation of expression of the pituitary-specific transcription factor GHF-1/Pit-1 by thyroid hormones through interference with promoter enhancer elements. Mol. Cell. Biol., 15, 6322–6330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sap J., Munoz,A., Damm,K., Goldberg,Y., Ghysdael,J., Leutz,A., Beug,H. and Vennstrom,B. (1986) The c-erb-A protein is a high-affinity receptor for thyroid hormone. Nature, 324, 635–640. [DOI] [PubMed] [Google Scholar]
- Schule R., Rangarajan,P., Kliewer,S., Ransone,L.J., Bolado,J, Yang,N., Verma,I.M. and Evans,R.M. (1990) Functional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell, 62, 1217–1226. [DOI] [PubMed] [Google Scholar]
- Seternes O.M., Johansen,B. and Moens,U. (1999) A dominant role for the Raf-MEK pathway in forskolin, 12-O-tetradecanoyl-phorbol acetate and platelet-derived growth factor-induced CREB (cAMP-responsive element-binding protein) activation, uncoupled from serine 133 phosphorylation in NIH 3T3 cells. Mol. Endocrinol., 13, 1071–1083. [DOI] [PubMed] [Google Scholar]
- Shaywitz A.J. and Greenberg,M.E. (1999) CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu. Rev. Biochem., 68, 821–861. [DOI] [PubMed] [Google Scholar]
- Tagami T., Park,Y. and Jameson,J.L. (1999) Mechanisms that mediate negative regulation of the thyroid-stimulating hormone alpha gene by the thyroid hormone receptor. J. Biol. Chem., 274, 22345–22353. [DOI] [PubMed] [Google Scholar]
- Tolón R.M., Castillo,A.I., Jiménez-Lara,A.M. and Aranda,A. (2000) Association with Ets-1 causes ligand- and AF2-independent activation of nuclear receptors. Mol. Cell. Biol., 20, 8793–8802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzagarakis-Foster C. and Privalsky,M.L. (1998) Phosphorylation of thyroid hormone receptors by protein kinase A regulates DNA recognition by specific inhibition of receptor monomer binding. J. Biol. Chem., 273, 10926–10932. [DOI] [PubMed] [Google Scholar]
- Umesono K. and Evans,R.M. (1989) Determinants of target gene specificity for steroid/thyroid hormone receptors. Cell, 57, 1139–1146. [DOI] [PubMed] [Google Scholar]
- Yang-Yen H.F., Chambard,J.C., Sun,Y.L., Smeal,T., Schmidt,T.J., Drouin,J. and Karin,M. (1990) Transcriptional interference between c-Jun and the glucocorticoid receptor: mutual inhibition of DNA binding due to direct protein–protein interaction. Cell, 62, 1205–1215. [DOI] [PubMed] [Google Scholar]
- Zhang J. and Lazar,M.A. (2000) The mechanism of action of thyroid hormones. Annu. Rev. Physiol., 62, 439–466. [DOI] [PubMed] [Google Scholar]
- Zhang J., Kalkum,M., Chait,B.T. and Roeder,R.G. (2002) The N-CoR-HDAC3 nuclear receptor corepressor complex inhibits the JNK pathway through the integral subunit GPS2. Mol. Cell., 9, 611–623. [DOI] [PubMed] [Google Scholar]
- Zhang X.K., Wills,K.N., Husmann,M., Hermann,T. and Pfahl,M. (1991) Novel pathway for thyroid hormone receptor action through interaction with jun and fos oncogene activities. Mol. Cell. Biol., 11, 6016–6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.F., Shen,X.Q. and Shemshedini,L. (1999) Ligand-activated retinoic acid receptor inhibits AP-1 transactivation by disrupting c-Jun/c-Fos dimerization. Mol. Endocrinol., 13, 276–285. [DOI] [PubMed] [Google Scholar]