

Abstract
Androgen receptor (AR) protein expression and function are critical for survival and proliferation of androgen-sensitive (AS) prostate cancer cells. Besides its ability to function as a transcription factor, experimental observations suggest that AR becomes a licensing factor for DNA replication in AS prostate cancer cells and thus must be degraded during each cell cycle in these cells to allow reinitiation of DNA replication in the next cell cycle. This possibility was tested by using the AS human prostate cancer cell lines, LNCaP, CWR22Rv1, and LAPC-4. These studies demonstrated that AR levels fluctuate both within and between various phases of the cell cycle in each of these AS lines. Consistent with its licensing ability, AR is degraded during mitosis via a proteasome-dependent pathway in these AS prostate cancer cells. In contrast, proteasome-dependent degradation of AR during mitosis is not observed in AR-expressing but androgen-insensitive human prostate stromal cells, in which AR does not function as a licensing factor for DNA replication. To evaluate mitotic degradation of AR in vivo, the same series of human AS prostate cancers growing as xenografts in nude mice and malignant tissues obtained directly from prostate cancer patients were evaluated by dual Ki-67 and AR immunohistochemistry for AR expression in mitosis. These results document that AR is also down-regulated during mitosis in vivo. Thus, AS prostate cancer cells do not express AR protein during mitosis, either in vitro or in vivo, consistent with AR functioning as a licensing factor for DNA replication in AS prostate cancer cells.
Keywords: cell cycle, prostate stroma
Despite aggressive androgen-ablation therapy, >30,000 men will die of prostate cancer this year in the United States (1). This therapeutic failure, however, does not mean that the androgen receptor (AR) is no longer engaged at the lethal stages of the disease. This conclusion is based on multiple experimental and correlative data. One of the most important is the realization that even in the lethal stages of the disease, AR is strongly expressed in most of metastatic tissues obtained from autopsy from androgen-ablation-failing patients (2). Consistent with the above results is the fact that the majority of human prostate cancer lines obtained from androgen-ablation-failing hosts (e.g., LNCaP, LAPC-4, LAPC-9, MDA-PC-2B, V-Cap, DuCap, CWR22Rv1, etc.) strongly express AR and AR-regulated genes such as PSA and hK2 (3). Furthermore, the AR locus on the X chromosome is frequently amplified in androgen-ablation-resistant prostate cancers (4). Numerous experimental studies have documented that interference with AR expression or function within prostate cancer cell lines at the level of mRNA or protein results in inhibition of proliferation and, eventually, cell death (5–9). In addition, recent studies designed to evaluate genetic changes as the disease acquires an androgen-ablation-resistant phenotype demonstrated that enhanced expression of AR mRNA and protein is the only consistently observed molecular change (10). These results refocus our attention on the role AR is playing in initiation and progression of prostate cancers.
During prostatic carcinogenesis there is a conversion from the stromal-cell-dependent paracrine to an autocrine mechanism of AR-stimulated growth (11). As part of this malignant conversion, AR undergoes a molecular switch from its ability to suppress proliferation of normal prostatic epithelia to directly stimulating the proliferation of prostate cancer cells (4, 11, 12). Such a molecular switch requires gain-of-function changes in AR signaling. Recent studies documented that one such gain-of-function event involves DNA rearrangement such that the androgen response element is translocated to confer androgen responsiveness upon promoters for select members of the ETS transcription factor family (13). Additional studies suggested that besides these malignancy-dependent transcriptional changes, AR becomes a licensing factor for DNA replication in proliferating cancer cells. This hypothesis is based on the observation that AR complexes with a number of licensing factors including hZimp7, hZimp10, and SWI/SNF-like BRG1-associated factor complexes at the origins of DNA replication in cancer cells (14, 15).
The known family of licensing factor proteins include Cdc6, Orc1–6, Cdt1, and Mcm2–7, which form a preinitiation complex at the origins of DNA replication during early G1 (16, 17). Formation of these prereplicative complexes licenses these sites to bind DNA polymerase and initiate DNA replication during S phase. Although initiation of DNA replication occurs in S phase, many of these members of the prereplicative complexes remain associated with the origins of replication during G2 to prevent reinitiation, thus allowing only one round of replication of DNA per cycle (16, 17). Therefore, these licensing factor proteins are degraded in mitosis or early G1 via mechanisms including ubiquitination and subsequent proteasomal degradation to allow reinitiation of DNA replication in the next cell cycle (16). These observations are relevant to androgen-sensitive (AS) prostate cancer cells because the androgen-stimulated proliferation of these cells can be blocked by coexposure to anti-androgens (i.e., AR antagonists) if given in early G1 before the formation of the prereplicative complexes that license DNA replication (18, 19). In contrast, if such anti-androgen coexposure is delayed until a later point in G1 after prereplicative complexes formation, proliferation is not prevented (18, 19). These results document that the major point of androgen regulation in AS prostate cancer cell proliferation occurs in early G1, at the point when licensing for DNA replication occurs. This finding is consistent with AR being a licensing factor for AS prostate cancer cells and would predict that as a licensing factor, it must be degraded in mitosis or early G1 of each cell cycle in these cells.
To test this prediction, AR protein expression throughout the cell cycle was evaluated in AS LNCaP, CWR22Rv1, and LAPC-4 human prostate cancer cells, where the primary function of AR is to promote cell growth. In contrast, AR does not regulate the growth of AR-expressing normal human prostate stromal cells (PrSCs). In these nonmalignant PrSCs, AR functions as a transcription factor and not as a licensing factor for DNA replication. In these cells, AR transcriptionally regulates the expression of paracrine growth factors (i.e., andromedins) needed for normal prostate epithelial growth (11). Thus, in these AR-expressing PrSCs, AR should not be degraded during mitosis. To test this hypothesis, AR expression throughout the cell cycle was evaluated in normal human PrSCs, and these results were compared with the above results of AR expression in LNCaP, CWR22Rv1, and LAPC-4 prostate cancer cells. These combined results document that AR fluctuates dynamically both within and between various phases of cell cycle and is degraded via proteasomal activity during mitosis in AS prostate cancer cells but not in PrSCs. These results are consistent with AR becoming a licensing factor for DNA replication in AS prostate cancer cells but not in normal human PrSCs.
Results and Discussion
Fluctuation of AR Protein both Within and Between Various Phases of Cell Cycle in AS Prostate Cancer Cells.
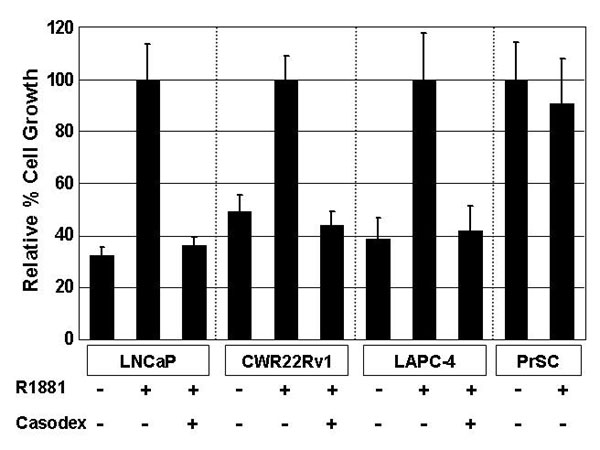
LNCaP, CWR22Rv1, and LAPC-4 human prostate cancer cells express AR protein at a level 102 higher than human PrSCs (Fig. 1A). The in vitro growth of these human prostate cancer lines is stimulated by 1.0 nM of synthetic androgen, R1881, and such a growth effect is reversed with 10 μM of AR antagonist, Casodex (see Fig. 6, which is published as supporting information on the PNAS web site). In contrast, AR does not regulate cell survival and/or proliferation in human PrSCs and, as a consequence, addition of androgens to the media does not affect their growth rate (Fig. 6). In exponentially growing tissue cultures of the prostate cell lines, >85% of cells are positive for nuclear expression of the Ki-67 proliferation marker, as reported (20). This finding is consistent with the observation that in these growing cultures, cells are constantly proliferating and enter G1 immediately after mitosis without exiting the cell cycle to enter G0. Thus, these in vitro cultures provide a useful model to determine the dynamics of AR fluctuation throughout the cell cycle of these prostate malignant and normal cells.
Fig. 1.

AR protein expression during cell cycling of AS prostate cancer cells. (A) AR protein is expressed strongly in CWR22Rv1, LNCaP, and LAPC-4, is very weakly expressed in human PrSCs, and is not expressed in PC3 and DU145 cells. (B–D) Dual-parameter FACS analyses for AR expression and DNA fluorescence analysis in PC3 (B), LNCaP (C), and CWR22Rv1 (D) prostate cancers cells. Results are presented as linear scale dot plots with DNA fluorescence (i.e., cell cycle stage) on the x axis and AR staining intensity on y axis. All analyzed cells were grown in culture in the presence of 1.0 nM R1881. PC3 expression pattern was used to set up a gate to identify specific (i.e., detectable) vs. nonspecific (i.e., nondetectable) staining for AR. This gate was then applied to all subsequent FACS analyses.
To determine AR levels as a function of cell cycle, dual-parameter FACS analysis was performed by using the AR-specific primary antibody along with a FITC-conjugated secondary antibody to measure AR expression and the DNA-binding dye Hoechst 33258 to measure DNA content as an indicator of a position in cell cycle. This Hoechst dye binds within the DNA minor groove, and the intensity of Hoechst staining is affected by DNA accessibility as defined by chromatin structure (21–23). Thus, the same amount of DNA gives a slightly different fluorescence output on the basis of its chromatin confirmation (21–23). In particular, during mitosis or immediately after, in early G1, the DNA is less accessible to Hoechst dye because of tight chromatin confirmation than during the later cell cycle phases, resulting in a slightly less-than-expected fluorescence on the basis of the DNA content. [For an example of the Hoechst binding effect, see Fig. 3A, in which Hoechst-stained phospho-histone H3+ (i.e., mitotic) cells exhibit <4 N DNA fluorescence.]
Fig. 3.

AR protein is not expressed in mitotic AS prostate cancer cells. (A) Flow sorting of exponentially growing LNCaP cells positive for phospho-histone H3 expression to select cells in mitosis. (B and C) Immunocytochemical analysis of AR expression in exponentially growing (control) LNCaP and LAPC-4 prostate cancer cells compared with phospho-histone H3+ (i.e., mitotic) sorted cells. Compared with exponentially growing (i.e., nonmitotic) AR+ LNCaP and LAPC-4 control cells (B Left and C Left), AR expression was undetectable in phospho-histone H3+ prostate cancer cells (B Center and Right and C Center and Right). (D) LNCaP cells were treated with MG-132 proteasome inhibitor for 6 h in the presence of R1881, flow sorted for phospho-histone H3 expression, and analyzed for AR expression by immunocytochemistry. Proteasome inhibition resulted in strong AR expression during mitosis.
A number of human prostate cancer lines, including PC3 and DU145 cells, do not express AR (Fig. 1A). In this report, PC3 cells were used to define the nonspecific gate for identification of positive, detectable AR expression (Fig. 1B). By using this gate, the cell cycle pattern of AR expression was evaluated in all three human prostate cancer cell lines, LNCaP, CWR22Rv1, and LAPC-4. AR stimulates cell growth, localizes to the origins of DNA replication (14, 15), and contains a number of gain-of-function mutations that broaden its ligand specificity and potentiate its function in LNCaP and CWR22Rv1 cells (3, 24). Although AR stimulates cell growth in LAPC-4 cells, these cells were documented to express wild-type AR (3). Dual-parameter FACS analysis documents that in each of these AS cell lines, AR protein levels fluctuate dynamically both during and between specific cell cycle phases [Fig. 1 C and D for LNCaP and CWR22Rv1 and Fig. 7 (which is published as supporting information on the PNAS web site) for LAPC-4 cells, respectively]. Evaluation of AR levels by FACS in these cycling cells documented that during the G1 phase, the AR protein level increases from undetectable to high expression. In particular, cells with undetectable AR expression have the lowest DNA signal as measured by Hoechst binding, which is consistent with these cells having just exited mitosis and entering early G1 (Fig. 1 C and D and Fig. 7). It is notable that during G1 progression, before DNA replication, AR expression increases proportionally to the increase in DNA fluorescence. This is because as a cell unwinds its chromosomes during early G1, the DNA becomes increasingly accessible to Hoechst fluorochrome, thus increasing the DNA fluorescence of these G1 cells without any increase in DNA content. This enhanced fluorescence documents that the increase in AR protein level occurs in early G1 before DNA replication. Thus, as the cells progress through G1, they increase their AR protein expression from an undetectable level in early G1 to a high level in late G1 (Fig. 1 C and D and Fig. 7). Subsequently, the AR level decreases at the G1/S border and then remains relatively constant throughout the S phase (Fig. 1 C and D and Fig. 7). Once in G2, the cells again increase their AR protein level until the G2/M border or mitosis, when AR is quickly down-regulated and returns to the original undetectable levels observed in the early G1 phase. Interestingly, similar cell cycle-dependent fluctuations in AR protein have been reported by Krishan et al. (25) using DNA/AR dual-parameter flow cytometry of nuclei isolated from human prostate cancer tissue.
AR Protein Decreases in Mitosis in AS Prostate Cancer Cells.
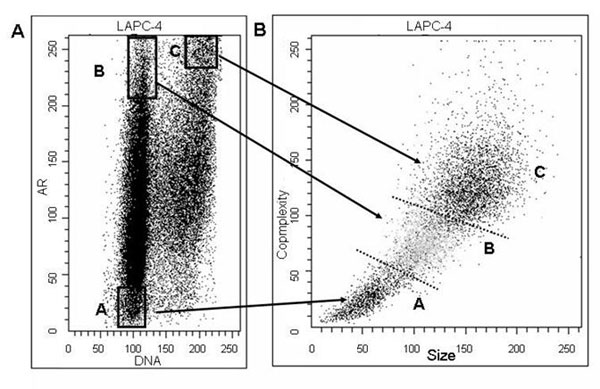
To further validate the above results, LNCaP and LAPC-4 cells were flow-sorted based on DNA fluorescence and AR protein expression. The sorted populations of cells were subsequently analyzed on the basis of cell size and internal complexity (Fig. 2A for LNCaP and Fig. 7A for LAPC-4). Fraction A cells were gated on the basis of low AR expression and lower DNA fluorescence. Fraction B cells were gated on the basis of moderately high levels of AR expression and increased DNA fluorescence. Finally, fraction C cells were gated on the basis of the highest level of AR expression and the G2 phase DNA content. Morphologic evaluation of the sorted cells that were stained by methylene blue (DNA stain) confirmed that they were indeed cells and not cell debris. It was observed that the cells in fraction A were smaller than the ones in fraction B, which were in turn smaller than the cells in fraction C. These size differences were confirmed by flow sorting analysis (Fig. 2B and Fig. 7B). In particular, forward-side scatter comparison of fraction A, B, and C cells documented that fraction B cells were larger and more complex than fraction A cells, whereas fraction C cells were larger and more complex than fraction B cells (i.e., A < B < C) (Fig. 2B and Fig. 7B). These findings are consistent with cells in fraction A being in the early phase of cell cycle (postmitotic/early G1), cells in fraction B being in a more advanced point of cell cycle (late G1/S border), and cells in fraction C being in late phase of cell cycle (late G2/M border).
Fig. 2.

AR protein decreases in mitosis in AS prostate cancer cells. (A) Exponentially growing LNCaP cells in the presence of 1 nM R1881 were analyzed by dual-parameter FACS analysis, and subsequent fractions of cells were sorted on the basis of AR expression and DNA fluorescence. (B) Forward-side scatter analysis of LNCaP cells shows that fraction B cells are significantly larger and more complex than fraction A cells, and fraction C cells are significantly larger and more complex than fraction B cells. (C–E) Western blot analyses and quantitation of AR and phospho-Rb protein expression in sorted A, B, and C fractions and exponentially growing LNCaP control cells. Quantitation of Western blot analysis is presented in D [folds of AR levels (normalized per actin)] and E [folds of phospho-Rb levels (normalized per actin)].
Collected cell fractions (i.e., fractions A, B, and C) were lysed to measure AR and phospho-Rb protein levels by Western blotting (Fig. 2C). The total lysates of fractionated LNCaP cells were immunoblotted with AR and phospho-Rb antibodies. As evident from Fig. 2 C–E, Western blotting confirmed that AR protein levels indeed were much lower in cells in fraction A than in fractions B and C (Fig. 2 C and D). Furthermore, in fractions B and C, a number of higher-molecular-weight AR-staining bands were present. Observation of such an “AR protein ladder” is consistent with AR being polyubiquitinated and degraded at those time points (26–28). This finding is biologically relevant because after these phases of the cycle (G1/S and G2/M borders), the AR levels subsequently decrease (Fig. 1 C and D). Such an AR ladder was not observed in lysates prepared from fraction A cells. Finally, examination of the levels of phospho-Rb protein in collected cells documented that fraction A cells express a much lower amount of this phosphorylated protein than cells in fractions B and C, which confirms that fraction A cells are postmitotic/early G1 cells, whereas cells in fractions B and C represent more advanced phases of the cell cycle (Fig. 2 C and E).
Cells in mitosis can be specifically flow sorted on the basis of expression of phospho-histone H3 (29). Therefore, to test whether, after reaching its maximum protein levels at the end of G2, AR is down-regulated during mitosis to return to undetectable AR expression at the beginning of G1, LNCaP and LAPC-4 cells were flow sorted by using the phospho-histone H3 fluorescently conjugated antibody to select for cells in mitosis (Fig. 3A). The recovered phospho-histone H3+ LNCaP and LAPC-4 cells were cytospun onto glass slides and analyzed by immunocytochemistry for AR expression, as shown in Fig. 3 B and C, respectively. Exponentially growing (i.e., nonmitotic) LNCaP and LAPC-4 cells were used as a positive control for AR expression, shown in Fig. 3 B Left and C Left. In contrast to these control cells, phospho-histone-H3-sorted mitotic LNCaP and LAPC-4 cells have undetectable expression of AR protein (Fig. 3 B Center and Right and C Center and Right). These data document that AR is down-regulated in mitosis, resetting the AR to undetectable levels in postmitotic/early G1 cells. This AR decrease in mitosis occurs regardless of whether the expressed AR is wild type, as in LAPC-4 cells, or mutated, as in LNCaP cells. These results agree with those reported by Cinar et al. (30), who used nocodazole to arrest LNCaP cells in mitosis and demonstrated that such mitotically arrested cells have no detectable AR levels.
Decreased AR in Mitosis Is Due to Proteasomal Degradation.
Proteasome-dependent degradation of licensing factors is the major mechanism of down-regulating these cell cycle effectors and resetting the cell for the next division (16). To test whether proteasomes are involved in AR down-regulation within AS prostate cancer cells, LNCaP cells were treated for 6 h with 5 μM of proteasome inhibitor (MG-132) and flow sorted by using the phospho-histone H3 antibody to isolate mitotic cells that were subsequently subjected to immunocytochemistry analysis with AR antibody. As documented in Fig. 3D, LNCaP cells treated with MG-132 dramatically up-regulated AR protein expression during mitosis. It is also notable that 6-h treatment of cells with MG132 proteasome inhibitor increases the fraction of LNCaP cells in mitosis from 0.8% to 2.4% (data not shown). Because mitosis is ≈1 h in prostate cancer cells (31), 4.8% of the cells should have completed mitosis during this 6-h observation period (i.e., 0.8% of cells in mitosis per h × 6 h). The fact that MG132 treatment results in 2.4% of the cells being in mitosis documents that proteasome inhibition retards mitotic progression. These findings demonstrate that proteasomal degradation is a major mechanism of AR down-regulation during mitosis and that interference with this down-regulation hinders the ability of cells to reset for another cycle. This finding provides an explanation for the growth inhibition response of these AR-sensitive prostate cancer cell lines when treated with proteasome inhibitors (32).
Normal PrSCs Do Not Degrade AR in Mitosis.
In contrast to AS prostate cancer cells, proliferating normal human AR-expressing PrSCs do not possess malignancy-dependent changes in AR signaling. In PrSCs, AR does not regulate growth but acts as a transcription factor and regulates the expression of andromedins (11). As a result, addition of androgen ligand does not affect the growth of PrSCs (Fig. 6). Also, unlike prostate cancer cells, when exponentially growing PrSCs reach confluency, they exit the cell cycle and enter a G0 state, as documented by their lack of immunologically detectable expression of Ki-67 (data not shown) and down-regulation in the expression of DNA replication licensing factors, such as MCM2 (Fig. 4A). Normal human PrSC lines (PrSC in Fig 4A, designated nos. 1–12) were established in our laboratory (11) and were documented to express a lower level of AR compared with prostate cancer cells (Fig. 1A). These PrSCs were arrested in G0 via contact inhibition for 1 week and then analyzed for their AR level. As documented in Fig. 4A, these PrSCs express easily detectable AR even when they are arrested in G0. To evaluate changes in AR expression during the cell cycle, proliferating PrSCs were analyzed by dual-parameter FACS analysis. As documented in Fig. 4B, PrSCs exhibited a monotonous linear increase in AR expression as they progressed throughout the cell cycle. In these PrSCs, such AR protein increase with cell cycle progression correlates with an increase in cell size. To confirm that AR is continuously expressed even during mitosis, AR expression was analyzed in phospho-histone-H3-expressing (i.e., mitotic) PrSCs. To do this, PrSCs were double stained by AR and phospho-histone H3 antibodies and analyzed by FACS. These results document that >90% of the PrSCs simultaneously express phospho-histone H3 and AR protein (data not shown). Thus, in normal PrSCs, the AR level is 1× in G0 and increases throughout the cell cycle in correlation with an increase in cell size until it reaches its peak of 2× at the G2/M border. When the cells divide, the G0 daughter cells are small and have a 1× AR protein level.
Fig. 4.

AR expression during cell cycling of androgen-insensitive prostate stromal cells. (A) Normal human PrSCs (PrSC cells nos. 1–4, 6, and 12) were exponentially growing or arrested in G0 in the presence of androgen by contact inhibition. Protein lysates were immunoblotted for the expression of AR and the DNA replication licensing factor MCM2. (B) Dual-parameter FACS analysis of proliferating PrSC cell no. 6 shows that AR levels fluctuate from 1× to 2× throughout the cell cycle.
Degradation of AR in Mitosis in AS Prostate Cancer Cells Is Not an in Vitro Artifact.
To confirm the in vitro observations that prostate cancer cells undergoing mitosis degrade AR to an undetectable level, we evaluated the in vivo expression of AR in mitotic cells of AS prostate cancer LNCaP, CWR22Rv1, and LAPC-4 xenografts grown in intact male nude mice or prostate cancer tissues directly from patients with localized disease. Dual staining of Ki-67 and AR in all three prostate cancer xenografts demonstrated that AR expression is undetectable during mitosis (Fig. 5). Additional studies demonstrated that although the mitotic index in prostate cancer tissue directly from patients with localized prostate cancer (n = 27) is <1%, such mitotic figures are consistently negative for AR protein. Mitoses were also consistently AR-negative in prostate cancer lymph node metastases (n = 30). These findings document that AR down-regulation in mitosis is not simply an in vitro tissue culture artifact but also occur in vivo in AS prostate cancers.
Fig. 5.

AR protein levels are down-regulated during mitosis in AS prostate cancer LNCaP (A), CWR22Rv1 (B), and LAPC-4 (C) xenografts grown in intact male nude mice. In these studies, tumor tissue sections were stained for AR (green) and the proliferation marker Ki-67 (red). Ki-67 is a well established marker for proliferation that is expressed in all parts of cell cycle except in proliferatively quiescent G0 or early G1 phases. Importantly, Ki-67 reacts with an interchromatin network during mitosis, thereby “painting” the chromosomes (36) and making it easy to identify mitotic cells (arrows). Consistent with the in vitro data, AR expression was greatly decreased in cells undergoing mitosis in LNCaP, CWR22Rv1, and LAPC-4 prostate cancer xenografts.
Conclusion
This study of fluctuation of the AR level during cell cycle progression in normal stromal vs. AS prostate cancer lines documents a fundamental difference in how the AR protein is regulated and functions in these cells. In cells in which AR acts only as a transcription factor, including normal human PrSCs, AR level increases from 1× at G0 to 2× just before division, subsequently returning to 1× in the daughter cell. In contrast, in AS prostate cancer cells, AR increases from an undetectable level in mitosis to a detectable level in early G1. Once detectable, AR levels increase in AS prostate cancer cells during G1, decrease and remain constant during S, and increase again in G2 before being degraded in mitosis to start the whole process over again. Such an AR cycle in these AS prostate cancers is consistent with AR being a licensing factor for DNA replication in malignant prostate cells. These findings emphasize the need for research on AR's effect on DNA replication in prostate cancer cells to supplement ongoing studies of AR's transcriptional activity during prostatic carcinogenesis.
Materials and Methods
Materials.
The synthetic androgen, R1881, was purchased from Perkin–Elmer (Boston, MA). Z-Leu-Leu-Leu-CHO (MG-132) was purchased from Biomol (Plymouth Meeting, PA). LNCaP and PC-3 human prostate cancer lines were purchased from American Type Culture Collection (Rockville, MD) (3). The CWR22Rv1 line was provided by J. W. Jacobberenger (Case Western Reserve University, Cleveland, OH) (3). PrSCs were previously derived in our laboratory (11). These lines were grown by serial passage in RPMI 1640 media containing 10% FBS (Invitrogen Life Technologies, Carlsbad, CA) and 1 nM R1881. LAPC-4 human prostate cancer line was provided by C. Sawyers (University of California, Los Angeles, CA) (3) and was grown is Iscove's media (Invitrogen) containing 10% FBS and 1 nM R1881. All cells were grown in 5% CO2/95% air humidified incubators at 37°C.
Human malignant prostate tissues were obtained through Institutional Review Board (IRB)-approved protocols. These tissues included 27 primary prostate cancers and 30 lymph node metastases, all from hormonally naïve prostate cancer patients, as described (31).
In Vitro Growth Assays.
Cell growth was measured by a 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay (CellTiter 96 Non-Radioactive Cell Proliferation Assay; Promega, Madison, WI) as described (33). Cell numbers for each sample were extrapolated from standard curves (absorbance vs. cell number) prepared for each cell line.
Western Blotting.
Western blotting was carried out on cell lysates equivalent to 105 cells per lane as described (20). The following primary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA): rabbit polyclonal AR (N-20) and goat polyclonal phospho-Rb antibody (directed to phospho- Ser-294/Thr-252 residues) (sc-16671). Mouse monoclonal β-actin antibody was purchased from Sigma (St. Louis, MO).
Immunocytochemistry.
AR immunocyochemical staining was performed as described (34) by using the AR (N-20) rabbit polyclonal antibody from Santa Cruz Biotechnology at 1:500 dilution.
Flow Cytometry.
Exponentially growing cells were trypsinized and washed with ice-cold PBS. Cells were repelleted between washes by mild centrifugation. Subsequently, the cells were resuspended in PBS and then fixed in final concentration of 80% ice-cold methanol at 4°C. Cells were blocked by resuspending the cell pellet in PBS supplemented with 2.5% FBS blocking solution followed by a 30-min incubation at 4°C. Subsequently, cells were stained with primary anti-AR (PG-21) (Upstate Biotechnology, Lake Placid, NY) or phospho-histone H3 (Alexa Fluor 488 Conjugate; Cell Signaling, Beverly, MA) antibodies in volume of 100 μl of blocking solution. Primary antibody incubation was carried out for 30 min at 4°C followed by a wash. Cells stained with primary anti-AR antibody were subjected to incubation with secondary goat anti-rabbit FITC-conjugated IgG (catalog no. AP307F; Chemicon, Temecula, CA) for AR for 30 min at 4°C in the dark followed by a wash, whereas phospho-histone-H3-stained cells did not require this step. Finally, all cell samples were resuspended in PBS containing EDTA and Hoechst dye and flow sorted by using a FACSVantage SE sorter (Becton Dickinson, San Jose, CA) or analyzed by using a LSR flow cytometer (Becton Dickinson). In carrying out flow cytometry analysis, cross-channel compensation was carefully monitored with appropriate controls. Similar procedure was followed to evaluate by FACS AR vs. phospho-histone H3 expression in 6S stromal cells. In this case, goat anti-rabbit IgG-F(ab′)2-PE-Cy5 conjugate antibody form Santa Cruz Biotechnology was used as a secondary antibody for AR.
In Vivo Growth of CWR22Rv1, LNCaP, and LAPC-4 Lines and Immunohistochemical Staining for AR and Ki-67 Expression.
CWR22Rv1, LNCaP, and LAPC-4 lines were inoculated and grown in intact male nude mice as described (35) according to protocols approved by the Johns Hopkins Animal Care and Use Committee. About 6 weeks after inoculation, the tumors were harvested and animals were killed by asphyxiation. These mouse tumor xenografts and tumor tissues obtained from prostate cancer patients were formalin-fixed in neutral buffered saline and mounted into paraffin blocks. Slides were then deparaffinized by heating at 60°C for 10 min and treated with xylene for 5 min (two times). Slides were subsequently hydrated by using an ethanol gradient and then placed in 0.01% Triton aqueous solution. After exposing slides to Antigen Unmasking Solution from Vector Laboratories (Carpinteria, CA), slides were steamed for 40 min. Deparaffinized and hydrated slides were washed in PBS and treated with 3% hydrogen peroxide for 5 min followed by a wash in PBS supplemented with 0.1% Tween 20 (PBST). By using the Biotin Blocking System (Dako, Carpinteria, CA) the slides were exposed to avidin, biotin, and protein blocking solutions for 10 min each with two PBST washes in between. Tissues were then treated with 1:100 dilution of AR rabbit polyclonal AR antibody (Santa Cruz Biotechnology) and Ki-67 mouse monoclonal antibody (Zymed Laboratories, South San Francisco, CA) in antibody dilution buffer from Ventana Medical Systems (Tucson, AZ) overnight at 4°C. After primary antibody binding, the slides were washed twice with PBST, and secondary biotinylated anti-mouse antibody from a VECTASTAIN kit was applied for 15 min according to the manufacturer's instructions (Vector Laboratories). Slides were rinsed with PBST and treated with primary streptavidin-biotin HRP complex for 15 min, again rinsed with PBST, and treated with biotinyl tyramide solution diluted in the aforementioned protein block solution 1:10 for 15 min. After an additional rinse with PBST, slides were treated with combined secondary streptavidin, Alexa Fluor 568 conjugate (1:100 dilution) from Molecular Probes (Eugene, OR) to stain Ki-67 in red and with anti-rabbit antibody Alexa Fluor 488 conjugate (1:50 dilution) from Molecular Probes to stain AR in green. This treatment was carried out for 15 min at room temperature. After a PBST rinse, the slides were treated with DAPI (1:10,000 dilution) (Sigma) for 1 min, coverslipped, and mounted. Subsequently, the slides were analyzed by confocal imaging.
Supplementary Material
Acknowledgments
We thank Mrs. Leslie Meszler, Mrs. Ada Tam, and Mr. Lee Blosser of the Johns Hopkins School of Medicine Flow Cytometry Facilities for technical assistance and FACS expertise. We also thank Mrs. Jessica Hicks for her assistance and expertise in immunohistochemistry. This work was supported by National Institutes of Health Grant R01 DK52645 (to J.T.I.).
Abbreviations
- AR
androgen receptor
- AS
androgen-sensitive
- PrSCs
prostate stromal cells
- PBST
PBS supplemented with 0.1% Tween 20.
Footnotes
The authors declare no conflict of interest.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Jemal A, Murray T, Ward E, Samuels A, Tiwari RC, Ghafoor A, Feuer EJ, Thun MJ. CA Cancer J Clin. 2005;55:10–30. doi: 10.3322/canjclin.55.1.10. [DOI] [PubMed] [Google Scholar]
- 2.Shah RB, Mehra R, Chinnaiyan AM, Shen R, Ghosh D, Zhou M, Macvicar GR, Varambally S, Harwood J, Bismar TA, et al. Cancer Res. 2004;64:9209–9216. doi: 10.1158/0008-5472.CAN-04-2442. [DOI] [PubMed] [Google Scholar]
- 3.van Bokhoven A, Varella-Garcia M, Korch C, Johannes WU, Smith EE, Miller HL, Nordeen SK, Miller GJ, Lucia MS. Prostate. 2003;57:205–225. doi: 10.1002/pros.10290. [DOI] [PubMed] [Google Scholar]
- 4.Litvinov IV, De Marzo AM, Isaacs JT. J Clin Endocrinol Metab. 2003;88:2972–2982. doi: 10.1210/jc.2002-022038. [DOI] [PubMed] [Google Scholar]
- 5.Haag P, Bektic J, Bartsch G, Klocker H, Eder IE. J Steroid Biochem Mol Biol. 2005;96:251–258. doi: 10.1016/j.jsbmb.2005.04.029. [DOI] [PubMed] [Google Scholar]
- 6.Yang Q, Fung KM, Day WV, Kropp BP, Lin HK. Cancer Cell Int. 2005;5:8. doi: 10.1186/1475-2867-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liao X, Tang S, Thrasher JB, Griebling TL, Li B. Mol Cancer Ther. 2005;4:505–515. doi: 10.1158/1535-7163.MCT-04-0313. [DOI] [PubMed] [Google Scholar]
- 8.Kuratsukuri K, Sugimura K, Harimoto K, Kawashima H, Kishimoto T. Prostate. 1999;41:121–126. doi: 10.1002/(sici)1097-0045(19991001)41:2<121::aid-pros6>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- 9.Zegarra-Moro OL, Schmidt LJ, Huang H, Tindall DJ. Cancer Res. 2002;62:1008–1013. [PubMed] [Google Scholar]
- 10.Chen CD, Welsbie DS, Tran C, Baek SH, Chen R, Vessella R, Rosenfeld MG, Sawyers CL. Nat Med. 2004;10:33–39. doi: 10.1038/nm972. [DOI] [PubMed] [Google Scholar]
- 11.Gao J, Arnold JT, Isaacs JT. Cancer Res. 2001;61:5038–5044. [PubMed] [Google Scholar]
- 12.Litvinov IV, Antony L, Dalrymple SL, Becker R, Cheng L, Isaacs JT. Prostate. 2006 doi: 10.1002/pros.20483. in press. [DOI] [PubMed] [Google Scholar]
- 13.Tomlins SA, Rhodes DR, Perner S, Dhanasekaran SM, Mehra R, Sun XW, Varambally S, Cao X, Tchinda J, Kuefer R, et al. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 14.Huang CY, Beliakoff J, Li X, Lee J, Sharma M, Lim B, Sun Z. Mol Endocrinol. 2005;19:2915–2929. doi: 10.1210/me.2005-0097. [DOI] [PubMed] [Google Scholar]
- 15.Sharma M, Li X, Wang Y, Zarnegar M, Huang CY, Palvimo JJ, Lim B, Sun Z. EMBO J. 2003;22:6101–6114. doi: 10.1093/emboj/cdg585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Takeda DY, Dutta A. Oncogene. 2005;24:2827–2843. doi: 10.1038/sj.onc.1208616. [DOI] [PubMed] [Google Scholar]
- 17.Kelly TJ, Brown GW. Annu Rev Biochem. 2000;69:829–880. doi: 10.1146/annurev.biochem.69.1.829. [DOI] [PubMed] [Google Scholar]
- 18.Bai VU, Cifuentes E, Menon M, Barrack ER, Reddy GP. J Cell Physiol. 2005;204:381–387. doi: 10.1002/jcp.20422. [DOI] [PubMed] [Google Scholar]
- 19.Cifuentes E, Croxen R, Menon M, Barrack ER, Reddy GP. J Cell Physiol. 2003;195:337–345. doi: 10.1002/jcp.10317. [DOI] [PubMed] [Google Scholar]
- 20.Uzgare AR, Isaacs JT. Cancer Res. 2004;64:6190–6199. doi: 10.1158/0008-5472.CAN-04-0968. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto A, Araki T, Fujimori K, Yamada M, Yamaguchi H, Izumi K, Matsumoto K. Histochemistry. 1989;92:65–68. doi: 10.1007/BF00495018. [DOI] [PubMed] [Google Scholar]
- 22.Holtfreter HB, Cohen N. Cytometry. 1990;11:676–685. doi: 10.1002/cyto.990110604. [DOI] [PubMed] [Google Scholar]
- 23.Jacobsen PB, Stokke T, Solesvik O, Steen HB. J Histochem Cytochem. 1988;36:1495–1501. doi: 10.1177/36.12.2461412. [DOI] [PubMed] [Google Scholar]
- 24.Chlenski A, Nakashiro K, Ketels KV, Korovaitseva GI, Oyasu R. Prostate. 2001;47:66–75. doi: 10.1002/pros.1048. [DOI] [PubMed] [Google Scholar]
- 25.Krishan A, Oppenheimer A, You W, Dubbin R, Sharma D, Lokeshwar BL. Clin Cancer Res. 2000;6:1922–1930. [PubMed] [Google Scholar]
- 26.Sheflin L, Keegan B, Zhang W, Spaulding SW. Biochem Biophys Res Commun. 2000;276:144–150. doi: 10.1006/bbrc.2000.3424. [DOI] [PubMed] [Google Scholar]
- 27.Faus H, Meyer HA, Huber M, Bahr I, Haendler B. Mol Cell Endocrinol. 2005;245:138–146. doi: 10.1016/j.mce.2005.11.011. [DOI] [PubMed] [Google Scholar]
- 28.Gaughan L, Logan IR, Neal DE, Robson CN. Nucleic Acids Res. 2005;33:13–26. doi: 10.1093/nar/gki141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Juan G, Traganos F, James WM, Ray JM, Roberge M, Sauve DM, Anderson H, Darzynkiewicz Z. Cytometry. 1998;32:71–77. doi: 10.1002/(sici)1097-0320(19980601)32:2<71::aid-cyto1>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- 30.Cinar B, De Benedetti A, Freeman MR. Cancer Res. 2005;65:2547–2553. doi: 10.1158/0008-5472.CAN-04-3411. [DOI] [PubMed] [Google Scholar]
- 31.Berges RR, Vukanovic J, Epstein JI, CarMichel M, Cisek L, Johnson DE, Veltri RW, Walsh PC, Isaacs JT. Clin Cancer Res. 1995;1:473–480. [PMC free article] [PubMed] [Google Scholar]
- 32.Williams S, Pettaway C, Song R, Papandreou C, Logothetis C, McConkey DJ. Mol Cancer Ther. 2003;2:835–843. [PubMed] [Google Scholar]
- 33.Litvinov IV, Antony L, Isaacs JT. Prostate. 2004;61:299–304. doi: 10.1002/pros.20187. [DOI] [PubMed] [Google Scholar]
- 34.Dalrymple S, Antony L, Xu Y, Uzgare AR, Arnold JT, Savaugeot J, Sokoll LJ, De Marzo AM, Isaacs JT. Cancer Res. 2005;65:9269–9279. doi: 10.1158/0008-5472.CAN-04-3989. [DOI] [PubMed] [Google Scholar]
- 35.Denmeade SR, Sokoll LJ, Dalrymple S, Rosen DM, Gady AM, Bruzek D, Ricklis RM, Isaacs JT. Prostate. 2003;54:249–257. doi: 10.1002/pros.10199. [DOI] [PubMed] [Google Scholar]
- 36.Saiwaki T, Kotera I, Sasaki M, Takagi M, Yoneda Y. Exp Cell Res. 2005;308:123–134. doi: 10.1016/j.yexcr.2005.04.010. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}