

Abstract
A cDNA coding for the human neuronal nicotinic α7 receptor subunit with Leu-248 mutated to threonine was expressed in Xenopus oocytes. When activated by acetylcholine (AcCho), the receptors expressed generated currents that had low desensitization, linear current–voltage relation, and high apparent affinity for both AcCho and nicotine. These characteristics are similar to those already described for the chick threonine-for-leucine-247 α7 nicotinic AcCho receptor (nAcChoR) mutant (L247Tα7). These properties were all substantially maintained when the human L248Tα7 mutant was transiently expressed in human Bosc 23 cells. Simultaneous whole-cell clamp and fluorescence measurements with the Ca2+ indicator dye Fura-2 showed that nicotine induced a Ca2+ influx in standard 2 mM Ca2+ solution. The average fractional Ca2+ current flowing through L248Tα7 nAcChoRs was 6.7%, which is larger than that flowing through muscle αβɛδ nAcChoRs (4.1%). The relative Ca2+ permeability, determined in oocytes in the absence of Cl−, was measured from the shift in reversal potential caused by increasing the external Ca2+ concentration from 1 to 10 mM. The human wild-type α7 nAcChoR was found to be more permeable than the L248Tα7 mutant to Ca2+. Our findings indicate that the Ca2+ permeability of the homomeric α7 nAcChoR is larger than that of the heteromeric neuronal nicotinic receptors studied to date and is possibly similar to that of the N-methyl-d-aspartate subtype of brain glutamate receptors.
For many years, it has been known that Ca2+ crosses the postjunctional membrane after activation of nicotinic acetylcholine receptors (nAcChoRs; refs. 1–5), and it is now well established that Ca2+ may permeate the pore of various types of neurotransmitter-gated receptors in the brain. Nevertheless, direct determinations of the transmitter-activated Ca2+ inflow lacked until the Ca2+ entry into the cells was monitored by a combination of electrophysiological and optical techniques with Ca2+ sensitive dyes (6–12). While trying to measure Ca2+ influx through nicotinic receptors, we and others found that the adult muscle nAcChoR channel is much more permeable than the embryonic muscle nAcChoR (11, 13) but less permeable than some neuronal nAcChoR channels (7, 11, 14).
The α7 nAcChoR subunit is expressed throughout the brain; however, it is still not entirely clear whether it is assembled into a full homomeric receptor, because it occurs in heterologous cell expression systems, or whether it is also made into heteromeric α7-containing receptors of uncertain stoichiometry, as has been suggested for the chick nervous system (15–18). The α7 nAcChoR is located at the presynaptic site (18) and, to a minor extent, possibly also at postsynaptic sites (19, 20). Furthermore, α7 nAcChoRs are believed to be involved in a variety of neurological disorders (21) and have the largest Ca2+ permeability among the family of nAcChoRs (18, 22–24). For these reasons, we thought it of interest to measure directly the Ca2+ permeability of the homomeric α7 nAcChoRs. Specifically, using combined patch-clamp techniques and Fura-2 fluorescence measurements, we determined the Ca2+ influx through agonist-activated α7 receptors in which Leu-248 was mutated into threonine (L248Tα7). Our experiments focused on the human L248Tα7 mutant because of its relatively large transient expression in Bosc 23 human cells, and because the mutant receptors desensitize much less than the wild-type (WT)α7 nAcChoRs, which desensitize so fast that it becomes very difficult to determine Ca2+ inflow by fluorescence. This L248Tα7 mutation is equivalent to the Leu-247 mutation of the chick α7 subunit, which, when expressed in Xenopus oocytes, forms homomeric mutant receptors that, in contrast to WT receptors, have a linear current–voltage (I–V) relation, higher affinity for AcCho, and less desensitization; also, in these chick L247Tα7 mutant receptors, curare and 5-hydroxytryptamine (5HT) become agonists instead of antagonists (25, 26).
Materials and Methods
Site-Directed Mutagenesis.
The human L248Tα7 mutant was created by site-directed mutagenesis with the QuickChange kit (Stratagene), according to the manufacturer's instructions. The oligonucleotide primers synthesized to generate the appropriate leucine mutation were (mutagenic sequence underlined) 5′-GGATAACAGTCTTAACTTCTCTTACCGTCTTCATG-3′ and its complement. The mutation site and the fidelity of the α7 M2 coding region were confirmed by DNA sequencing.
Electrophysiology in Oocytes.
At 2–4 days after injection, membrane currents were recorded in voltage-clamped oocytes by using two microelectrodes filled with 2.5 M potassium acetate. Oocytes were placed in a recording chamber and perfused continuously (10–11 ml/min; 0.1 ml volume of chamber) with oocyte Ringer's solution at room temperature (20–22°C). Unless otherwise specified, oocytes were voltage-clamped at −60 mV. Recordings were digitized at 50–200 Hz and stored on a computer for subsequent analysis by using pclamp 6.0.2 and clampex routines (Axon Instruments, Foster City, CA). The concentration producing an EC50 of AcCho/nicotine and the Hill coefficient nH were estimated as described (26, 27). I–V relationships for WTα7 nAcChoR, which showed fast desensitizing AcCho currents, were determined by applying AcCho at 3-min intervals, 5 s after stepping the holding potential from −50 mV to the desired voltage. I–V relationships for mutant α7 receptors, which generate slowly decaying AcCho currents, were constructed using 2-s voltage ramps in the range of +40 mV to −50 mV during agonist application. The decay of current amplitude during the ramp was negligible, and the corresponding control ramps were subtracted. I–V relationships derived with voltage steps gave values of reversal potential (Vrev) similar to those determined with voltage ramps. The relative Ca2+ vs. Na+ permeability (PCa/PNa) was calculated by an extended Goldman–Hodgkin–Katz equation (see refs. 3 and 11), considering only Na+, K+, and Ca2+ and assuming the contribution of other ions to be negligible (see Oocyte Solutions). Vrevs were determined in a Cl−-free external solution with a standard agar bridge as the reference electrode. Activities, not concentrations, of ions were used for the calculations. The activity coefficients have been described (8).
Expression of nAcChoRs in Bosc 23 Cells.
The full-length cDNAs encoding mouse muscle nAcChoR α-, β-, ɛ-, and δ-subunits were cloned in the simian virus 40-based pSM expression vector. Transient transfections were carried out in the cell line Bosc 23 as described (28–30). Subunit cDNAs were added to 35-mm dishes in equivalent amounts (2 μg of L248Tα7 subunit cDNA and 1 μg each of all other subunits) as described (28–30). More than the 50% of the cells died after transfection with L248Tα7 subunit cDNA, and the survival increased to ≈90% when the cells were incubated with the nicotinic antagonist methyllycaconitine (MLA, 2 μM). Cells were used for experiments 36–48 h after transfection.
Electrophysiology in Bosc 23 Human Cells.
Whole-cell patch-clamp recordings of AcCho- or nicotine-induced currents (IAcCho and Inic, respectively) were performed on cells continuously superfused with control or agonist solutions via independent tubes positioned 50–100 μm from the patched cell and connected to a fast exchanger system. Recordings were performed as described (28–30). Unless otherwise indicated, cells were voltage-clamped at −50 mV. Both nH and EC50 were calculated as for oocytes. To measure Vrev in cells expressing L248Tα7 nAcChoRs, voltage ramps (from −120 to +40 mV; 0.5-s duration) were applied during nicotine superfusion, 5–10 s after the peak of the responses. The relative PCa/PCs was estimated as described (11).
Fluorescence Measurements.
Fluorescence determinations were made with a conventional system driven by axon imaging workbench software (Axon Instruments), exciting at 340 nm and 380 nm and monitoring emission at 510 nm. All optical parameters and digital camera settings were maintained throughout to avoid nonhomogenous data. Records of fluorescent signals and membrane currents were synchronized, and images were acquired and stored on a personal computer and analyzed off-line. The rise in cytosolic Ca2+ concentration induced by nicotine was expressed as the ratio of time-resolved fluorescence variation over the basal fluorescence (ΔF/F) and averaged from an ellipsoidal domain approximating the cell shape. For determining the fractional Ca2+ current (Pf), the fluorescence emission elicited by an excitation wavelength of 380 nm (F380) was used (8, 10, 12, 31).
The usual protocol was as follows. Transfected cells were incubated (37°C) with the membrane-permeant Fura-2 acetoxymethyl ester (4 μM; Molecular Probes) for 45 min in DMEM. nAcChoR-expressing cells were identified as those generating a transient fluorescence after a brief nicotine application. These cells were then whole-cell patch clamped with electrodes filled with Fura-2 (cell-impermeant; 250 μM; Molecular Probes). Determinations were carried out after the establishment of a stable value of basal fluorescence. Cells with high basal F340/F380 ratio values (>2 a.u. in our conditions) and/or low basal F380 values (<100 a.u.) were discarded.
To evaluate Pf, the ratio of fluorescence increase (F) over the total charge (Q) was calculated within the first 2.5 s after the onset of the Inic response. The ratio F/Q was defined as:
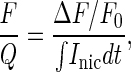 |
1 |
where dt is the time. Each F/Q point was obtained by measuring the charge entering the cell at each fluorescence acquisition time. We used only F/Q points that were measured immediately after the onset of the nicotine-induced response and that had a linear relationship (see Fig. 3B), indicating that the Ca2+ buffering capability of Fura-2 was not saturated.
Figure 3.

Fractional Ca2+ current (Pf) through αβɛδ nAcChoRs determined from simultaneous recordings of whole-cell currents and fluorescent transients. (A Upper) Whole-cell currents elicited by nicotine (100 μM; horizontal bars) in an αβɛδ nAcChoR-transfected Bosc 23 cell equilibrated in normal (2 mM) or in isotonic (110 mM) external Ca2+ solutions. The cell was internally dialyzed with an N-methyl-d-glucamine-based solution containing 250 μM Fura-2 (see Materials and Methods). Holding potential = −50 mV. (A Lower) Simultaneous recordings (●, 2 mM Ca2+; ○, isotonic Ca2+) of fluorescent transients (expressed as ΔF/F) at 380-nm excitation wavelength. Note decrease of ΔF/F reflecting the rise of intracellular free Ca2+ concentration. Black, shaded, and hatched areas (A Upper), drawn as an example, represent the electric charge entering the cell from the onset of the response to the time at which the corresponding fluorescence values were recorded. Current traces and fluorescence signals sharing same time scale are aligned. Note that the delays between the onset of the current and the fluorescence increase (A Left, 570 ms; A Right, 524 ms) are very similar to those reported elsewhere under similar conditions (7, 8). (B) Linear relationship between fluorescence (expressed as ΔF/F) and charge (Q) entering the cell (same cell described in A). The Pf value, calculated after normalizing (in percentage) the slope (calculated by linear regression) obtained in 2 mM Ca2+ medium (●) to the slope in isotonic Ca2+ (○), is 4.3%.
Pf was determined by normalizing the F/Q ratio obtained in standard medium containing 2 mM Ca2+ (2Ca2+) to the F/Q ratio in isotonic 110 mM Ca2+ medium (110Ca2+):
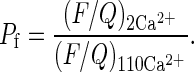 |
2 |
The F/Q ratio value used in determining Pf was represented by the slope of the linear regression best fitting the F/Q plot (see Fig. 3).
In cells expressing L248Tα7 nAcChoRs, the shift to isotonic Ca2+ medium produced large fluorescence signals that were blocked by 0.5 μM MLA (see also ref. 27). Therefore, fluorescence signals elicited by activation of mutant α7-transfected Bosc 23 cells were calibrated by using the mean F/Q values determined in αβɛδ-transfected cells in isotonic Ca2+.
Oocyte Solutions.
The standard oocyte Ringer contained (in mM): NaCl, 82.5; KCl, 2.5; CaCl2, 2.5; MgCl2, 1; and Hepes-NaOH, 5 (pH 7.4). To determine the Vrev shift caused by different Ca2+ concentrations, a modified Cl−-free oocyte Ringer's solution was used to inhibit the endogenous Ca2+-activated Cl− currents (32). This solution contained (in mM): Na+-gluconic acid, 82.5; K+-gluconic acid, 2.5; and Hepes-NaOH, 5 (pH 7.4). Oocytes were incubated in the Cl−-free solution with 2 mM Ca2+ (gluconic acid) for about 1 day before the electrophysiological recordings, which were performed with a final Ca2+ concentration of 1 mM or 10 mM. When necessary, the solution osmolarity was adjusted with sucrose.
Bosc 23 Cell Solutions.
The standard external medium used to measure Pf values contained (in mM): NaCl, 140; KCl, 2.5; CaCl2, 2; MgCl2, 2; Hepes-NaOH, 10; and glucose, 10 (pH 7.3). F/Q calibrations were performed in an external isotonic Ca2+ medium containing (in mM): CaCl2, 110 and Hepes-Ca(OH)2, 10 (pH 7.3). Cells were dialyzed with an internal solution containing (in mM): N-methyl-d-glucamine, 140; Hepes, 10; and Fura-2, 0.25 (adjusted to pH 7.3 with HCl; ref. 7). Vrev shift determinations were performed in cells equilibrated in the following solution (in mM): CsCl, 150; Hepes-CsOH, 10; and CaCl2, 1 or 10 (pH 7.3). Cells were then dialyzed with an internal medium having the following composition (in mM): CsCl, 145; Hepes-CsOH, 10; and 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetate, 5 (pH 7.3).
Results
Properties of Human L248Tα7 nAcChoRs Expressed in Oocytes.
To determine whether the L248T mutation alters the human WTα7 receptors, as it does chick L247Tα7 nAcChoR, oocytes injected with the L248Tα7 subunit cDNA were exposed to either AcCho or nicotine, and their responses to the agonists were studied. Voltage-clamped oocytes responded to both AcCho and nicotine with an inward current whose peak amplitude depended on agonist concentration. At 300 nM, AcCho elicited a current with mean peak amplitude of −1.1 μA (13 oocytes, four donors; range = −0.18 μA to −3.1 μA), whereas in the same oocytes, 50 nM nicotine elicited an Inic whose amplitude was −1.2 μA (range = −0.1 μA to −3.4 μA; Fig. 1). The currents generated by the mutant receptors were better maintained, indicating a reduced rate of desensitization, compared with those of the WTα7 human receptors (Fig. 1). The amplitudes of IAcCho and Inic increased linearly with hyperpolarization (Fig. 1C). Furthermore and in agreement with our previous work on chick L247Tα7 nAcChoRs (26), 5HT (500 μM) gave rise to large and long-lasting inward currents (Fig. 1A). The AcCho dose-response as well the nicotine dose-response relationships (Fig. 1B) gave EC50 and nH values (Table 1) similar to those of chick α7 mutant expressed in oocytes (26, 33), indicating that the agonist affinity of the receptor is maintained across species.
Figure 1.

Human L248Tα7 nAcChoRs have low desensitization, high ligand affinity, and I–V linearity. (A) Sample currents evoked by AcCho, nicotine, and 5HT in an oocyte injected with human L248Tα7 cDNA (Left traces) and by AcCho in another oocyte injected with the human WTα7 cDNA (Right traces). Holding potential = −60 mV. Note faster IAcCho decay in the oocyte expressing WTα7 receptors. (B) AcCho (●) and nicotine (○) dose-response relationships in oocytes expressing L248Tα7 nAcChoRs. Peak currents were normalized to those evoked by 50 μM AcCho (1.1 μA) or 5 μM nicotine (1.3 μA). Each point represents a mean ± SEM (n = 7; two donors). See Table 1 for the best fitting parameters. (C) I–V ramp (duration 2 s) in an oocyte (representative of four experiments; one donor) expressing L248Tα7 mutant in the presence of 200 nM nicotine. Note Inic linearity. Vrev = −12.8 mV.
Table 1.
Best-fitting Hill equation parameters from oocytes or human Bosc 23 cells expressing L248Tα7 mutant receptors
| Agonist |
Xenopus oocyte
|
Bosc 23 cell
|
||
|---|---|---|---|---|
| EC50, nM (no.) | nH | EC50, nM (no.) | nH | |
| AcCho | 230 (7) | 0.98 | 186 (8) | 3.5 |
| Nicotine | 30 (7) | 1.0 | 80 (8) | 1.2 |
See text for details.
It is known that the input resistance of oocytes injected with the chick L247T α7 mutant is much less than that of oocytes injected with WTα7 subunit cDNAs (34). This fact and the fact that the competitive nAcChoR antagonist MLA increases the membrane resistance indicate the presence of “spontaneous” active chick α7 mutant channels. In oocytes expressing human L248Tα7 nAcChoRs, MLA (5 μM) again induced an outward current (720 ± 82 nA; seven oocytes, one donor; mean ± SEM), which was of the same order of magnitude as that recorded in oocytes expressing chick L247Tα7 nAcChoRs (34).
All together, these findings show that equivalent mutations within the leucine ring of the channel domain of chick and human α7 subunits induce similar changes in the properties of the α7 nAcChoRs expressed. Additional experiments were performed in Bosc 23 human cells to determine whether the human L248Tα7 nAcChoR would maintain its characteristic functional and pharmacological behavior in a different cell expression system.
Properties of Human L248Tα7 nAcChoRs Expressed in a Human Cell Line.
In the transfected Bosc cells, 200 nM AcCho elicited an inward current with a mean peak amplitude of −430 pA (n = 8; range = −12 pA to −1.22 nA), whereas at 100 nM, nicotine Inic averaged −520 pA (n = 12; range = −170 pA to −1.4 μA). Both currents were maintained during long-lasting agonist applications (up to 20 s) and increased linearly with hyperpolarization, showing clear rectification at positive potentials (Fig. 2). As for the chick α7 mutant receptors, although 5HT (500 μM) gave rise to an inward current, this current was much smaller. The mean amplitude of the 5HT-current was −74 pA (range = −50 to −113 mV; n = 5; Fig. 2). The apparent AcCho and nicotine affinities in Bosc 23 cells were similar to those obtained in oocytes (Fig. 2B and Table 1). Considered together, these findings indicate that agonist-activated L248Tα7 nAcChoRs have similar, though not identical, characteristics in human Bosc 23 cells and in Xenopus oocytes.
Figure 2.

L248Tα7 expresses functional nAcChoRs in Bosc 23 cells. (A) Typical whole-cell currents elicited by the agonists indicated. Horizontal bars represent timing of drug applications. Holding potential = −50 mV. (B) AcCho (●; eight cells) and nicotine (○; eight cells) dose-response relationships. Solid lines show fitted Hill equations, and parameters are reported in Table 1. Peak currents were normalized to those evoked by 0.5 μM AcCho (0.76 ± 0.30 nA) or 5 μM nicotine (0.72 ± 0.17 nA). (C) Averaged I–V ramp (duration 0.5 s) from five cells, during sustained responses to nicotine (100 nM). Note marked current rectification at positive potentials.
Fractional Ca2+ Current of αβɛδ nAcChoR.
To determine whether the Pf estimated under our experimental conditions was the same as that reported previously (11, 13), we measured the Pf of αβɛδ nAcChoRs. Nicotine (100 μM), used to avoid possible fluorescence signals caused by the activation of native muscarinic AcChoRs, evoked inward currents and fluorescence transients in αβɛδ subunit cDNA-transfected Bosc 23 cells, both in standard and in isotonic Ca2+ medium. By using the experimental protocol described (see Materials and Methods), the measured Pf of αβɛδ nAcChoR (Fig. 3) was 4.1 ± 0.8% (mean ± SD; n = 9), which matches the Pf value determined previously by laser confocal microscopy with the Ca2+ indicator dye Fluo-3 (4.2%; see ref. 11).
Fractional Ca2+ Current of L248Tα7 nAcChoR.
Bosc 23 cells transiently transfected with WTα7 subunit cDNA were unresponsive to nicotine, or when responsive (7 of 48 cells examined), they elicited small Inic (< 50 pA), suggesting that WTα7 nAcChoRs were expressed only sparsely in Bosc 23 cells, were nonfunctional, or were desensitized very rapidly. In contrast, applications of nicotine (100 nM) to Bosc 23 cells transiently transfected with the L248Tα7 mutant subunit cDNA and loaded with Fura-2 in an N-methyl-d-glucamine medium elicited relatively large currents (−460 pA; range = −60 to −1,400 pA; n = 15) and fluorescence transients (ΔF/F = −0.36 ± 0.06; n = 8) in the 2 mM Ca2+ standard medium. By using the same protocol as for the muscle nAcChoRs of Bosc cells, the Pf was 6.7 ± 1.6% (see Fig. 4), which is significantly larger than Pf values found in this study (Fura-2; Student's t test, P < 0.005) and than the Pf values previously determined in the same cell expression system (Fluo-3; ref. 11) for heteromeric nAcChoRs (Pf < 5).
Figure 4.

Nicotine-activated human L248Tα7 mutant receptors show high permeability to Ca2+. (A) Whole-cell current elicited by nicotine in a L248Tα7 cDNA-transfected Bosc 23 cell equilibrated in normal (2 mM Ca2+) external solution (Upper) and fluorescence decrease during the response (Lower). Delay between current and fluorescence signal = 780 ms. (B) Linear relationship between ΔF/F and Q in the same cell. Pf = 6.5%, calculated as the ratio between the slope obtained in standard medium (in this specific case, 0.34 nC−1) and the averaged slope value (5.26 nC−1) measured in isotonic Ca2+ in cells expressing muscle nAcChoR (e.g., Fig. 3B).
Relative Ca2+ Permeability of WT vs. Mutant α7 nAcChoRs.
Because functional WTα7 nAcChoR could not be studied well in transiently transfected Bosc 23 cells, we injected the human WTα7 or mutant subunit cDNAs into Xenopus oocytes and determined the relative Ca2+ permeabilities of the nAcChoRs by measuring Vrev of nicotine-induced currents when the external Ca2+ was changed from 1 mM to 10 mM. To prevent contamination of the measurements by activation of endogenous Ca2+-activated Cl− currents, we removed Cl− from both the recording medium and the oocytes themselves by incubating them for 22–30 h in Cl−-free medium. The shift in Vrev (ΔVrev) determined by best fitting the mean I–V relationships (see Fig. 5) was 14.3 mV for WTα7 nAcChoR (11 oocytes, four donors) and 10.1 mV for α7 mutant (12 oocytes, four donors), whereas the respective estimated PCa/PNa values were 6.6 and 4.0. These results suggest a larger Ca2+ inflow through WTα7 nAcChoRs vs. α7 mutant receptors. These values are considerably different from those obtained after acute exposure of the oocytes to the Cl−-free medium (ΔVrev = 22 mV; four oocytes, one donor) and from values reported previously (ΔVrev = 29 mV; ref. 23). This discrepancy probably reflects differences in the amounts of Cl− withdrawn from both external and internal media. In contrast, using the same protocol, we measured the ΔVrev in oocytes injected with αβγδ nAcChoR subunit cDNAs. In this case, the ΔVrev was 1.6 mV (seven oocytes, one donor; Fig. 5), a value that is compatible with previously reported values (11, 13, 23).
Figure 5.

Vrev shifts indicate larger permeability to Ca2+ of human WTα7 nAcChoR vs. either α7 mutant or αβγδ nAcChoR. (A) Inic–V relationships of oocytes injected with WTα7 cDNA and equilibrated in Cl−-free medium (see Materials and Methods) in the presence of 1 mM Ca2+ (●) or 10 mM Ca2+ (○). Solid lines represent second-order polynomial fits. Each point represents a mean ± SEM (11 oocytes, four donors). Vrev = −6.44 mV in 1 mM Ca2+ and +7.82 mV in 10 mM Ca2+. (Inset) Sample currents evoked by nicotine (200 μM; horizontal bar) during potential steps (Upper traces) from −50 mV to −20 or 0 mV. (B) Averaged I–V ramps in the presence of nicotine (200 nM) from oocytes (12 oocytes, four donors) injected with L248Tα7 cDNA at the indicated Ca2+ concentrations (in mM). Vrev = −2.38 mV in 1 mM Ca2+ and +7.7 mV in 10 mM Ca2+. (Inset) Averaged I–V ramps from oocytes expressing muscle αβγδ nAcChoRs (seven oocytes, one donor). Vrev = −1.4 mV in 1 mM Ca2+ and +0.2 mV in 10 mM Ca2+.
To investigate whether the lower relative Ca2+ permeability of L248Tα7 nAcChoR determined in oocytes was maintained in Bosc 23 cells, we measured ΔVrev in L248Tα7-transfected cells by using nicotine at 100 nM concentration, when the external Ca2+ concentration was changed from 1 to 10 mM. In four cells examined, ΔVrev was 6.3 ± 3.0 mV (Vrev = + 0.7 mV at 1 mM Ca2+ and + 7 mV at 10 mM Ca2+), which, according to the constant field theory, would give a PCa/PCs ratio of 3.6. This value is compatible with the PCa/PNa value calculated in oocytes, considering PCs ≈ PNa, as described for α7-containing nAcChoRs in hippocampal neurons (35).
Discussion
Cholinergic neurotransmission via nicotinic receptors is widely distributed throughout both the peripheral and central nervous systems. In particular, nAcChoRs composed of α7 subunits are strategically located not only at presynaptic sites, where they modulate neurotransmitter release at central synapses (18, 19), but also at postsynaptic sites where they generate depolarizing currents (20). It has been proposed that dysfunction of the central cholinergic system, and especially that of α7 subunit-dependent neurotransmission, may be associated with some severe neurological disorders, such as Parkinson's and Alzheimer's diseases, Tourette's syndrome, and schizophrenia (see ref. 21 for a review). Finally, α7 nAcChoRs are believed to be highly Ca2+ permeable (9, 22–24, 35); because of the very wide range of actions played by intracellular Ca2+, a study of Ca2+ permeability will help us to understand the physiopathological role of α7-containing nAcChoRs in the brain.
To date, the relative Ca2+ permeability of α7 nAcChoR was studied in the Xenopus oocyte by measuring the ΔVrev caused by increasing the external Ca2+ concentration and by applying a simple model of ion permeation, assuming that there is no interaction among ions passing through the channel. The results obtained were not very consistent (23, 36) and differ importantly from those presented herein because, at least in part, of the different external solutions used and of the differing degrees of Cl− withdrawal from internal and external media. Altogether, the shifts in Vrev values that we found point strongly to a higher Ca2+ influx through α7 nAcChoR compared with that in heteromeric muscle or neuronal nicotinic receptors. These shifts represent a starting point and hopefully a stimulus for further and more direct determinations by using alternative experimental approaches.
In addition to the ΔVrev protocol commonly used to determine relative ionic permeabilities, we report herein on simultaneous measurements of fluorescence and whole-cell current made in Bosc 23 human cells expressing recombinant human L248Tα7 mutant receptors. The α7 mutant was selected as a tool to estimate better the fractional Ca2+ currents (i.e., the proportion of whole-cell current carried by Ca2+), because, similar to the WTα7 nAcChoR, the α7 mutant receptors are highly permeable to Ca2+ (22–24, 36); however, unlike the WTα7 nAcChoR, the α7 mutant receptors show very little desensitization in the presence of the transmitter (26) and generate longer lasting currents that make the determinations of many functional parameters much more reliable.
We show herein that some properties of the human L248Tα7 nAcChoR are the same when the receptors are expressed in Xenopus oocytes or in human Bosc 23 cells. However, the Inic rectification at positive membrane potentials is more marked in the human cell expression system; and dose-response relations of nicotine vs. AcCho were different in oocytes and in Bosc 23 cells, indicating that the α7 receptors can be functionally modulated by the host cell type, similar to other neuronal nAcChoRs (37, 38). Furthermore, our findings on the Ca2+ permeability of L248Tα7 nAcChoR channels show that their fractional Ca2+ current is 6.7%. This Pf is larger than Pf values determined for muscle nAcChoRs or neuronal heteromeric nAcChoRs such as α3β4, α4β4, and α4β2 (11) but is in agreement with theoretical predictions of Ca2+ permeability reported elsewhere (22). We also found that the PCa/PCs value (3.6) measured in Bosc 23 cells expressing human L248Tα7 nAcChoR is comparable to the PCa/PNa value (4.0) obtained in Xenopus oocytes, indicating that the Ca2+ permeability is retained in both expression systems and that WTα7 nAcChoR is more permeable than the mutant α7 to Ca2+, as shown by a 65% increase of PCa/PNa in WTα7 nAcChoR vs. the α7 mutant. All these results indicate that the WTα7 nAcChoR may have a Ca2+ permeability in the same range as that of the N-methyl-d-aspartate receptors (8–12%; refs. 8 and 10). This finding is in agreement with a previous estimate of the α7 nAcChoR Ca2+ permeability in HEK-293 cells (39), although the reported value of Pf (≈20%) was only assumed and not determined directly. However, ΔVrev measurements in hippocampal neurons suggest a lower Ca2+ permeability of α7-containing nAcChoRs than that of N-methyl-d-aspartate receptors (35).
In conclusion, our findings, considered together with those reported in the literature, indicate that α7 nAcChoRs, along with N-methyl-d-aspartate receptors, provide an important route for Ca2+ entry into neurons via synaptic actions. This observation suggests that, because of their high Ca2+ permeability, the α7-containing receptors may be involved in synaptic plasticity and in the pathogenesis of various central nervous system diseases that may follow a cytosolic Ca2+ overload (40, 41).
Acknowledgments
We thank Drs. Francesca Grassi and Fabio Ruzzier for critical reading of the manuscript. This work was supported by grants from the Ministero Università Ricerca Scientifica e Tecnologica (to F.E.) and the National Science Foundation (to R.M.). The WTα7 subunit cDNA was a gift from Janssen (Belgium).
Abbreviations
- AcCho
acetylcholine
- nAcChoR
nicotinic AcCho receptor
- IAcCho and Inic
AcCho- and nicotine-activated current
- WT
wild type
- L248Tα7
threonine-for-leucine-248 human α7
- I–V
current–voltage
- 5HT
5-hydroxytryptamine
- MLA
methyllycaconitine
- Vrev
reversal potential
- PCa
PCs, and PNa, permeability of calcium, cesium, and sodium
- ΔF/F
ratio of time-resolved fluorescence variation over the basal fluorescence
- Pf
fractional Ca2+ current
- Q
total charge
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.050582497.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.050582497
References
- 1.Takeuchi N. J Physiol. 1963;167:141–155. doi: 10.1113/jphysiol.1963.sp007137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bregestovski P D, Miledi R, Parker I. Nature (London) 1979;279:638–639. doi: 10.1038/279638a0. [DOI] [PubMed] [Google Scholar]
- 3.Lewis C A. J Physiol (London) 1979;286:417–445. doi: 10.1113/jphysiol.1979.sp012629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Adams D J, Dwyer T M, Hille B. J Gen Physiol. 1980;75:493–510. doi: 10.1085/jgp.75.5.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Katz B, Miledi R. J Physiol (London) 1969;203:689–706. doi: 10.1113/jphysiol.1969.sp008887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miledi R, Parker I, Schalow G. J Physiol (London) 1980;300:197–212. doi: 10.1113/jphysiol.1980.sp013158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vernino S, Rogers M, Radcliffe K A, Dani J A. J Neurosci. 1994;14:5514–5524. doi: 10.1523/JNEUROSCI.14-09-05514.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burnashev N, Zhou Z, Neher E, Sakmann B. J Physiol (London) 1995;485:403–418. doi: 10.1113/jphysiol.1995.sp020738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rogers M, Dani J A. Biophys J. 1995;68:508–509. doi: 10.1016/S0006-3495(95)80211-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bollman J H, Helmchen F, Borst J G G, Sakmann B. J Neurosci. 1998;18:10409–10419. doi: 10.1523/JNEUROSCI.18-24-10409.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ragozzino D, Barabino B, Fucile S, Eusebi F. J Physiol (London) 1998;507:749–757. doi: 10.1111/j.1469-7793.1998.749bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhou Z, Neher E. Pflügers Arch. 1993;425:511–517. doi: 10.1007/BF00374879. [DOI] [PubMed] [Google Scholar]
- 13.Villarroel A, Sakmann B. J Physiol (London) 1996;496:331–338. doi: 10.1113/jphysiol.1996.sp021688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sands S B, Barish M E. Brain Res. 1991;560:38–42. doi: 10.1016/0006-8993(91)91211-i. [DOI] [PubMed] [Google Scholar]
- 15.Keyser K, Britto L, Schopfer R, Whiting P, Cooper J, Conroy W, Brozozozwska-Prechtl A, Karten H, Lindstrom J. J Neurosci. 1993;13:442–454. doi: 10.1523/JNEUROSCI.13-02-00442.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yu C R, Role L W. J Physiol (London) 1998;15:651–665. doi: 10.1111/j.1469-7793.1998.651bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gotti C, Fornasari D, Clementi F. Prog Neurobiol. 1997;53:199–237. doi: 10.1016/s0301-0082(97)00034-8. [DOI] [PubMed] [Google Scholar]
- 18.MacDermott A B, Role L W, Siegelbaum S A. Annu Rev Neurosci. 1999;22:443–485. doi: 10.1146/annurev.neuro.22.1.443. [DOI] [PubMed] [Google Scholar]
- 19.Role L W, Berg D K. Neuron. 1996;16:1077–1085. doi: 10.1016/s0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]
- 20.Frazier C, Buhler A, Weiner J, Dunwiddie T. J Neurosci. 1998;18:8228–8235. doi: 10.1523/JNEUROSCI.18-20-08228.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindstrom J M. Mol Neurobiol. 1997;15:193–222. doi: 10.1007/BF02740634. [DOI] [PubMed] [Google Scholar]
- 22.Gerzanich V, Wang F, Kuryatov A, Lindstrom J. J Pharmacol Exp Ther. 1998;286:311–320. [PubMed] [Google Scholar]
- 23.Séguéla P, Wadiche J, Dineley-Miller K, Dani J A, Patrick J W. J Neurosci. 1993;13:596–604. doi: 10.1523/JNEUROSCI.13-02-00596.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Devillers-Thiery A, Galzi J L, Bertrand S, Changeux J P, Bertrand D. NeuroReport. 1992;3:1001–1004. doi: 10.1097/00001756-199211000-00014. [DOI] [PubMed] [Google Scholar]
- 25.Bertrand D, Devillers-Thiery A, Revah F, Galzi J L, Hussy N, Mulle C, Bertrand S, Ballivet M, Changeux J P. Proc Natl Acad Sci USA. 1992;89:1261–1265. doi: 10.1073/pnas.89.4.1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Palma E, Mileo A M, Eusebi F, Miledi R. Proc Natl Acad Sci USA. 1996;93:11231–11235. doi: 10.1073/pnas.93.20.11231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palma E, Maggi L, Miledi R, Eusebi F. Proc Natl Acad Sci USA. 1998;95:10246–10250. doi: 10.1073/pnas.95.17.10246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fucile S, Mileo A M, Grassi F, Salvatore A M, Alemà S, Eusebi F. Eur J Neurosci. 1996;8:2564–2570. doi: 10.1111/j.1460-9568.1996.tb01550.x. [DOI] [PubMed] [Google Scholar]
- 29.Ragozzino D, Fucile S, Giovannelli A, Grassi F, Mileo A M, Ballivet M, Alemà S, Eusebi F. Eur J Neurosci. 1997;9:480–488. doi: 10.1111/j.1460-9568.1997.tb01625.x. [DOI] [PubMed] [Google Scholar]
- 30.Fucile S, Matter J M, Erkman L, Ragozzino D, Barabino B, Grassi F, Alemà S, Ballivet M, Eusebi F. Eur J Neurosci. 1998;10:172–178. doi: 10.1046/j.1460-9568.1998.00001.x. [DOI] [PubMed] [Google Scholar]
- 31.Frings S, Seifert R, Godde M, Kaupp U B. Neuron. 1995;15:169–179. doi: 10.1016/0896-6273(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 32.Miledi R. Proc R Soc London Ser B. 1982;215:491–497. doi: 10.1098/rspb.1982.0056. [DOI] [PubMed] [Google Scholar]
- 33.Revah F, Bertrand D, Galzi J L, Devillers-Thièry A, Mulle C, Hussy N, Bertrand S, Ballivet M, Changeux J P. Nature (London) 1991;353:846–849. doi: 10.1038/353846a0. [DOI] [PubMed] [Google Scholar]
- 34.Maggi L, Palma E, Miledi R, Eusebi F. Mol Psychiatry. 1998;3:350–355. doi: 10.1038/sj.mp.4000392. [DOI] [PubMed] [Google Scholar]
- 35.Castro N G, Albuquerque E X. Biophys J. 1995;68:516–524. doi: 10.1016/S0006-3495(95)80213-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bertrand D, Galzi J L, Devillers-Thiéry A, Bertrand S, Changeux J P. Proc Natl Acad Sci USA. 1993;90:6971–6975. doi: 10.1073/pnas.90.15.6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fucile S, Barabino B, Palma E, Grassi F, Limatola C, Mileo A M, Alemà S, Ballivet M, Eusebi F. NeuroReport. 1997;28:2433–2436. doi: 10.1097/00001756-199707280-00005. [DOI] [PubMed] [Google Scholar]
- 38.Lewis T M, Harkness P C, Sivilotti L G, Colquhoun D, Millar N S. J Physiol (London) 1997;505:299–306. doi: 10.1111/j.1469-7793.1997.299bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Delbono O, Golapalakrishnan M M, Renganathan M, Monteggia L M, Messi M L, Sullivan J P. J Pharmacol Exp Ther. 1997;280:428–438. [PubMed] [Google Scholar]
- 40.Lee J M, Zipfel G J, Choi D W. Nature (London) 1999;399,Suppl.:A7–A14. doi: 10.1038/399a007. [DOI] [PubMed] [Google Scholar]
- 41.Dunnet S B, Bjorklund A. Nature (London) 1999;399,Suppl.:A32–A39. doi: 10.1038/399a032. [DOI] [PubMed] [Google Scholar]