

Abstract
A de novo 687-amino-acid residue polypeptide with a regular 32-amino-acid repeat sequence, (GA)3GY(GA)3GE(GA)3GH(GA)3GK, forms large β-sheet assemblages that exhibit remarkable folding properties and, as well, form fibrillar structures. This construct is an excellent tool to explore the details of β-sheet formation yielding intimate folding information that is otherwise difficult to obtain and may inform folding studies of naturally occurring materials. The polypeptide assumes a fully folded antiparallel β-sheet/turn structure at room temperature, and yet is completely and reversibly denatured at 125°C, adopting a predominant polyproline II conformation. Deep ultraviolet Raman spectroscopy indicated that melting/refolding occurred without any spectroscopically distinct intermediates, yet the relaxation kinetics depend on the initial polypeptide state, as would be indicative of a non-two-state process. Thermal denaturation and refolding on cooling appeared to be monoexponential with characteristic times of ∼1 and ∼60 min, respectively, indicating no detectable formation of hairpin-type nuclei in the millisecond timescale that could be attributed to nonlocal “nonnative” interactions. The polypeptide folding dynamics agree with a general property of β-sheet proteins, i.e., initial collapse precedes secondary structure formation. The observed folding is much faster than expected for a protein of this size and could be attributed to a less frustrated free-energy landscape funnel for folding. The polypeptide sequence suggests an important balance between the absence of strong nonnative contacts (salt bridges or hydrophobic collapse) and limited repulsion of charged side chains.
INTRODUCTION
The molecular self-assembly of the constituent building blocks of nature, such as amino acids, have found frequent applications in modern nanotechnology and have led researchers to develop biomimics of these processes (1–7). The problems of protein folding, self-assembly, and sequence-structure relationships are as important in nanotechnology as in biology. Genetic engineering affords a plethora of options for the examination of these fundamental problems, as well as for the development of novel materials with the desired functionalities (8). It is not surprising that a great variety of small polypeptides have been utilized both for modeling protein folding and development of novel nanomaterials during the last decades. In the case of biological studies, small model systems can mimic the folding properties of major secondary structural motifs of globular proteins including α-helices and β-sheets, and -turns (9–15). However, tertiary structural elements, which play an important role in globular proteins (16–23), could not be modeled in such small polypeptide-based systems. The widely recognized importance of the role of tertiary contacts in protein folding (16) has led to the development of model systems based on small proteins (24–28). Another important phenomenon, postulated to influence the kinetics and the pathway of protein folding, is nonspecific, Flory-type coil-to-global-protein collapse with changes in temperature or solvent quality (e.g., denaturant concentration) (29,30). Nonspecific collapse might result in nonnative contacts that roughen the free-energy landscape funnel and slow the folding process (31–33). In the case of α-helical proteins, molecular dynamic simulations (27,34,35) predicted (36,37) that helix formation and native tertiary packing occur simultaneously, an observation that was verified experimentally (24,26,36–38). In contrast, for proteins composed of β-sheets, the initial collapse precedes the formation of secondary structures (15,39,40). Again, small polypeptide systems cannot be used for modeling this process. In contrast, with large polypeptides, which exhibit a well-defined 3D structure in a folded state, a model for the folding process requires a surrogate where nonspecific collapse is invoked in the first stage. Spiro and co-workers (41) have recently investigated the kinetics of α-helix-to-β-sheet transition of poly-L-lysine using time-resolved deep ultraviolet (UV) resonance Raman (DUVRR) spectroscopy. They demonstrated that sheet formation takes minutes and is clearly distinct from submicrosecond melting of helices that result in the polyproline II conformation.
Our long-term strategy for studying protein folding, with particular focus on aggregation and fibrillation phenomena, is based on 1), the development of a family of large polypeptides that would form fibrils and show reversible folding, and 2), establishment of a structural relationship between polypeptide sequence and folding properties. Model compounds such as the materials described below are not only simple by virtue of their highly repetitive nature but also enable dissection of various contributions to the folding and fibrillation processes. Genetic engineering affords excellent opportunities to verify the mechanism of folding and fibrillation by tuning selectively the primary polypeptide sequence and testing the influence of those changes on kinetic and equilibrium properties. Our recent study (42) has demonstrated that a de novo, genetically engineered 687-residue peptide with 32 amino acid repeats utilizing carbamylate lysines, GH6[(GA)3GY(GA)3GE(GA)3GH(GA)3GK]21GAH6 (YEHK21), based on the pioneering work of Tirrell (43), self-assembles into a well defined antiparallel β-sheet structure (Fig. 1) stabilized by intramolecular hydrogen bonds. YEHK21 forms a gelatinous phase in solution and fibril-type ribbons (44) on well defined, hydrophobic surfaces. Here we report on equilibrium and kinetics of the reversible thermal denaturation of YEHK21. Being well folded to a single “native” conformation initially, YEHK21 was completely and reversibly denatured on heating. YEHK21 was demonstrated to be an excellent model for large β-sheet proteins, known to form well organized fibrillar aggregates.
FIGURE 1.

Two and a half repeats of (GA)3GY(GA)3GE(GA)3GH(GA)3GK constituent polypeptide unit of YEHK21 (42).
EXPERIMENTAL PROCEDURES
Materials
Poly(L-glutamic acid) (PGA, 17,000 mol wt; Sigma, St. Louis, MO), poly(L-lysine) (PLL, 28,200 mol wt, Sigma), tripeptide H-Gly-Tyr-Gly-OH (Bachem), NaCl, Na2HPO4, NaH2PO4, urea, and imidazole (all from Aldrich Chemical, Milwaukee, WI) were used as received.
Expression and purification of YEHK21
Construction of the expression vector was described in detail elsewhere (42,45). The overnight preculture (30 mL of TB media containing kanamycin, 50 μg/mL; chloramphenicol 34 μg/mL; and glucose, 3.6 mg/mL) was used to inoculate 800 mL of 2xYT media containing kanamycin, 50 μg/mL, and chloramphenicol, 34 μg/mL. The cultures were grown to an initial OD600 = 1.5 and polypeptide expression was started by the addition of IPTG to a final concentration of 1 mM and allowed to incubate for 3–4 h (45). Cells were harvested by centrifugation (3500 × g, 20 min at 4°C). The cell pellets were resuspended in water (15 mL per 800 mL of cell culture) and stored at −20°C.
Cell lysis was performed by freeze-thaw sonication for 20 min. in the presence of benzonase (2 μL per 800 mL culture) and PMSF (phenylmethanesulfonyl fluoride), 2 mM with subsequent incubation for 30 min at room temperature. For protein denaturation and chemical modification, urea was added (19.2 g per 800 mL culture, 8 M at final concentration) and the sample was incubated in a boiling water bath for 4 h with occasional mixing. The resulting solution was centrifuged for 45 min at 15,000 × g at 20°C and the supernatant was diluted twofold with 10 mM imidazole, 8 M urea, phosphate-buffered saline (PBS) solution (0.1 M phosphate buffer, pH 7.4, 0.5 M NaCl at final concentration) and then was recentrifuged for 45 min at 15,000 × g at 20°C.
The recovered supernatant was applied to a Ni-NTA column preequilibrated with 40 mL of 10 mM imidazole in 8 M urea and PBS buffer. The column was washed with 80 mL of 20 mM imidazole in 8 M urea and PBS solution, and then was eluted with 80 mL portions of 300 and 500 mM imidazole in 8 M urea and PBS. Most of the desired polypeptide was found in the 300-mM fraction.
The eluent containing the polypeptide was dialyzed against doubly distilled H2O at 4°C using a dialysis membrane with a 3500-Da molecular mass cutoff. After 4 days of dialysis, formation of a gelatinous phase within the solution was observed. The gelatinous phase was separated by centrifugation at 15,000 × g for 45 min at 4°C and subsequent decantation of the supernatant. The polypeptide aggregates were rendered more soluble by adding to the gel an equal volume of water followed by intensive vortex mixing. An aqueous phase containing a soluble polypeptide fraction was separated from the remaining gelatinous phase by centrifugation at 15,000 × g for 45 min at 4°C. This solution (referred to as the initial YEHK21 solution hereafter) was used for the reported studies.
Expression and purification of polypeptides were analyzed by SDS-PAGE and western blotting using SuperSignal West HisProbe kit (Pierce Biotechnologies, Rockford, IL). The concentrations of the polypeptide in solutions were determined by UV absorption spectroscopy using a 276-nm tyrosine band (1280 M−1 cm−1) (46).
Raman measurements
Deep UV Raman instrumentation has been described in detail elsewhere (47). Briefly, a 197-nm laser beam (∼1 mW, Indigo-S laser system, Coherent, Santa Clara, CA) was focused into either 1), a temperature-controlled open stream (0.5-mm diameter) of circulating solution, or 2), a spinning Suprasil NMR tube (5-mm outer diameter, 0.38 mm wall thickness) containing 150-μl solution. To avoid NMR tube sample heating, the solution was also mixed using a magnetic stirrer. Scattered radiation was collected in backscattering geometry, dispersed using a home-built double monochromator, and detected with a liquid-nitrogen-cooled CCD camera (Roper Scientific). For steady-state measurements, the accumulation time for every spectrum was 6 min. GRAMS/AI (7.01) software was used for Raman spectroscopic data processing. The contribution of Suprasil and water was quantitatively subtracted.
Fluorescence, UV absorption, and CD spectroscopy
Fluorescence spectra were measured in a 1-cm rectangular quartz cell with a magnetic stirrer using a Jobin Yvon Fluoromax-3 spectrofluorometer (Jobin Yvon, Edison, NJ). Typically, 275 nm excitation, with 2-nm excitation and 4-nm emission slits, a 1-nm data interval, and a 0.5-s integration time were used for fluorescence measurements. UV absorption spectra were measured in 1-mm quartz cell using Hewlett-Packard HP 8452 diode array spectrophotometer. Far-UV circular dichroism (CD) spectra were measured in 0.05-cm temperature-controlled quartz cell using Jasco J-720 spectropolarimeter (Tokyo, Japan). A bandwidth of 1 nm, scan speed of 100 nm/min, resolution of 0.5 nm, and response time of 4 s were utilized. Five accumulations were averaged normally. The absorbance of the sample in the CD cell did not exceed 1.5, which was well within the recommended absorbance range (<3). The photomultiplier voltage was recorded in each run and did not exceed 600 V, which was also within the recommended range (not to exceed 900 V for a 4-s response time).
Kinetic measurements
Two types of kinetic experiment were performed. A temperature jump with an ∼2-s time resolution was utilized using a solution circulator with a heat exchanger connected to two thermal baths. First, a circulating solution of YEHK21 was held at room temperature using one thermal bath. Raman spectra, measured in the open stream immediately after the heat exchanger, were recorded continuously with an accumulation time of 20 s. Switching from the room-temperature bath to a high-temperature bath (85°C) resulted in the desired temperature jump. Twenty-second Raman spectra were recorded until no further changes were evident in the Raman spectra. Melting kinetics of YEHK21 induced by a 25–85°C temperature jump was studied using this approach.
The kinetics of YEHK21 refolding on thermal denaturation and subsequent cooling was found to be slow. This slow process allowed conservation of substantial quantities of polypeptide by using a spinning NMR tube for Raman measurements. A YEHK21 solution (2 mL in a boiling-proof Eppendorf tube) was heated to the desired “melting temperature” for 5 min and then cooled to a “recovery temperature”. After various recovery times, 150-μL samples were withdrawn, placed into an NMR tube, and cooled to room temperature for immediate Raman measurements (see Results for further justification of this approach).
Raman and CD spectra of homopolypeptides
Deep UV resonance Raman and far-UV CD spectra of pure secondary structure conformations, β-sheet and unordered, were obtained using homopolypeptides, PGA, and PLL. Completely unordered PGA was obtained in aqueous solution at 25°C and pH 7.1 (48, 49). A freshly prepared, pH 7.0 PLL solution (0.5 mg/mL) was used as an unordered PLL sample (50). A β-sheet-rich (59%) conformation of PLL was prepared by incubating a PLL solution at 52°C and pH 11.3 for 3 hr (48). The spectra of pure β-sheet conformation were obtained by numerically subtracting the spectra of unfolded PLL from the spectra of the β-sheet-rich solution of PLL. Concentrations of both homopolypeptides were 0.5 mg/mL. Sodium perchlorate (150 mM) was added to the solutions as an internal standard.
RESULTS
Polypeptide characterization
YEHK21 charge state
Increased ionic strength was found to reduce the solubility of YEHK21. Therefore, no buffer was used for YEHK21 solutions dialyzed with distilled water. As a result, the solution pH remained within the range of 6.5 ± 0.5. Based on the pKa values for isolated amino acid residues, all tyrosine and carbamylated lysine residues should remain uncharged, all glutamic acid side chains (total of 21) should be deprotonated, but the extent of histidine protonation might vary. However, no difference in YEHK21 behavior was found when the pH was varied between 6 and 7 by addition of HCl or NaOH. The latter might indicate that either 1), the pKa of the YEHK21 histidines was different relative to that of an isolated amino acid residue, as the extent of histidine protonation did not change in the pH range between 6 and 7; or 2), the extent of histidine protonation does not influence the YEHK21 behavior. In future studies, this question will be resolved by monitoring the extent of histidine protonation, for example, by measuring 229-nm excited resonance Raman spectra of histidine (51).
Deep UV Raman spectra
After 4 weeks at 4°C, the initial YEHK21 solution showed no changes in the fluorescence, Raman, and UV absorption spectra. Fig. 2 shows 197-nm excited Raman spectra of gelatinous YEHK21 and the initial room-temperature YEHK21 solution. The spectra were dominated by contributions from the amide chromophore and tyrosine as indicated in Fig. 2. Amide I mode (Am I) consists of carbonyl C=O stretching, with a small contribution from C-N stretching and N-H bending. Amide II and Amide III bands involve significant C-N stretching, N-H bending, and C-C stretching. The Cα-H bending vibrational mode involves Cα-H symmetric bending and C-Cα stretching (48). Deep UV resonance Raman spectra are widely recognized as being sensitive to the changes in the protein/polypeptide secondary structure due to the coupling of various amide vibrational modes (41,48,52–54).
FIGURE 2.

The 197-nm excited Raman spectra of YEHK21 in gelatinous phase and in solution measured at 25°C and 90°C.
The DUVRR spectra of a gelatinous phase and the initial YEHK21 solution shown in Fig. 2 were practically identical, indicating that the secondary structural composition of the polypeptide was the same in these two samples. The shape of the amide component of Raman spectra of the gelatinous phase and the initial room-temperature solution suggests the dominant contribution of β-sheet conformation (see Discussion section for details).
Fluorescence spectra
It is known that tyrosine fluorescence is sensitive to the local environment and is therefore often used as a natural biomarker (46). The fluorescence spectrum of YEHK21 was very similar to the spectrum of a tyrosine tripeptide (H-Gly-Tyr-Gly-OH) in water (Fig. 3). In the latter spectra, the tyrosine residues of the polypeptide were fully exposed to water and do not interact with any charged amino acid residue or another tyrosine. The fluorescence spectrum did not change over 4 weeks if the YEHK21 sample was kept at 4°C. Similarly, the absorption spectrum of the YEHK21 solution in the wavelength range of the ∼280-nm peak of tyrosine did not change when the solution was kept at room temperature. However, dramatic changes were observed for the fluorescence spectrum of the room-temperature sample: the emission peak became smaller and broader (Fig. 3). Although work is in progress in our laboratory to understand the cause of these changes in YEHK21 fluorescence, the work reported in this study utilized only solutions stored at 4°C.
FIGURE 3.

The 275-nm excited fluorescence spectra of YEHK21 and tyrosine. The fluorescence of a YEHK21 sample kept at 4°C does not change for at least 4 weeks, whereas storage at room temperature resulted in a weaker and broader fluorescence peak. The fluorescence spectrum of tyrosine (as a part of tripeptide) aqueous solution was normalized with that of YEHK21 for comparison.
Far-UV CD spectra
The initial far-UV CD spectra of YEHK21 (Fig. 4), consisted of a strong trough at ∼206 nm and a peak at ∼197 nm. The overall shape of this curve resembles qualitatively the far-UV CD signature of the β-sheet conformation of polypeptides and proteins. For comparison, the far-UV CD spectra of poly-L-lysine β-sheet and unordered conformations are also shown together (Fig. 4, lower). Although the far-UV CD data strongly indicated that YEHK21 adopted a predominantly β-sheet conformation in the initial solution, traditional deconvolution programs do not provide good approximations of YEHK21 secondary structure, presumably because globular protein databases were used in the comparison of CD spectra (55).
FIGURE 4.

(Upper) Far-UV CD spectra of YEHK21 measured at room temperature (initial solution), at 80°C and 95°C. (Lower) Far-UV CD spectra of poly-L-lysine in β-sheet and unordered form.
Temperature effect
Steady-state studies
The temperature-induced changes in the YEHK21 far-UV CD spectrum were consistent with β-sheet melting (Fig. 4, upper). The CD spectrum measured at 95°C resembled the CD spectrum of unordered poly-L-lysine. DUVRR spectra of YEHK21 were also measured at various temperatures between 25 and 90°C using the open stream of a flow cell (see Experimental Procedure). Multiple spectra were measured at each temperature to assure that the equilibrium had been attained. The spectra measured at ∼5, 10, and 30 min after a temperature change were practically identical. Substantial changes in the spectra were evident at temperatures exceeding 80°C. For example, the Raman spectra of YEHK21 measured at 25 and 90°C are shown in Fig. 2. The observed spectral changes were consistent with those expected for the melting of a β-sheet structure and the formation of unordered conformation (47,56,57). In particular, the sharp Am I peak indicative of a β-sheet conformation broadened at high temperatures. A quantitative analysis of the spectra targeting the evaluation of the polypeptide secondary structure content was complicated by the temperature dependence of the basis spectra of pure secondary structural elements including random coil (58,59) and β-sheet (57). To avoid these complications, we took advantage of the slow recovery process (discussed below) of the Raman spectrum at room temperature after heating the YEHK21 sample. Using this approach, the polypeptide solution was heated for 5 min to various temperatures between 25 and 125°C and cooled quickly to room temperature, after which two consecutive DUVRR spectra were measured using a quartz NMR tube. No noticeable differences were observed between spectra in these pairs, indicating that no changes occurred during the spectral measurements. Any changes in DUVRR spectra obtained using this approach could be directly attributed to the temperature-induced structural rearrangements of the polypeptide without any contribution from the temperature-induced changes in the basis Raman spectra of secondary structural elements (since all spectra were measured at the same temperature). Raman spectra of YEHK21 measured at room temperature after a brief heating to various temperatures (selected spectra are shown in Fig. 5) were used to evaluate the melting of the YEHK21 polypeptide.
FIGURE 5.

DUVRR Raman spectra of YEHK21 measured at room temperature after a brief heating to various temperatures. The calculated spectra (see Discussion), as well as difference spectra between the measured and calculated ones, are also shown for 35 and 85°C. The near overlap of the calculated and measured spectra are indicative of the high quality of the model.
Kinetic studies
The kinetics of YEHK21 β-sheet melting initiated by a temperature jump was measured in an open stream using a flow cell with a heat exchanger. DUVRR spectra of YEHK21 were accumulated and recorded at 85°C every 20 s after a temperature jump from 25°C. The spectral changes were similar to those found for the steady-state Raman spectra measured at various temperatures (Fig. 6). In Fig. 7 a, the decrease in intensity of the Am I band with time after a temperature jump can be seen. The kinetic data fit the monoexponential function (Eq. 1) with the characteristic time, τ, of ∼1 min:
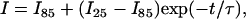 |
(1) |
where I, I85, and I25 are the Am I Raman peak intensity at delay time t.
FIGURE 6.

Kinetics of YEHK21 melting and folding initiated with temperature change. (a) DUVRR spectra of YEHK21 measured 20, 60, and 340 s after the 25–85°C temperature jump. The spectra were measured in an open solution stream. The accumulation time was 20 s. (b and c) The difference spectra shown were obtained by subtracting DUVRR spectra of YEHK21 measured 20 and 340 s, respectively, after the temperature jump from that measured before the jump. (d) The difference between the room-temperature DUVRR spectra of YEHK21 obtained for the initial folded polypeptide and that briefly heated to 125°C and cooled to room temperature (see Fig. 5). (g) DUVRR spectra of YEHK21 measured at room temperature 1, 60, and 240 min after a 5-min exposure at 100°C. The spectra were measured in an NMR tube. The accumulation time was 60 s. The spectrum of the initial folded polypeptide (black) is shown for comparison. (e and f) The difference spectra shown were obtained by subtracting the DUVRR spectrum of the initial folded YEHK21 from those measured at room temperature 60 and 240 min, respectively, after a brief exposure at 100°C.
FIGURE 7.

(a) Intensity of the Amide I band in the DUVRR spectrum of YEHK21 as a function of time after the temperature jump from 25°C to 85°C (selected spectra and the experimental conditions are given in Fig. 6 a). (b) The percent contribution of the folded conformation to the DUVRR spectra of YEHK21 measured at various stages of folding at room temperature after a brief exposure to 100°C (selected spectra and the experimental conditions are given in Fig. 6 g).
Our preliminary study showed that the YEHK21 Raman spectrum was completely or partially restored if the polypeptide was cooled after a brief (5-min) treatment at high temperature. The higher the treatment temperature, the less complete was the recovery. For example, upon heating the polypeptide to 85°C, the complete recovery of the spectrum was observed after ∼6 h at room temperature (data not shown). Eighty percent recovery was found for the sample kept at room temperature for 4 h after a brief heating to 100°C (Fig. 7 b). The recovery kinetics were found to be approximately monoexponential, with the characteristic time of ∼60 min.
For a comparison of melting and recovery kinetics at the same temperature, the YEHK21 Raman spectrum recovery was also measured at 85°C after heating to 100°C. Although the spectral changes were small in this case, the recovery occurred in the range of tens of minutes, which was still slower than the melting kinetics at 85°C. There were some indications that the recovery kinetics, as well as the completeness of recovery, depend to some extent on the heating time and the age of the sample, and therefore the extent of intermolecular aggregation.
DISCUSSION
A variety of novel experimental methods, including 2D solid-state NMR spectroscopy, electron and IR microscopy, and x-ray and neutron scattering, have been utilized in attempts to understand amyloid fibril structure at a molecular level (60–65). These methods require deposition of fibrils. Far-UV CD spectroscopy is a conventional technique for the determination of protein secondary structure and for characterization of protein structural rearrangements both under equilibrium conditions (66) and in real-time kinetic experiments (21,67,68). Far-UV CD spectra shown in Fig. 4 resemble qualitatively a reversible melting of β-sheet conformation of YEHK21 on heating and subsequent incubation at room temperature. However, the “unusual” shape of the spectrum obtained for folded YEHK21 samples complicates the quantitative analysis of the CD data. Traditional deconvolution programs did not provide good approximations of YEHK21 secondary structure transformations because globular protein databases were used to compare CD spectra (55). At this point, one can only speculate about the origin of an unusual shape and the position of the ∼206-nm trough in the CD spectrum (Fig. 4). In addition to the general suggestion that the CD of the YEHK21 polypeptide in the folded state is different relative to a β-sheet conformation of a global protein, at least two other hypotheses are possible. The difference found in CD spectra could be attributed to a strong contribution from either 1), tyrosine and/or histidine side chains in the formation of π-stacked arrays (Fig. 1), or 2), a strong contribution to the spectrum from the regularly arranged peptide turns. Since the CD phenomenon results from coupling of magnetic and electric dipole transition moments of coupled electronic transitions (69), stacked structures, which facilitate coupling of the dipole transition moments, might result in an intense CD signal. Additional studies will be required for understanding the origin of YEHK21 CD spectra and retrieving structural information about the polypeptide.
In contrast to CD, the Raman scattering phenomenon is based on short-range interactions. Consequently, regularly stacking of chromophores might result in no substantial contribution to the additive Raman scattering of individual chromophores. In contrast to CD, a Raman spectrum of the polypeptide backbone results from independent contributions of each peptide bond (70–72). As discussed below, the Raman spectroscopic signature of folded YEHK21 polypeptide backbone is very similar to a typical spectrum of homopolypeptide β-sheets and allows for quantitative characterization of YEHK21 structural rearrangements.
Resonance Raman scattering from amide chromophores makes the major contribution to Raman spectra of proteins under deep UV excitation (41,48,53,73–76). The amide chromophore Raman signature is extraordinarily sensitive to the polypeptide backbone conformation and provides direct quantitative information about the secondary structure of proteins (41,48,53,74,77,78). In the first approximation, a linear combination of the basis DUVRR spectra obtained for pure secondary structure elements, α-helix, β-sheet, or random coil, could be used to evaluate the secondary-structure composition of polypeptides and proteins from deep UV Raman spectra (48). The accuracy and validity of structural information retrieved from DUVRR spectra are determined primarily by the quality and completeness of the set of basis spectra. Chi et al. (48) determined the average pure α-helix, β-sheet, and random coil DUVRR spectra from the amide resonance Raman spectra of 13 proteins with well-known x-ray crystal structures. This set of basis spectra has allowed the determination of the protein secondary structure (48). In the case of homopolypeptides, DUVRR spectra have been reported to be somewhat different from the protein spectra (57,58). In particular, the fact that homopolypeptide amide Raman bands are narrower than those of proteins can be attributed to the greater structural inhomogeneity of proteins due to a wide range of amino acid residues. It was not surprising that the attempt to fit the YEHK21 Raman spectra using the average protein basis set gave unsatisfactory results (data not shown). Consequently, a detailed analysis of YEHK21 Raman spectra was required to retrieve quantitative information about polypeptide structural transformations.
DUVRR spectra of YEHK21 polypeptide backbone
Consistent with our fluorescence results, where we proposed that the tyrosine side chains were fully exposed to the solvent, we propose that the tyrosine contribution to the resonance Raman spectra of YEHK21 could be similar to the Raman spectrum of tyrosine tripeptide in aqueous solution. Moreover, if the contribution of tyrosine to the resonance Raman spectra of YEHK21 Raman spectra does not change with polypeptide heating, the tyrosine Raman bands could be used as an internal standard for normalization of YEHK21 Raman spectra. It is worth noting here that the use of common internal standards such as perchlorate or sulfate ions were not practical in this study because the YEHK21 peptide precipitated when perchlorate or sulfate were added in quantities sufficient to reach concentrations of ∼0.1 M.
Melted YEHK21
Fig. 8 shows the room temperature DUVRR spectrum of YEHK21 shortly after heating to 125°C. The Raman spectrum of an aqueous solution of the tyrosine tripeptide (Fig. 8) was quantitatively subtracted from the YEHK21 spectrum to separate the contribution of the polypeptide backbone. One could appreciate a similarity between the latter spectrum and the spectra of unordered homopolypeptides, such as a 20-amino-acid, mainly alanine, peptide or PGA (Fig. 8 and Table 1). The spectra of YEHK21 backbone and that of the alanine peptide are especially close. Asher et al. (59) have recently reported that polyproline II (PPII) is a dominating conformation of a thermally unfolded alanine peptide. It has been concluded that the Amide III3 UV Raman peak located at ∼1245 cm−1 (Table 1) is a characteristic signature of this conformation (57). In contrast, unordered PGA and PLL have been reported to form a mixture of two predominant conformations, polyproline II and an extended 2.51-helix (57). A characteristic UV Raman signature of the 2.51-helix, the Amide III3 peak at ∼1270 cm−1, was evident as a shoulder in the Raman spectrum of an unordered PGA (Fig. 8 and Table 1). There was no indication of the presence of this peak in the spectra of thermally denatured YEHK21, polyalanine (57) or poly-L-lysine (41,57) (Fig. 8). The observed close similarity between the Raman spectra of thermally unfolded YEHK21 and the alanine peptide enables us to make three conclusions. First, the YEHK21 polypeptide adopted a completely unordered conformation (no residual β-sheet structure) after heating to 125°C and remained disordered upon rapid cooling to room temperature. Second, our tactic of retrieving the Raman spectrum of a polypeptide backbone by subtracting the spectrum of tyrosine tripeptide in aqueous solution from the spectrum of YEHK21 was well justified. The latter approach was further supported by the observation that the tyrosine contribution was not at all evident in the difference spectra (Fig. 6, curves b–f) suggesting that this contribution did not change as a result of YEHK21 unfolding. Third, it was deduced that the thermally unfolded YEHK21 adopted a PPII conformation without any noticeable contribution from the extended 2.51-helix. Complete thermal denaturation and reversible folding make YEHK21 an excellent model for studying the folding mechanism of β-sheet proteins.
FIGURE 8.

DUVRR spectra of unordered polypeptides. (Top to bottom) The 197-nm excited room-temperature Raman spectrum of YEHK21 melted at 125°C and the Raman spectrum of tyrosine; the Raman spectrum of melted YEHK21 backbone obtained as a difference between the spectrum of melted YEHK21 and the spectrum of tyrosine; the 204-nm DUVRR spectrum of 20-amino acid mainly alanine peptide adopted from Lednev et al. (58), previously reported (59) to be dominated by PPII conformation; the 197-nm DUVRR spectrum of PGA adopting PPII and extended 2.51-helix conformations (59).
TABLE 1.
Amide UV Raman bands (cm−1) of unordered polypeptides
| YEHK21 denatured | PGA unfolded (this work) | PGA unfolded (57), PPII + extended 2.51-helix | AP unfolded (57), essentially PPII | |
|---|---|---|---|---|
| Am I | 1667 | 1672 | 1673 | 1659 |
| Am II | 1556 | 1560 | 1555 | 1548 |
| CαH(1) | 1392 | 1394 | 1396 | 1399 |
| CαH(2) | 1377 | 1360 weak | 1359 | 1377 broad |
| Am III2 | 1299 | 1315 | 1319 | 1311 |
| ∼1295 | 1298 | |||
| Am III3 (2.51-helix) | 1266 shoulder | 1269 | NA | |
| Am III3 (PPII) | 1246 | 1243 | 1240 | 1247 |
AP, alanine peptide.
The major discrepancy between the Raman spectra of the polyglutamic acid and unordered YEHK21, where the sequence was dominated by the AG repeats, occurred in the area of CαH bending mode. This is not surprising since the resonance Raman band associated with CαH bending vibration is very sensitive to the polypeptide sequence. In fact, the CαH bending band in the unordered YEHK21 Raman spectrum is more like the corresponding band in the spectrum reported for the unordered mainly alanine peptide (Fig. 8).
Folded YEHK21
Fig. 1 depicts schematically the major structural motif of folded YEHK21 consistent with all data available at this moment (42,45). Although a β-sheet conformation dominates the proposed structure, the contribution of turn-type conformations to the spectra might be as high as 15–30% depending on the turn type and the number of amide chromophores involved. Differing turn motifs can be assumed by β-hairpins as reflected in the number of amino acids per turn and the number of hydrogen bonds formed between the distal strands (79,80) with a consequence that the faces of β-sheet may be differentiated by the orientation of the methyl groups of constituent strands toward a single face of the sheet. It has been previously shown that closely related constructs poly((AG)3EG), poly((AG)3YG) and poly((AG)3KG) form amphiphilic β-sheet structures via the intermediacy of γ-turns to redirect the β-strand subunits (81). If a γ-turn-containing structure is assumed for YEHK21, as shown in Fig. 1, the amphiphilic character of the resultant β-sheet would be consistent with the observed bilayer formation (42). There is very little information about DUVRR spectral signature of turns (82,83). This is mainly due to the relatively low abundance of turns in proteins and the difficulty in preparing a stable turn using a small peptide.
Fig. 9 compares the DUVRR spectra of a folded YEHK21 and a pure β-sheet conformation obtained for a homopolypeptide, polylysine, and a lysozyme. Aromatic amino acid contributions to the spectra (tyrosine in the case YEHK21, phenylalanine and tyrosine for lysozyme) were quantitatively subtracted. There was an obvious similarity between all three spectra especially in the areas of the Am I, Am II, and Am III bands (Table 2). The substantial differences evident in the area of CαH bending mode might be attributed to the nature of amino acid residues forming β-sheet conformation.
FIGURE 9.

DUVRR spectra of ordered, mainly β-sheet, polypeptide backbones. (Top to bottom) The 197-nm excited Raman spectrum of mainly β-sheet lysozyme backbone obtained from the lysozyme fibril Raman spectrum (56) by subtracting the contribution of aromatic amino acid residues. DUVRR spectrum of folded YEHK21 backbone obtained from the spectrum of folded YEHK21 (Fig. 5) by subtracting the contribution of tyrosine. DUVRR spectrum of β-sheet PLL (see Results).
TABLE 2.
Amide UV Raman bands (cm−1) of β-sheet-rich polypeptides
| YEHK21 | Lysozyme fibrils (56) | PLL, pure β-sheet spectrum | PLL-PGA mixture (57), ∼60% β-sheet, ∼40% unfolded | PLL-PGA mixture (57), pure β-sheet spectrum | |
|---|---|---|---|---|---|
| Am I | 1665 | 1674 | 1671 | 1668 | 1672 |
| Am II | 1555 | 1558 | 1561 | 1548 | 1550 |
| CαH(1) | 1391 | 1400 | 1405 | 1402 | 1403 |
| CαH(2) | 1374 | 1371 shoulder | 1350 | 1381 | 1379 weak |
| Am III2 | 1316 | 1285 shoulder | (1350) | 1290 | |
| 1310 | |||||
| Am III3 | 1243 | 1233 | 1237 | 1239 | 1228 |
As evident from Figs. 8 and 9 and Tables 1 and 2, the deep UV Raman spectroscopic signatures of polypeptide backbones in disordered and β-sheet conformations are strikingly different. The shape of the Am I Raman peak (narrow for β-sheet and broad for unordered conformations) and the relative intensities of Am I and Am II peaks could be used to distinguish the β-conformation of a polypeptide backbone. A comparative analysis of the Raman spectra (Fig. 9 and Table 2) indicates that the secondary structure of naive YEHK21 is dominated by a β-sheet conformation. These Raman spectroscopic data confirmed the conclusion derived from our earlier transmission electron microscopy results (42) that the initial YEHK21 was completely folded, with very few if any unordered regions. In particular, deposition from solution onto a highly oriented pyrolytic graphite surface followed by atomic force and transmission electron microscopy studies (42) indicated the presence of fibrils with very few if any unordered (not involved in the fibril structure) polypeptide domains. From this viewpoint, YEHK21, a 687-residue peptide, folds into a single “native” state stabilized by internal hydrogen bonds.
Melting curve
The absence of reliable basis DUVRR spectra for the YEHK21 β-sheet and turns made it impossible to retrieve complete quantitative information about structural rearrangements of the polypeptide during thermal denaturation. Instead, we utilized three spectra including two YEHK21 backbone Raman spectra obtained at room temperature for 1), the initial sample (Fig. 9); 2), the sample treated at 125°C (Fig. 8); and 3), the Raman spectrum of tyrosine (Fig. 8) as a set of basis spectra to which the Raman spectra measured for the polypeptide solutions treated at various temperatures (Fig. 5) can be fitted. The first spectrum represents completely folded YEHK21 composed mainly of β-sheet and turn contributions. The second spectrum represented an unordered YEHK21 in the predominant PPII conformation. We chose to use the tyrosine contribution as a separate basis spectrum because this allowed us to utilize tyrosine as an internal standard.
Fig. 5 shows the fitting results of Raman spectra of YEHK21 treated at various temperatures. In addition to experimental spectra and best linear combinations of three basis spectra, the residual spectra are also shown for the temperatures of 35° and 85°C. One could appreciate that the fitting was very good and that allowed several conclusions to be drawn. Firstly, these fitting results provide further support for our suggestion that the tyrosine contribution to the Raman spectrum of YEHK21 did not change as a result of heating the polypeptide to various temperatures. Second, two conformations that could be preliminarily described as folded and unfolded dominate in various proportions at different points on the melting curve. Melting of the predominantly β-sheet/turn conformation resulted directly in unfolded polypeptide with the dominant PPII conformation. No contribution from any of the intermediate states was evident independent of the treatment temperature. The two-state behavior found for YEHK21 melting does not necessarily result from an all-or-none transition. For example, a 20-amino-acid residue consisting mainly of alanine has exhibited a two-state-like melting transition from the α-helical state to the predominate PPII conformation without any noticeable intermediates based on DUVRR spectra (58,59). At the same time, isotope labeling has validated the proposal for a gradual melting with temperature of the alanine peptide helices instead of an all-or-none transition (84). Chemometric treatments of electrospray ionization mass spectra and DUVRR spectra have been utilized to establish the presence of the all-or-none transition during thermal denaturation of lysozyme (M. Xu, V. A. Shashilov, V. V. Ermolenkov, L. Fredriksen, and I. K. Lednev, unpublished data). The utilization of this method for testing the thermal denaturation of YEHK21 is currently in progress in our laboratory.
The fitting results of the Raman spectra of YEHK21 treated at various temperatures allows determination of the relative proportions of the YEHK21 polypeptide in the folded and unfolded states. The corresponding melting curve is shown in Fig. 10. As a first approximation, the data was fitted by Eq. 1 to determine the thermodynamic parameters associated with the unfolded↔folded equilibrium,
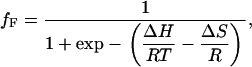 |
(2) |
where fF is the fraction of folded polypeptide after treatment at temperature T and R is the gas constant. This equation is normally applied to two-state processes only and then only when the effect of heat capacity change ΔCp on the enthalpy of folding is negligible over the narrow temperature range of the transition (86). The curve in Fig. 10 shows the least-squares best fit of Eq. 2 to the melting data, which yields R2 > 0.996. The obtained thermodynamic parameters were ΔH = 135 ± 14 kJ/mol and ΔS = 370 ± 40 J/(mol K), where a standard error represented ±2σ. Since the YEHK21 melting discussed below is most probably not a two-state process, the estimated thermodynamic parameters should be considered as preliminary and interpreted with caution. We plan a more detailed study of YEHK21 melting thermodynamics in the near future, including the estimation of ΔCp using scanning calorimetry and the effect of denaturants on the melting curve and folding/unfolding kinetics.
FIGURE 10.

YEHK21 melting curve. The solid line represents the best fit with Eq 2.
Denaturation and refolding kinetics
The relaxation of YEHK21 to a new equilibrium conformation, after the temperature jump from 25° to 85°C, occurs monoexponentially, with a characteristic time of ∼1 min (Fig. 7 a). Transient DUVRR spectra measured at various delay times after the temperature change were accumulated for 20 s, with the result that the spectra were quite noisy (Fig. 6 a). The fitting of these spectra with the three basis spectra, utilized for the fitting of the room-temperature steady-state spectra obtained for samples treated at various temperatures, would not be justified since the basis spectra obtained for completely folded and completely unfolded polypeptide could be temperature-dependent (57,58). It is important that the changes in the Raman spectrum of YEHK21 with time (Fig. 6, difference spectra b and c) were consistent with those during polypeptide melting (Fig. 6, difference spectrum d).
The incubation of unfolded YEHK21 at room temperature resulted in folding, as was evident from the difference spectra presented in Fig. 6 (curves e and f). Since the recovery kinetics were relatively slow, the spectra were accumulated for 1 min. These transient spectra were fit well by a linear combination of the three basis spectra, the room-temperature folded and unfolded YEHK21 backbone and tyrosine spectra. The contribution of the folded conformation was evaluated from the fitting of the transient spectra as a function of the incubation time at room temperature (Fig. 7 b). The solid curve represents the best fit by a monoexponential function, with a characteristic time of ∼60 min. This suggests that the folding process is much slower than unfolding. As was noted in Results, the incubation was also effected at 85°C for a sample heated briefly to 100°C. Although the degree of folding was relatively small, the characteristic time was much longer than the melting time at the same temperature. Consequently, the YEHK21 thermal folding-unfolding process could not be described as a simple two-state model. The same characteristic time is expected for relaxation processes occurring at the same temperature regardless of the initial state of the system.
Although 1-min melting and 60-min folding seem like slow processes, much longer folding kinetics would be expected (87) for a protein of size comparable to that of YEHK21 (687-amino-acid residues). The faster folding dynamics of YEHK21 relative to a typical globular protein could be attributed to a less frustrated free-energy landscape funnel for YEHK21 folding. The YEHK21 polypeptide is mainly composed of electrically neutral amino acid residues, with only glutamic acids being charged. This structure might result in an important balance between the absence of strong nonnative contacts (salt bridges or hydrophobic collapse) and limited repulsion of negatively charged side chains. Alternatively, a relatively fast refolding of YEHK21 can be attributed to the presence of residual β-sheet structures (nuclei) surviving thermal denaturation that preceded the refolding experiments. The presence of prearranged nucleating β-sheet regions could influence both the rate and the degree of folding. More detailed description of the YEHK21 folding mechanism will require additional investigation, including MD simulations. Speculation at this point must be based on published information on related systems. An accurate prediction of the protein folding rate has typically required consideration of the contact order of the 3D protein structure (88,89). Alternatively, a method has been suggested for prediction of the folding rates for proteins and short peptides from their primary structure without any information on the 3D folding (89). In the latter approach, the folding rates have been estimated from sequence-dependent secondary-structure predictions and sequence length. Theoretically, the protein chain length is determined by a number of links between folded units (not the number of amino acid residues) that substantially reduces the effective length of the folding chain (90). For example, a high helical content has been noted to accelerate folding of two-state proteins due to a rapid and independent formation of folded segments (89,91); local helix folding in a globular protein has been reported to occur on a nanosecond timescale (22). A similar approach has not been realized for β-sheet domains in proteins, largely because internally stable and therefore rapidly folding β-hairpins could not be predicted based on an amino acid sequence (89). Nevertheless, it has been well documented that the formation of local structures, such as helices, hairpins, and loops, can be orders of magnitude more rapid than the rate-limiting step in protein folding (92). Consequently, a simple hypothetical mechanism of YEHK21 folding predicated upon multiple nucleation (since the chain length exceeds 100 amino acid residues (93)) and spontaneous formation of β-sheet segments could explain the relatively fast folding found for YEHK21. Although the β-sheet folding is often a slower process than the α-helix formation, the characteristic time is still in the microsecond time range (11,12). It is evident from the data presented in Fig. 7 that there was no noticeable change in the β-sheet content during the first micro- and even milliseconds of YEHK21 thermal denaturation and reverse folding. Most probably, there is no substantial contribution from the fast spontaneous formation of isolated β-sheet segments at early stages of folding. Such isolated β-sheet segments could form as a result of nonlocal interactions, perhaps simply as a result of the fact that the stabilizing hydrogen bonds are between residues distant in sequence space (94).
CONCLUSIONS
Amyloid formation can be considered self-assembly into a very well defined structure, e.g., one-dimensional crystallization (95,96). This process can occur at as low as nanomolar concentrations, indicating that specific molecular recognition elements mediate this process (95). The investigation of small peptide fragments initiating the fibrillation is therefore an important and trendy approach to the study of fibrillation mechanisms. However, tertiary structural elements, which play an important role in globular proteins (16–23), could not be modeled in such small polypeptide-based systems. Our long-term strategy for studying protein folding, with particular focus on aggregation and fibrillation phenomena, is based on the development of a family of large polypeptides that would form fibrils and show reversible folding, and establishment of a structural relationship between polypeptide sequence and folding properties. To the best of our knowledge, YEHK21 is the first example of a large polypeptide genetically engineered from repetitive units, which forms an extended β-sheet conformation and a fibril-type structure. The thermal reversibility of β-sheet folding demonstrated in this study illustrates the promise of this system for investigations of the mechanism of folding and fibrillation. We used deep UV Raman spectroscopy to characterize the secondary structure of folded and thermally denatured conformations of YEHK21. By demonstrating efficient, reversible β-sheet folding and fibrillation of YEHK21, we established the basis for studying the detailed mechanism of fibrillation that utilizes all the potential of genetic engineering.
A de novo 687-amino acid residue polypeptide, YEHK21, has been designed to self-assemble into antiparallel β-sheet structures (42). Deposition from the solution onto a highly oriented pyrolytic graphite surface followed by atomic force and transmission electron microscopy studies (42) indicated the presence of fibrils with very little if any unordered (not involved in the fibril structure) parts of polypeptide. From this viewpoint, YEHK21, a 687-residue peptide, folds into a single “native” state stabilized by internal hydrogen bonds. At the same time, heating of YEHK21 resulted in complete and reversible denaturation that makes this system an interesting model for studying the folding mechanism of globular β-sheet proteins. In particular, as a large polypeptide with numerous “native” tertiary contacts, YEHK21 exhibited folding/unfolding dynamics that are quite different from those found for small β-sheet-forming peptides.
Although the far-UV CD spectra resembled qualitatively a melting of the YEHK21 β-sheet conformation on heating and subsequent incubation at room temperature, the “unusual” shape of the CD spectra complicated structural interpretation of CD data. In contrast to CD, the Raman scattering phenomenon is based on short-range interactions. Consequently, a regular stacking of chromophores, which strongly influence CD spectra, should result in no substantial contribution to the additive Raman scattering of individual chromophores. The Raman spectroscopic signature of folded YEHK21 polypeptide backbone is very similar to a typical spectrum of homopolypeptide β-sheet conformation and therefore facilitated the use of Raman spectroscopy for quantitative characterization of YEHK21 structural rearrangements. Raman data established that heating of YEHK21 to 125°C resulted in complete denaturation of the β-sheet and formation of a predominant PPII conformation. The YEHK21 Raman spectra obtained for various heating points were found to fit well to a linear combination of spectra of completely folded, predominantly β-sheet, and PPII conformational spectral signatures. No noticeable contribution of an intermediate state between these two forms was found. This did not necessarily indicate an all-or-none transition and/or a two-state reaction mechanism. Most probably, the denatured portion of a polypeptide molecule quickly adopted PPII conformation, leaving a small (spectroscopically insignificant) intermediate area between folded and PPII conformations. Both the tyrosine fluorescence spectrum and tyrosine Raman signature were unchanged in the course of YEHK21 thermal denaturation. Moreover, the tyrosine fluorescence spectrum of YEHK21 was practically identical to that of a model tripeptide and was indicative of the complete exposure of tyrosine side chains to water in both folded and unfolded forms of YEHK21.
Thermal denaturation of YEHK21 initiated by the 25–85°C temperature jump appeared to be monoexponential, with a characteristic time of ∼1 min. The folding of heat denatured YEHK21 on cooling of the solution followed monoexponential kinetics as well. However, folding was ∼2 orders of magnitude slower than melting, indicating that the folding mechanism is not reproduced well by a simple two-state model. No noticeable formation of a β-sheet conformation in subminutes was found. This indicated that no hairpin-type local structures were formed on this timescale. This is in contrast to short β-sheet peptides, which have been reported to fold in microseconds (11,12). The absence of fast spontaneous formation of isolated β-sheet segments at early stages of folding could be attributed to nonlocal “nonnative” interactions. Thus, YEHK21 folding dynamics agree well with a general property of β-sheet proteins; that is, initial collapse precedes secondary structure formation (15,39,40).
Although 1-min melting and 60-min folding are superficially slow processes, much longer folding kinetics would be expected (87) for a protein with a size comparable to YEHK21 (687-amino-acid residues). The faster folding dynamics of YEHK21 relative to a typical globular protein could be attributed to a less frustrated free-energy landscape funnel for YEHK21 folding. The YEHK21 polypeptide is mainly composed of electrically neutral amino acid residues with only glutamic acids being charged. This structure might provide an important balance between the absence of strong nonnative contacts (salt bridges or hydrophobic collapse) and limited repulsion of negatively charged side chains. Alternatively, a relatively fast refolding of YEHK21 could be the result of residual β-sheet structures (nuclei) that might survive thermal denaturation preceding the refolding experiments. Further work will be required to elucidate a detailed folding mechanism for YEHK21. In particular, we will evaluate 1), the possibility of YEHK21 folding via an initial-stage Flory-type coil-to-global collapse (29), and 2), the role of intermolecular aggregation under folding conditions. The important outcome of the study we have presented is that this very large genetically engineered protein exhibits remarkable folding properties and offers a novel platform for study of β-sheet and fibrillar structures.
Acknowledgments
I.K.L. acknowledges Research Corporation grant RI1229 for financial support. J.T.W. acknowledges support of this work by the Microelectronics Advanced Research Corporation (MARCO), the Defense Advanced Research Programs Agency (DARPA), and the New York State Office of Science, Technology and Academic Research (NYSTAR) through the Interconnect Focus Center (IFC) and the Materials, Structures, and Devices (MSD) Center.
Vladimir V. Ermolenkov is on leave from the B. I. Stepanov Institute of Physics, National Academy of Sciences of Belarus, Minsk, Belarus.
References
- 1.Mershin, A., B. Cook, L. Kaiser, and S. Zhang. 2005. A classic assembly of nanobiomaterials. Nat. Biotechnol. 23:1379–1380. [DOI] [PubMed] [Google Scholar]
- 2.Zhao, X., and S. Zhang. 2004. Fabrication of molecular materials using peptide construction motifs. Trends Biotechnol. 22:470–476. [DOI] [PubMed] [Google Scholar]
- 3.Lamm, M. S., K. Rajagopal, J. P. Schneider, and D. J. Pochan. 2005. Laminated morphology of nontwisting beta-sheet fibrils constructed via peptide self-assembly. J. Am. Chem. Soc. 127:16692–16700. [DOI] [PubMed] [Google Scholar]
- 4.Aggeli, A., M. Bell, N. Boden, L. M. Carrick, and A. E. Strong. 2003. Self-assembling peptide polyelectrolyte beta-sheet complexes form nematic hydrogels. Angew. Chem. Int. Ed. Engl. 42:5603–5606. [DOI] [PubMed] [Google Scholar]
- 5.Maskarinec, S. A., and D. A. Tirrell. 2005. Protein engineering approaches to biomaterials design. Curr. Opin. Biotechnol. 16:422–426. [DOI] [PubMed] [Google Scholar]
- 6.Langer, R., and D. A. Tirrell. 2004. Designing materials for biology and medicine. Nature. 428:487–492. [DOI] [PubMed] [Google Scholar]
- 7.Clark, J., E. M. Singer, D. R. Korns, and S. S. Smith. 2004. Design and analysis of nanoscale bioassemblies. Biotechniques. 36:992–996, 998–1001. [DOI] [PubMed] [Google Scholar]
- 8.Sun, S., R. Brem, H. S. Chan, and K. A. Dill. 1995. Designing amino acid sequences to fold with good hydrophobic cores. Protein Eng. 8:1205–1213. [DOI] [PubMed] [Google Scholar]
- 9.Chakrabartty, A., T. Kortemme, and R. L. Baldwin. 1994. Helix propensities of the amino acids measured in alanine-based peptides without helix-stabilizing side-chain interactions. Protein Sci. 3:843–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sanford, A. R., K. Yamato, X. Yang, L. Yuan, Y. Han, and B. Gong. 2004. Well-defined secondary structures. Eur. J. Biochem. 271:1416–1425. [DOI] [PubMed] [Google Scholar]
- 11.Munoz, V., P. A. Thompson, J. Hofrichter, and W. A. Eaton. 1997. Folding dynamics and mechanism of β-hairpin formation. Nature. 390:196–199. [DOI] [PubMed] [Google Scholar]
- 12.de Alba, E., J. Santoro, M. Rico, and M. A. Jimenez. 1999. De novo design of a monomeric three-stranded antiparallel β-sheet. Protein Sci. 8:854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Perczel, A., I. Jakli, B. M. Foxman, and G. D. Fasman. 1996. A search for the ideal type I β-turn. Biopolymers. 38:723–732. [DOI] [PubMed] [Google Scholar]
- 14.Thakur, A. K., and R. Kishore. 2000. Stabilization of a novel β-turn-like motif by nonconventional intramolecular hydrogen-bonding interactions in a model peptide incorporating β-alanine. Biopolymers. 53:447–454. [DOI] [PubMed] [Google Scholar]
- 15.Sheinerman, F. B., and C. L. Brooks, 3rd. 1998. Molecular picture of folding of a small α/β protein. Proc. Natl .Acad. Sci. USA. 95:1562–1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Clementi, C., A. E. Garcia, and J. N. Onuchic. 2003. Interplay among tertiary contacts, secondary structure formation and side-chain packing in the protein folding mechanism: all-atom representation study of protein L. J. Mol. Biol. 326:933–954. [DOI] [PubMed] [Google Scholar]
- 17.Deechongkit, S., H. Nguyen, E. T. Powers, P. E. Dawson, M. Gruebele, and J. W. Kelly. 2004. Context-dependent contributions of backbone hydrogen bonding to β-sheet folding energetics. Nature. 430:101–105. [DOI] [PubMed] [Google Scholar]
- 18.Uversky, V. N., V. P. Kutyshenko, N. Protasova, V. V. Rogov, K. S. Vassilenko, and A. T. Gudkov. 1996. Circularly permuted dihydrofolate reductase possesses all the properties of the molten globule state, but can resume functional tertiary structure by interaction with its ligands. Protein Sci. 5:1844–1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dill, K. A., S. Bromberg, K. Yue, K. M. Fiebig, D. P. Yee, P. D. Thomas, and H. S. Chan. 1995. Principles of protein folding: a perspective from simple exact models. Protein Sci. 4:561–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brooks, C. L., 3rd, M. Gruebele, J. N. Onuchic, and P. G. Wolynes. 1998. Chemical physics of protein folding. Proc. Natl. Acad. Sci. USA. 95:11037–11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen, E., R. A. Goldbeck, and D. S. Kliger. 1997. Nanosecond time-resolved spectroscopy of biomolecular processes. Annu. Rev. Biophys. Biomol. Struct. 26:327–355. [DOI] [PubMed] [Google Scholar]
- 22.Gilmanshin, R., S. Williams, R. H. Callender, W. H. Woodruff, and R. B. Dyer. 1997. Fast events in protein folding: relaxation dynamics of secondary and tertiary structure in native apomyoglobin. Proc. Natl. Acad. Sci. USA. 94:3709–3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Petrey, D., and B. Honig. 2000. Free energy determinants of tertiary structure and the evaluation of protein models. Protein Sci. 9:2181–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mayor, U., N. R. Guydosh, C. M. Johnson, J. G. Grossmann, S. Sato, G. S. Jas, S. M. Freund, D. O. Alonso, V. Daggett, and A. R. Fersht. 2003. The complete folding pathway of a protein from nanoseconds to microseconds. Nature. 421:863–867. [DOI] [PubMed] [Google Scholar]
- 25.Ahmed, Z., I. A. Beta, A. V. Mikhonin, and S. A. Asher. 2005. UV-resonance Raman thermal unfolding study of Trp-cage shows that it is not a simple two-state miniprotein. J. Am. Chem. Soc. 127:10943–10950. [DOI] [PubMed] [Google Scholar]
- 26.Vu, D. M., J. K. Myers, T. G. Oas, and R. B. Dyer. 2004. Probing the folding and unfolding dynamics of secondary and tertiary structures in a three-helix bundle protein. Biochemistry. 43:3582–3589. [DOI] [PubMed] [Google Scholar]
- 27.Garcia, A. E., and J. N. Onuchic. 2003. Folding a protein in a computer: an atomic description of the folding/unfolding of protein A. Proc. Natl. Acad. Sci. USA. 100:13898–13903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Alonso, D. O., and V. Daggett. 2000. Staphylococcal protein A: unfolding pathways, unfolded states, and differences between the B and E domains. Proc. Natl. Acad. Sci. USA. 97:133–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gillespie, B., and K. W. Plaxco. 2004. Using protein folding rates to test protein folding theories. Annu. Rev. Biochem. 73:837–859. [DOI] [PubMed] [Google Scholar]
- 30.Thirumalai, D., and D. K. Klimov. 1999. Deciphering the timescales and mechanisms of protein folding using minimal off-lattice models. Curr. Opin. Struct. Biol. 9:197–207. [DOI] [PubMed] [Google Scholar]
- 31.Fersht, A. 1999. Structure and Mechanism in Protein Folding. W. H. Freeman, New York.
- 32.Dill, K. A., and H. S. Chan. 1997. From evinthal to pathways to funnels. Nat. Struct. Biol. 4:10–19. [DOI] [PubMed] [Google Scholar]
- 33.Kuhlman, B., and D. Baker. 2004. Exploring folding free energy landscapes using computational protein design. Curr. Opin. Struct. Biol. 14:89–95. [DOI] [PubMed] [Google Scholar]
- 34.Brooks, C. L., 3rd. 2002. Protein and peptide folding explored with molecular simulations. Acc. Chem. Res. 35:447–454. [DOI] [PubMed] [Google Scholar]
- 35.Cardenas, A. E., and R. Elber. 2003. Kinetics of cytochrome C folding: atomically detailed simulations. Proteins. 51:245–257. [DOI] [PubMed] [Google Scholar]
- 36.Uzawa, T., S. Akiyama, T. Kimura, S. Takahashi, K. Ishimori, I. Morishima, and T. Fujisawa. 2004. Collapse and search dynamics of apomyoglobin folding revealed by submillisecond observations of α-helical content and compactness. Proc. Natl. Acad. Sci. USA. 101:1171–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akiyama, S., S. Takahashi, T. Kimura, K. Ishimori, I. Morishima, Y. Nishikawa, and T. Fujisawa. 2002. Conformational landscape of cytochrome c folding studied by microsecond-resolved small-angle x-ray scattering. Proc. Natl. Acad. Sci. USA. 99:1329–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ferguson, N., and A. R. Fersht. 2003. Early events in protein folding. Curr. Opin. Struct. Biol. 13:75–81. [DOI] [PubMed] [Google Scholar]
- 39.Shea, J. E., J. N. Onuchic, and C. L. Brooks, 3rd. 2002. Probing the folding free energy landscape of the Src-SH3 protein domain. Proc. Natl. Acad. Sci. USA. 99:16064–16068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kimura, T., T. Uzawa, K. Ishimori, I. Morishima, S. Takahashi, T. Konno, S. Akiyama, and T. Fujisawa. 2005. Specific collapse followed by slow hydrogen-bond formation of β-sheet in the folding of single-chain monellin. Proc. Natl. Acad. Sci. USA. 102:2748–2753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jiji, R. D., G. Balakrishnan, Y. Hu, and T. G. Spiro. 2006. Intermediacy of poly(L-proline) II and β-strand conformations in poly(L-lysine) β-sheet formation probed by temperature-jump/UV resonance Raman spectroscopy. Biochemistry. 45:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Topilina, N. I., S. Higashiya, N. Rana, V. V. Ermolenkov, C. Kossow, A. Carlsen, S. C. Ngo, C. C. Wells, E. T. Eisenbraun, K. A. Dunn, I. K. Lednev, R. E. Geer, A. E. Kaloyeros, and J. T. Welch. 2006. Bilayer fibril formation by genetically engineered polypeptides: preparation and characterization. Biomacromolecules. 7:1104–1111. [DOI] [PubMed] [Google Scholar]
- 43.Cantor, E. J., E. D. Atkins, S. J. Cooper, M. J. Fournier, T. L. Mason, and D. A. Tirrell. 1997. Effects of amino acid side-chain volume on chain packing in genetically engineered periodic polypeptides. J. Biochem. (Tokyo). 122:217–225. [DOI] [PubMed] [Google Scholar]
- 44.Aggeli, A., I. A. Nyrkova, M. Bell, R. Harding, L. Carrick, T. C. McLeish, A. N. Semenov, and N. Boden. 2001. Hierarchical self-assembly of chiral rod-like molecules as a model for peptide β-sheet tapes, ribbons, fibrils, and fibers. Proc. Natl. Acad. Sci. USA. 98:11857–11862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Higashiya, S., S. C. Ngo, K. S. Bousman, X. Jin, J. T. Welch, R. P. Cunningham, E. T. Eisenbraun, R. E. Geer, and A. E. Kaloyeros. 2003. Efficient biological construction of repetitive polypeptides for interconnect applications by block copolymerization. Polym. Prepr. (Am. Chem. Soc., Div. Polym. Chem.) 44:679–680. [Google Scholar]
- 46.Demchenko, A. P. 1986. Ultraviolet Spectroscopy of Proteins. Springer-Verlag, Berlin.
- 47.Lednev, I. K., V. V. Ermolenkov, W. He, and M. Xu. 2005. Deep-UV Raman spectrometer tunable between 193 and 205 nm for structural characterization of proteins. Anal. Bioanal. Chem. 381:431–437. [DOI] [PubMed] [Google Scholar]
- 48.Chi, Z., X. G. Chen, J. S. Holtz, and S. A. Asher. 1998. UV resonance Raman-selective amide vibrational enhancement: quantitative methodology for determining protein secondary structure. Biochemistry. 37:2854–2864. [DOI] [PubMed] [Google Scholar]
- 49.Asher, S. A., A. Ianoul, G. Mix, M. N. Boyden, A. Karnoup, M. Diem, and R. Schweitzer-Stenner. 2001. Dihedral psi angle dependence of the amide III vibration: a uniquely sensitive UV resonance Raman secondary structural probe. J. Am. Chem. Soc. 123:11775–11781. [DOI] [PubMed] [Google Scholar]
- 50.Fandrich, M., and C. M. Dobson. 2002. The behavior of polyamino acids reveals an inverse side chain effect in amyloid structure formation. EMBO J. 21:5682–5690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu, Q., F. Li, W. Wang, M. H. Hecht, and T. G. Spiro. 2002. UV Raman monitoring of histidine protonation and H-(2)H exchange in plastocyanin. J. Inorg. Biochem. 88:381–387. [DOI] [PubMed] [Google Scholar]
- 52.Asher, S. A. 2001. Ultraviolet Raman spectrometry. In Handbook of Vibrational Spectroscopy. John Wiley & Sons, New York. 557–571.
- 53.Copeland, R. A., and T. G. Spiro. 1987. Secondary structure determination in proteins from deep (192–223-nm) ultraviolet Raman spectroscopy. Biochemistry. 26:2134–2139. [DOI] [PubMed] [Google Scholar]
- 54.Hudson, B., and L. Mayne. 1986. Ultraviolet resonance Raman spectroscopy of biopolymers. Methods Enzymol. 130:331–350. [DOI] [PubMed] [Google Scholar]
- 55.Wilson, D., R. Valluzzi, and D. Kaplan. 2000. Conformational transitions in model silk peptides. Biophys. J. 78:2690–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xu, M., V. V. Ermolenkov, W. He, V. N. Uversky, L. Fredriksen, and I. K. Lednev. 2005. Lysozyme fibrillation: deep UV Raman spectroscopic characterization of protein structural transformation. Biopolymers. 79:58–61. [DOI] [PubMed] [Google Scholar]
- 57.Mikhonin, A. V., N. S. Myshakina, S. V. Bykov, and S. A. Asher. 2005. UV resonance Raman determination of polyproline II, extended 2.5(1)-helix, and β-sheet psi angle energy landscape in poly-L-lysine and poly-L-glutamic acid. J. Am. Chem. Soc. 127:7712–7720. [DOI] [PubMed] [Google Scholar]
- 58.Lednev, I. K., A. S. Karnoup, M. C. Sparrow, and S. A. Asher. 1999. Alpha-helix peptide folding and unfolding activation barriers: a nanosecond UV resonance Raman study. J. Am. Chem. Soc. 121:8074–8086. [Google Scholar]
- 59.Asher, S. A., A. V. Mikhonin, and S. Bykov. 2004. UV Raman demonstrates that α-helical polyalanine peptides melt to polyproline II conformations. J. Am. Chem. Soc. 126:8433–8440. [DOI] [PubMed] [Google Scholar]
- 60.Tycko, R. 2004. Progress toward a molecular-level structural understanding of amyloid fibrils. Curr. Opin. Struct. Biol. 14:96–103. [DOI] [PubMed] [Google Scholar]
- 61.Hiramatsu, H., Y. Goto, H. Naiki, and T. Kitagawa. 2004. Core structure of amyloid fibril proposed from IR-microscope linear dichroism. J. Am. Chem. Soc. 126:3008–3009. [DOI] [PubMed] [Google Scholar]
- 62.Torok, M., S. Milton, R. Kayed, P. Wu, T. McIntire, C. G. Glabe, and R. Langen. 2002. Structural and dynamic features of Alzheimer's Aβ peptide in amyloid fibrils studied by site-directed spin labeling. J. Biol. Chem. 277:40810–40815. [DOI] [PubMed] [Google Scholar]
- 63.Harper, J. D., C. M. Lieber, and P. T. Lansbury, Jr. 1997. Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer's disease amyloid-β protein. Chem. Biol. 4:951–959. [DOI] [PubMed] [Google Scholar]
- 64.Nelson, R., M. R. Sawaya, M. Balbirnie, A. O. Madsen, C. Riekel, R. Grothe, and D. Eisenberg. 2005. Structure of the cross-β spine of amyloid-like fibrils. Nature. 435:773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sambashivan, S., Y. Liu, M. R. Sawaya, M. Gingery, and D. Eisenberg. 2005. Amyloid-like fibrils of ribonuclease A with three-dimensional domain-swapped and native-like structure. Nature. 437:266–269. [DOI] [PubMed] [Google Scholar]
- 66.Fasman, G. D., editor. 1996. Circular Dichroism and the Conformational Analysis of Biomolecules. Plenum, New York.
- 67.Goldbeck, R. A., R. M. Esquerra, and D. S. Kliger. 2002. Hydrogen bonding to Trp b37 is the first step in a compound pathway for hemoglobin allostery. J. Am. Chem. Soc. 124:7646–7647. [DOI] [PubMed] [Google Scholar]
- 68.Chen, E., T. Gensch, A. B. Gross, J. Hendriks, K. J. Hellingwerf, and D. S. Kliger. 2003. Dynamics of protein and chromophore structural changes in the photocycle of photoactive yellow protein monitored by time-resolved optical rotatory dispersion. Biochemistry. 42:2062–2071. [DOI] [PubMed] [Google Scholar]
- 69.Nakanishi, K., N. Berova, and R. W. Woody, editors. 1994. Circular Dichroism: Principles and Applications. VCH, New York.
- 70.Mikhonin, A. V., and S. A. Asher. 2005. Uncoupled peptide bond vibrations in α-helical and polyproline II conformations of polyalanine peptides. J. Phys. Chem. B. 109:3047–3052. [DOI] [PubMed] [Google Scholar]
- 71.Mix, G., R. Schweitzer-Stenner, and S. A. Asher. 2000. Uncoupled adjacent amide vibrations in small peptides. J. Am. Chem. Soc. 122:9028–9029. [Google Scholar]
- 72.Ozdemir, A., I. K. Lednev, and S. A. Asher. 2002. Comparison between UV Raman and circular dichroism detection of short α helices in Bombolitin III. Biochemistry. 41:1893–1896. [DOI] [PubMed] [Google Scholar]
- 73.Zhao, X., R. Chen, C. Tengroth, and T. G. Spiro. 1999. Solid-state tunable kHz ultraviolet laser for Raman applications. Appl. Spectrosc. 53:1200–1205. [Google Scholar]
- 74.Holtz, J. S. W., R. W. Bormett, Z. Chi, N. Cho, X. G. Chen, V. Pajcini, S. A. Asher, L. Spinelli, P. Owen, and M. Arrigoni. 1996. Applications of a new 206 5-nm continuous-wave laser source: UV Raman determination of protein secondary structure and CVD diamond material properties. Appl. Spectrosc. 50:1459. [Google Scholar]
- 75.Chi, Z., and S. A. Asher. 1998. UV resonance Raman determination of protein acid denaturation: selective unfolding of helical segments of horse myoglobin. Biochemistry. 37:2865–2872. [DOI] [PubMed] [Google Scholar]
- 76.Chi, Z., and S. A. Asher. 1999. Ultraviolet resonance Raman examination of horse apomyoglobin acid unfolding intermediates. Biochemistry. 38:8196–8203. [DOI] [PubMed] [Google Scholar]
- 77.Austin, J. C., K. R. Rodgers, and T. G. Spiro. 1993. Protein structure from ultraviolet resonance Raman spectroscopy. Methods Enzymol. 226:374–396. [DOI] [PubMed] [Google Scholar]
- 78.Asher, S. A., C. H. Munro, and Z. Chi. 1997. UV lasers revolutionize Raman spectroscopy. Laser Focus World. 33:99–109. [Google Scholar]
- 79.de Alba, E., M. A. Jiménez, and M. Rico. 1997. Turn residue sequence determines β-hairpin conformation in designed peptides. J. Am. Chem. Soc. 119:175–183. [Google Scholar]
- 80.Sibanda, B. L., T. L. Blundell, and J. M. Thornton. 1989. Conformation of β-hairpins in protein structures. A systematic classification with applications to modeling by homology, electron density fitting and protein engineering. J. Mol. Biol. 206:759–777. [DOI] [PubMed] [Google Scholar]
- 81.Parkhe, A. D., S. J. Cooper, E. D. Atkins, M. J. Fournier, T. L. Mason, and D. A. Tirrell. 1998. Effect of local sequence inversions on the crystalline antiparallel β-sheet lamellar structures of periodic polypeptides: implications for chain-folding. Int. J. Biol. Macromol. 23:251–258. [DOI] [PubMed] [Google Scholar]
- 82.Mikhonin, A. V., S. V. Bykov, N. S. Myshakina, and S. A. Asher. 2006. Peptide secondary structure folding reaction coordinate: correlation between UV Raman amide III frequency, Psi Ramachandran angle, and hydrogen bonding. .J Phys. Chem. B. 110:1928–1943. [DOI] [PubMed] [Google Scholar]
- 83.Vass, E., M. Hollosi, F. Besson, and R. Buchet. 2003. Vibrational spectroscopic detection of β- and γ-turns in synthetic and natural peptides and proteins. Chem. Rev. 103:1917–1954. [DOI] [PubMed] [Google Scholar]
- 84.Ianoul, A., A. Mikhonin, I. K. Lednev, and S. A. Asher. 2002. UV resonance Raman study of the spatial dependence of α-helix unfolding. J. Phys. Chem. A. 106:3621–3624. [Google Scholar]
- 85.Reference deleted in proof.
- 86.Swint, L., and A. D. Robertson. 1993. Thermodynamics of unfolding for turkey ovomucoid third domain: thermal and chemical denaturation. Protein Sci. 2:2037–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Naganathan, A. N., and V. Munoz. 2005. Scaling of folding times with protein size. J. Am. Chem. Soc. 127:480–481. [DOI] [PubMed] [Google Scholar]
- 88.Plaxco, K. W., K. T. Simons, and D. Baker. 1998. Contact order, transition state placement and the refolding rates of single domain proteins. J. Mol. Biol. 277:985–994. [DOI] [PubMed] [Google Scholar]
- 89.Ivankov, D. N., and A. V. Finkelstein. 2004. Prediction of protein folding rates from the amino acid sequence-predicted secondary structure. Proc. Natl. Acad. Sci. USA. 101:8942–8944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Thirumalai, D. 1995. From minimal models to real proteins: Time scales for protein folding kinetics. Journal de Physique I. 5:1457–1467. [Google Scholar]
- 91.Mirny, L., and E. Shakhnovich. 2001. Protein folding theory: from lattice to all-atom models. Annu. Rev. Biophys. Biomol. Struct. 30:361–396. [DOI] [PubMed] [Google Scholar]
- 92.Eaton, W. A., V. Munoz, S. J. Hagen, G. S. Jas, L. J. Lapidus, E. R. Henry, and J. Hofrichter. 2000. Fast kinetics and mechanisms in protein folding. Annu. Rev. Biophys. Biomol. Struct. 29:327–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Poland, D., and H. D. Scheraga. 1970. Theory of Helix-Coil Transitions in Biopolymers. Academic Press, New York.
- 94.Capaldi, A. P., and S. E. Radford. 1998. Kinetic studies of β-sheet protein folding. Curr. Opin. Struct. Biol. 8:86–92. [DOI] [PubMed] [Google Scholar]
- 95.Reches, M., and E. Gazit. 2004. Amyloidogenic hexapeptide fragment of medin: homology to functional islet amyloid polypeptide fragments. Amyloid. 11:81–89. [DOI] [PubMed] [Google Scholar]
- 96.Jarrett, J. T., and P. T. Lansbury, Jr. 1993. Seeding “one-dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 73:1055–1058. [DOI] [PubMed] [Google Scholar]