

Abstract
A generic statistical mechanical model is presented for the self-assembly of chiral rod-like units, such as β-sheet-forming peptides, into helical tapes, which with increasing concentration associate into twisted ribbons (double tapes), fibrils (twisted stacks of ribbons), and fibers (entwined fibrils). The finite fibril width and helicity is shown to stem from a competition between the free energy gain from attraction between ribbons and the penalty because of elastic distortion of the intrinsically twisted ribbons on incorporation into a growing fibril. Fibers are stabilized similarly. The behavior of two rationally designed 11-aa residue peptides, P11-I and P11-II, is illustrative of the proposed scheme. P11-I and P11-II are designed to adopt the β-strand conformation and to self-assemble in one dimension to form antiparallel β-sheet tapes, ribbons, fibrils, and fibers in well-defined solution conditions. The energetic parameters governing self-assembly have been estimated from the experimental data using the model. The 8-nm-wide fibrils consist of eight tapes, are extremely robust (scission energy ≈200 kBT), and sufficiently rigid (persistence length l̃fibril ≈ 20–70 μm) to form nematic solutions at peptide concentration c ≈ 0.9 mM (volume fraction ≈0.0009 vol/vol), which convert to self-supporting nematic gels at c > 4 mM. More generally, these observations provide a new insight into the generic self-assembling properties of β-sheet-forming peptides and shed new light on the factors governing the structures and stability of pathological amyloid fibrils in vivo. The model also provides a prescription of routes to novel macromolecules based on a variety of self-assembling chiral units, and protocols for extraction of the associated energy changes.
Prospects for the large-scale production of low-cost peptides by genetic engineering (1) open up new opportunities for exploiting protein-like self-assembly as a route to novel biomolecular materials (2–5). In this context, the small-oligopeptide route has distinct processing advantages over the use of longer polypeptides. Previously, we have demonstrated that oligopeptides can be designed to self-assemble into micrometer-long β-sheet tapes (6). We now wish to show that, as a consequence of the amino acid chirality, an entire hierarchy of twisted self-assembling macromolecular structures is accessible, with tapes as the most primitive form: ribbons, fibrils, and fibers. These polymers are shown to give rise to nematic fluids and gels at concentrations determined by the characteristic flexibility and length of each type of polymer.
The type of molecular assembly we discuss and exemplify here arises not only in the context of desirable engineered biomaterials, but also in pathological self-assembly of mis-folded proteins, when the aggregated assemblies are referred-to as “amyloids.” A very wide class of proteins may be induced into producing the tape-fibril-fiber sequence of structures (7), and a generic role of high-energy intermediate states has been suggested (8). The nature of the intermediate states and the factors determining the size of the intermediate structures remain puzzles.
We present a theoretical model that enables the morphology and properties of these self-assembling structures to be predicted from the molecular parameters of the peptide monomers. Protocols are described to extract the peptide–peptide interaction energies that govern the stabilities of the self-assembled structures, their average sizes, and the concentration ranges over which they are observable. The finite width and helicity of the fibrils is shown to stem from a competition between the free energy gain from attraction between ribbons and the penalty because of elastic distortion of the intrinsically twisted ribbons on incorporation into a growing fibril. Fibers are stabilized in a similar way. In the final section, we compare the self-assembly of our two model peptides with the theory.
Materials and Methods
Preparation of P11-I and P11-II (molecular masses 1,498 Da and 1,594 Da, respectively; purity >95%) as well as transmission electron microscopy (TEM) and far-UV circular dichroism (CD) techniques have been described elsewhere (9). Primary structure of P11-I: CH3CO-Gln-Gln-Arg-Gln-Gln-Gln-Gln-Gln-Glu-Gln-Gln-NH2. Primary structure of P11-II: CH3CO-Gln-Gln-Arg-Phe-Gln-Trp-Gln-Phe-Glu-Gln-Gln-NH2. β-AP (1–40) peptide (ref. 10; purity > 98%) was purchased from ICN.
Theoretical Model of Self-Assembling Chiral Rod-Like Units.
We start by considering a peptide in a β-strand conformation as a chiral rod-like unit, with complementary donor and acceptor groups aligned on opposing sides, and having chemically different upper and lower surfaces (Fig. 1Aa). In common with many successful approaches in statistical soft-matter physics, this represents a single step of coarse-graining from atomic detail to the nano-scale. We will find that the chiral unit is able to undergo one-dimensional self-assembly in solution and to form the hierarchical set of structures depicted in Fig. 1A at concentrations depending on the values of a small set of coarse-grained interaction energies, ɛj. Generally, an isolated monomer in solution will be in a different conformation (Fig. 1Ab), with lower free energy than in the rod-like state: the corresponding conformational free energy change is ɛtrans. The rod-like “monomers” self-assemble via recognition between complementary donor and acceptor groups, to form long twisted tapes (Fig. 1Ac): the association free energy change is ɛtape per intermonomer bond. The tape twist stems from the chirality of the monomers (Fig. 1Aa, e.g., right-handed in the case of peptides, because of the l-chirality of naturally occurring amino acids) which gives rise to a left-handed twist around the long axis of the tape (Fig. 1Ac). The tape has distinct faces (denoted white and black in Fig. 1Ac) whenever the upper and lower surfaces of the monomer are chemically different (we suppose that the black sides of the monomers have higher affinity for each other than for the white sides). The differences in the chemical structures of the two faces of the tape and in their affinity to the solvent give rise to a cylindrical curvature, causing the tape to curl into a helical configuration (Fig. 1Ac′), with helical pitch htape and radius rtape given by:
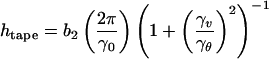 |
1 |
and
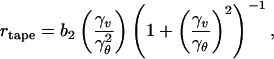 |
2 |
where γv and γθ are, respectively, the tape bend and twist angles (in radians) per monomer rod, along the tape and b2 is the distance between adjacent rods in the tape.
Figure 1.

Model of hierarchical self-assembly of chiral rod-like units. (A) Local arrangements (c–f) and the corresponding global equilibrium conformations (c′–f′) for the hierarchical self-assembling structures formed in solutions of chiral molecules (a), which have complementary donor and acceptor groups, shown by arrows, via which they interact and align to form tapes (c). The black and the white surfaces of the rod (a) are reflected in the sides of the helical tape (c), which is chosen to curl toward the black side (c′). The outer sides of the twisted ribbon (d), of the fibril (e), and of the fiber (f) are all white. One of the fibrils in the fiber (f′) is drawn with darker shade for clarity. (e and f) The front views of the edges of fibrils and fibers, respectively. Geometrical sizes (the numbers in parentheses show the values of the corresponding geometric sizes for P11-I and P11-II peptides, based on x-ray diffraction data and molecular modeling): interrod separation in a tape b2 (b2 = 0.47 nm); tape width, equal to the length of a rod, b1 (b1 = 4 nm); interribbon distance in the fibril, α (α = 1.6–2 nm for P11-I, and α = 2–2.4 nm for P11-II). (B) Phase diagram of a solution of twisted ribbons that form fibrils. The scaled variables are as follows: relative helix pitch of isolated ribbons hribbon/α, and relative side-by-side attraction energy between ribbons ɛ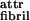 /ɛ*fibril [ɛ*fibril ≡ (2π2b2/α2) ktwist; see the text and Fig. 1 Ad and Ae′ for notations]. The areas divided by the thick lines reveal the conditions where ribbons, fibrils, and infinite stacks of completely untwisted ribbons are stable. The dotted lines are lines of stability for fibrils containing p ribbons (p are written on the lines); kbend/ktwist = 0.1.
/ɛ*fibril [ɛ*fibril ≡ (2π2b2/α2) ktwist; see the text and Fig. 1 Ad and Ae′ for notations]. The areas divided by the thick lines reveal the conditions where ribbons, fibrils, and infinite stacks of completely untwisted ribbons are stable. The dotted lines are lines of stability for fibrils containing p ribbons (p are written on the lines); kbend/ktwist = 0.1.
One face of the tape (colored black in Fig. 1Ac) is expected to be less soluble than the other (i.e., black is more hydrophobic if the solvent is water). This chemical anisotropy results in intertape attraction and hence in double tape (ribbon, Fig. 1Ad) formation (ɛ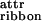 per peptide). Both faces of the ribbon are identical (white in Fig. 1Ad) and are characterized by a saddle curvature. Hence, the ribbon does not bend, and its axis is straight at equilibrium (Fig. 1Ad′). The white sides of the ribbons are, in turn, mutually attractive (via a typically smaller energy ɛ
per peptide). Both faces of the ribbon are identical (white in Fig. 1Ad) and are characterized by a saddle curvature. Hence, the ribbon does not bend, and its axis is straight at equilibrium (Fig. 1Ad′). The white sides of the ribbons are, in turn, mutually attractive (via a typically smaller energy ɛ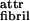 per pair of interacting peptides) leading to stacking of ribbons into fibrils (Fig. 1Ae). Furthermore, the ends of the rods decorating the edges of the fibrils can also be mutually attractive, causing fibrils to entwine into fibers (Fig. 1Af), stabilized by attraction energy ɛ
per pair of interacting peptides) leading to stacking of ribbons into fibrils (Fig. 1Ae). Furthermore, the ends of the rods decorating the edges of the fibrils can also be mutually attractive, causing fibrils to entwine into fibers (Fig. 1Af), stabilized by attraction energy ɛ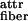 .
.
All of the self-assembling structures in Fig. 1A are left-handed twisted because of chirality of the rod-like monomer. If the ribbons were not twisted, an unlimited growth of fibril and fiber widths would be expected. Instead, when twisted ribbons aggregate into stacks, fibrils with well-defined widths are formed. Fibers are formed in a similar way from twisted fibrils, but again to well-defined widths. The inherent chiral twist is the key to understanding the finite fibril and fiber widths. Indeed, to aggregate, twisted objects must bend and adjust their twist in response to the packing constraints imposed by its twisted neighbours. Hence, there is an elastic energy cost, ɛelast, that must be compensated for, by the gain in attraction energy (coming from ɛ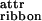 , ɛ
, ɛ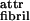 , and ɛ
, and ɛ ) on stacking. The distortion energy ɛelast is higher for thicker stacks. This ɛelast serves to stabilize the widths of fibrils and fibers. Thus, the fibril width is determined by a balance between the gain in attraction energy (coming from ɛ
) on stacking. The distortion energy ɛelast is higher for thicker stacks. This ɛelast serves to stabilize the widths of fibrils and fibers. Thus, the fibril width is determined by a balance between the gain in attraction energy (coming from ɛ ) associated with ribbon stacking, and the elastic cost on the ribbons associated with fibril formation. Assuming that the ribbon contour length is fixed and the deformations are weak, from symmetry arguments we find that this cost is
) associated with ribbon stacking, and the elastic cost on the ribbons associated with fibril formation. Assuming that the ribbon contour length is fixed and the deformations are weak, from symmetry arguments we find that this cost is
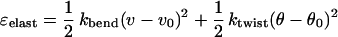 |
3 |
(per unit length of each ribbon in the fibril), where ν and θ are the local curvature and the local twist strength of the ribbon within fibril, θ0 = 2π/hribbon is the equilibrium value of twist strength of an isolated ribbon, whereas its equilibrium bend strength is zero (ν0 = 0), and kbend,ktwist are the ribbon elastic constants (11). For a ribbon a distance ρ from the central axis of a fibril, it is straightforward to show that ν = γ2ρ/(1 + γ2ρ2), θ = γ/(1 + γ2ρ2), where γ = 2π/hfibril, where hfibril is the fibril's helical pitch. The thicker the fibril is, the larger the typical ρ are, and hence the higher the cost ɛelast. The net energy gain ɛfibril per peptide in a fibril,
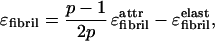 |
4 |
has a maximum at some p (p is the number of ribbons in the fibril). Hence, a well-defined width of fibrils arises, corresponding to this optimal p (11).
The “state diagram” of possible aggregate structures calculated by using this model, and seeking the structure of minimum free energy in each case, is shown in Fig. 1B. Fibrils with finite diameter are seen to be stable for a wide range of values of ɛ̃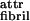 provided that the intrinsic pitch hribbon of the lone ribbon strongly exceeds the interribbon gap a in the fibril. For low ɛ
provided that the intrinsic pitch hribbon of the lone ribbon strongly exceeds the interribbon gap a in the fibril. For low ɛ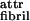 , the ribbons do not stack into fibrils. For high ɛ
, the ribbons do not stack into fibrils. For high ɛ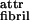 , the ribbons form infinite aggregates (sheet-like crystallites) in which the ribbons are completely untwisted. The optimum number p of stacked ribbons per fibril, and hence the fibril diameter, increases with hribbon and ɛ
, the ribbons form infinite aggregates (sheet-like crystallites) in which the ribbons are completely untwisted. The optimum number p of stacked ribbons per fibril, and hence the fibril diameter, increases with hribbon and ɛ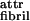 . This increase is usually accompanied by an increase in hfibril.
. This increase is usually accompanied by an increase in hfibril.
The concentration ranges over which the various self-assembled structures are observable, their contour lengths, and abruptness of interstructure transformations with concentration are determined by the energy parameters ɛj. For example, if ɛtrans is high enough, (ɛtrans > 4, all energies here are measured in kBT units) and ɛribbon is small (≤1), the single tapes emerge abruptly at
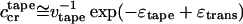 |
5 |
and their typical aggregation number is
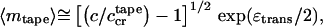 |
6 |
if c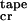 < c < c
< c < c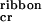 (c is the total peptide concentration and νtape is the “freedom” volume of the bonds forming the tape). Next, given that the tape bend and twist are not very high, i.e., ɛelast (cf. Eq. 3) is small enough, the net ribbon energy
(c is the total peptide concentration and νtape is the “freedom” volume of the bonds forming the tape). Next, given that the tape bend and twist are not very high, i.e., ɛelast (cf. Eq. 3) is small enough, the net ribbon energy
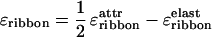 |
7 |
is positive. Hence, at concentration
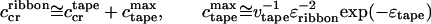 |
8 |
the ribbons emerge; above c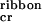 , the population of peptide in single tapes saturates at c
, the population of peptide in single tapes saturates at c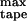 , and all extra peptide goes into ribbons; simultaneously the average aggregation number of ribbons grows as
, and all extra peptide goes into ribbons; simultaneously the average aggregation number of ribbons grows as
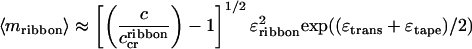 |
9 |
whereas the length of tapes saturates at
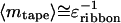 |
10 |
The formulae (Eqs. 5, 6, 8, 9, and 10) are asymptotic. The detailed theory generalizing the classic isodesmic model (12) has been published separately (13).
To realize sequentially the entire hierarchy of structures in Fig. 1A, with increasing monomer concentration, it is essential that ɛtape ≫ kBT ≫ ɛribbon ≫ ɛfibril ≫ ɛfiber. These are the net energies gained per one peptide inside the corresponding structures as compared with a peptide inside the structure of the previous level); otherwise, some structures may not appear.
Results and Discussion
To illustrate the predictions of our theoretical model, and to demonstrate how the ɛj can be measured for real self-assembling systems, we shall consider the behavior of two rationally designed peptides, P11-I and P11-II (Table 1, which is published as supporting information on the PNAS web site, www.pnas.org). P11-I is based on a sequence of glutamine (Gln) residues, whose side-chains are believed to interact strongly in water (14), presumably via hydrophobic and complementary hydrogen bonding interactions. Arginine (Arg) and glutamate (Glu) residues have been placed in positions 3 and 9, to provide molecular recognition between adjacent antiparallel β-strand peptides in tape-like aggregates, to prevent random peptide association. These favorable intermolecular side-chain interactions, coupled with the cooperative intermolecular hydrogen bonding between peptide backbones, will result in high scission energy ɛtape, thus promoting β-sheet tape formation (Fig. 1Ac). Furthermore, one side (“black”) of the tape will be lined by the CONH2 groups of Gln whereas its other side (“white”) will be lined by the CONH2, the guanido, and the COOH groups of Gln, Arg, and Glu, respectively. At low pH in particular, there will also be a net positive charge per peptide. The high hydrophilicity of both surfaces of the tape, combined with the electrostatic repulsion between positively charged surfaces, will result in very small ɛ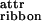 and ɛ
and ɛ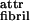 energies compared with kBT, thus promoting predominantly single tape formation for low enough peptide concentration in acidic solutions.
energies compared with kBT, thus promoting predominantly single tape formation for low enough peptide concentration in acidic solutions.
At very low concentrations, P11-I is predominantly in monomeric random coil conformation (Figs. 1Ab and 2a). At higher concentrations, c ≥ 0.01 mM, it forms semiflexible tapes (Fig. 2b) with a width W ≈ 4 nm, equal to the expected length of an 11-residue peptide in a β-strand conformation, and persistence length l̃ < 0.3 μm. The different chemical nature of the two sides of the tape seems to cause it to bend and twist simultaneously, resulting in curly tapes with a left-handed twist, a helical pitch htape ≈ (30 ± 15) nm, and a radius rtape ≈ 5 nm (Fig. 2b). At c ≥ 1 mM, loose ribbons are also observed, with l̃ ≈ 0.3–1 μm, and hribbon ≈ (50 ± 20) nm. These values in conjunction with the theoretical model were used to derive the magnitudes of the bend γν = 3° and twist γθ = 3° angles for the single tapes and the ribbons (Table 1).
Figure 2.

Self-assembly of P11-I. (a) Far-UV CD spectra as a function of peptide concentration. The solutions were prepared by mixing the dry peptide with the required volume of water adjusted to pH = 2 with phosphoric acid. Data were collected with 1-month-old solutions stored at 20°C. For interpretation of the CD spectra, see the text and legend of Fig. 4 a and b. (b) Negatively stained TEM image of single “curly” tapes, reminiscent of Fig. 1Ac′; the scale bar = 50 nm. (c) Plot of the β-sheet fraction in solution (black circles) as a function of total peptide concentration, based on the CD data (the mean residue ellipticity [θ] at 219 nm is taken as a linear function of the β-sheet fraction in solution). The solid line is the fit of the data with the single tape theory. The best-fit values of the energetic parameters ɛtrans and ɛtape, which were chosen to comply with the concentration dependence of the CD data and with the observed lengths of tapes at c = 5 mM, are shown in the panel. (d) Theoretical concentration dependence of the average number 〈m〉 of peptides per single tape (dotted line) and in ribbons (dash-dot line), based on the energetic parameters derived from the fit (c). Minimum number of peptides in tapes is two, and in ribbons is four. The predicted lengths of tapes and ribbons are in agreement with the observed lengths in the TEM pictures for the same peptide concentration.
Aqueous solutions of P11-I tapes produce FTIR spectra with absorption maxima in amide I′ at 1630 and 1690 cm−1, demonstrative of a predominantly antiparallel β-sheet structure. They also exhibit characteristic β-sheet CD spectra (15) with minimum and maximum ellipticities at 218 nm and 195 nm, respectively (Fig. 2a). The fraction of the peptide in β-sheet tapes starts to grow abruptly at a critical concentration c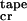 ≈ 0.008 mM (Fig. 2c). The two-state transition from random-coil to β-sheet with increasing concentration has an isodichroic point at 211 nm (Fig. 2a). We treated ɛtrans and ɛtape as fitting parameters and were able to describe well the growth of the β-sheet CD band with concentration (solid line in Fig. 2c). The best-fit energy values obtained are in Fig. 2c. The ɛtrans energy results in the nucleated growth of tapes, manifested by a “sudden” onset of β-sheet tapes' formation at c
≈ 0.008 mM (Fig. 2c). The two-state transition from random-coil to β-sheet with increasing concentration has an isodichroic point at 211 nm (Fig. 2a). We treated ɛtrans and ɛtape as fitting parameters and were able to describe well the growth of the β-sheet CD band with concentration (solid line in Fig. 2c). The best-fit energy values obtained are in Fig. 2c. The ɛtrans energy results in the nucleated growth of tapes, manifested by a “sudden” onset of β-sheet tapes' formation at c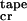 . By using these values of energetic parameters, this single tape model predicts a mean tape contour length for a given peptide concentration, which agrees well with the observed range of contour lengths in the TEM images for the same concentration. At c
. By using these values of energetic parameters, this single tape model predicts a mean tape contour length for a given peptide concentration, which agrees well with the observed range of contour lengths in the TEM images for the same concentration. At c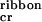 ≈ 1 mM, loose ribbons start appearing, implying a weak attraction between tapes. This attraction may be mediated by multiple, cooperative, complementary hydrogen bonding between the —CONH2 groups of glutamine side-chains, which line completely one of the two polar sides of the tapes. van der Waals forces are likely to be involved, too. From the value of c
≈ 1 mM, loose ribbons start appearing, implying a weak attraction between tapes. This attraction may be mediated by multiple, cooperative, complementary hydrogen bonding between the —CONH2 groups of glutamine side-chains, which line completely one of the two polar sides of the tapes. van der Waals forces are likely to be involved, too. From the value of c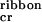 , we estimate that the ribbons are stabilized by ɛribbon = (0.0035 ± 0.0015) kBT (see Theoretical Model). Fibrils (Fig. 1Ae′) are not observed up to c = 25 mM; hence, ɛfibril < 10−3 kBT.
, we estimate that the ribbons are stabilized by ɛribbon = (0.0035 ± 0.0015) kBT (see Theoretical Model). Fibrils (Fig. 1Ae′) are not observed up to c = 25 mM; hence, ɛfibril < 10−3 kBT.
To increase the tendency of the peptide to associate into ribbons, we need to increase the magnitude of ɛribbon either by decreasing ɛ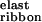 or by increasing ɛ
or by increasing ɛ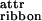 . The latter can be achieved by addition of salts or of appropriate cosolvents, but more elegantly by replacing the glutamines at positions 4, 6, and 8 by phenylalanine, tryptophan, and phenylalanine, respectively. This new peptide, P11-II (Table 1), will form β-tapes with a hydrophobic “adhesive” stripe running along one side of the tape, which will promote their association into ribbons in water. At c ≥ 0.1 mM in water, P11-II is indeed found to form long, stable semiflexible β-sheet ribbons with a width of 2–4 nm, which fits with the expected cross section of ≈2 × 4 μm2 of these ribbons, and a persistence length ε ≈ 1 μm, (Fig. 3a). At c ≥ 0.6 mM, a second transition from ribbons to fairly rigid fibrils is observed (Fig. 3 b and c). The fibrils have a well-defined screw-like structure with typical minimum and maximum widths W1 ≈ 4 nm and W2 ≈ 8 nm, respectively. At even higher concentrations still, a third structural transition takes place, and fibers are detected, typically comprised of two entwined fibrils (Fig. 3d). The sequence of these structural transitions is also supported by distinctive far- and near-UV CD spectra, corresponding to P11-II monomers, ribbons, and fibrils (supporting Fig. 4 a and b).
. The latter can be achieved by addition of salts or of appropriate cosolvents, but more elegantly by replacing the glutamines at positions 4, 6, and 8 by phenylalanine, tryptophan, and phenylalanine, respectively. This new peptide, P11-II (Table 1), will form β-tapes with a hydrophobic “adhesive” stripe running along one side of the tape, which will promote their association into ribbons in water. At c ≥ 0.1 mM in water, P11-II is indeed found to form long, stable semiflexible β-sheet ribbons with a width of 2–4 nm, which fits with the expected cross section of ≈2 × 4 μm2 of these ribbons, and a persistence length ε ≈ 1 μm, (Fig. 3a). At c ≥ 0.6 mM, a second transition from ribbons to fairly rigid fibrils is observed (Fig. 3 b and c). The fibrils have a well-defined screw-like structure with typical minimum and maximum widths W1 ≈ 4 nm and W2 ≈ 8 nm, respectively. At even higher concentrations still, a third structural transition takes place, and fibers are detected, typically comprised of two entwined fibrils (Fig. 3d). The sequence of these structural transitions is also supported by distinctive far- and near-UV CD spectra, corresponding to P11-II monomers, ribbons, and fibrils (supporting Fig. 4 a and b).
Figure 3.

Aggregate structures and liquid crystalline phase behavior observed in solutions of P11-II in water with increasing c (log scale). The electron micrographs (a) of ribbons (c = 0.2 mM), and (b) of fibrils (c = 6.2 mM) were obtained with a 4-month-old solution after platinum rotary shadowing. The observed micrometer-long contour length may be limited by multiple ruptures of the fibrils during preparation of the samples for TEM imaging. Higher resolution TEM images of ribbons were also obtained by using negatively stained samples. Micrographs c (c = 6.2 mM) and d (c = 6.2 mM) were obtained with a 1-month-old solution after uranyl acetate negative staining. CD and Fourier transfer IR (FTIR) have confirmed that the fibrils are made of β-sheet structures. X-ray diffraction data have also shown arcs corresponding to 0.47 nm periodicity, consistent with the expected interstrand distance in a β-sheet (unpublished data). The TEM micrographs show the principle aggregate structures whose populations cj = fjc (fj is the fraction of peptides incorporated in the jth structure) change with peptide concentration, as depicted in e. The curves in e were calculated with the generalized model described in the text (see also Fig. 4d). The aggregation behavior of the peptide, probed by using time-resolved fluorescence anisotropy and CD of filtered solutions, is fully consistent with the expectations of the model. The polarizing optical micrograph (f) shows the thick thread-like texture observed for a solution with c = 3.7 mM in a 0.2-mm pathlength microslide. (g) A self-supporting birefringent gel (c = 6.2 mM) in an inverted 10-mm o.d. glass tube, viewed between crossed polarizers. The scale bars in a, b, c, and d = 100 nm, in f = 100 μm.
Focusing on the behavior at low concentrations, we see that P11-II is predominantly in the monomeric random coil conformation (Fig. 1Ab), whereas the fraction of peptide in β-sheet structures starts to grow abruptly at c ≈ 0.07 mM (Fig. 4 a and c). We treated ɛtrans and ɛtape as fitting parameters, and were able to describe well the growth of the β-sheet CD band with concentration. However, a single tape model yields a mean tape length of about 20 nm at c = 0.2 mM (Fig. 4d), much shorter than the observed length ≥ 500 nm (Fig. 3a). It is possible, however, to describe the CD data (solid line in Fig. 4c) and simultaneously to predict the occurrence of these long aggregates (Fig. 3a) by inclusion of the third energetic parameter ɛribbon associated with ribbon (double tape) formation (Fig. 1d). These long aggregates then turn out to be double tapes rather than single ones (Fig. 4 c and d). The CD spectra as a function of c have no isodichroic point (Fig. 4a), further supporting that more than two states, i.e., peptide monomers, β-tapes, and ribbons, are involved in the conformational transition. The best-fit energy values obtained are: ɛtrans = (3 ± 1) kBT, ɛtape = (24.5 ± 1.0) kBT, and ɛribbon = (0.6 ± 0.3) kBT.
The estimated ɛtrans is higher for P11-I than for P11-II (Table 1). Although both peptides have the same length, they may have different propensity to form a random coil in the monomeric state, which could account for this difference in ɛtrans. The magnitude of ɛtape is also higher for P11-I than for P11-II, which indicates that the intermolecular glutamine side-chain interactions between P11-I peptides are more efficient at promoting self-assembly compared with intermolecular aromatic side-chain interactions between P11-II peptides. ɛribbon is at least two orders of magnitude lower for P11-I compared with P11-II, as predicted by peptide design. This difference explains the shorter (by one order of magnitude) length of P11-I ribbons compared with P11-II ones (Table 1). It also accounts for the one order of magnitude difference in critical concentrations for ribbon formation between the two peptides. This low magnitude of ɛribbon results in stabilization of single, curly β-tapes in a wide range of P11-I concentrations. In contrast, P11-II tapes are not observed because they convert to ribbons as soon as they are 3–4 peptides long, at very low concentration.
The formation of fibrils (Fig. 3b) at higher concentrations of P11-II implies the presence of a weaker attraction between the polar sides of P11-II ribbons (ɛ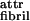 , Fig. 1e′). From the concentration at which they appear, we calculate ɛfibril = (2.0 ± 0.3)⋅10−4 kBT. Despite this attraction, the fibril dispersions are stable and the fibril diameter is finite (rather than growing indefinitely). Furthermore, the fibril width W1 corresponds to the expected length of an 11-residue β-strand, whereas the thickness W2 corresponds to roughly four ribbons (i.e., eight single tapes, each tape with a thickness of ca. 1 nm) per fibril, and is concentration independent (at least from 0.6 to 7 mM). Scanning TEM mass per unit length measurements also supports that the fibrils are made typically of eight tapes (A.A., M.B., N.B., L.C., and D. Holmes, unpublished data). The energy required to break such a fibril, scission energy ɛsc, is ɛsc = 8 ɛtape ≈ 200 kBT (comparable to covalent bond energies!), and is much higher than that of a single ribbon ɛsc = 2 ɛtape ≈ 50 kBT. This high ɛsc results in fibrils of extraordinary predicted equilibrium average length Lfibril ≈ 108 km!, compared with Lribbon ≈ 1 μm, for c = 6 mM (Fig. 4d).
, Fig. 1e′). From the concentration at which they appear, we calculate ɛfibril = (2.0 ± 0.3)⋅10−4 kBT. Despite this attraction, the fibril dispersions are stable and the fibril diameter is finite (rather than growing indefinitely). Furthermore, the fibril width W1 corresponds to the expected length of an 11-residue β-strand, whereas the thickness W2 corresponds to roughly four ribbons (i.e., eight single tapes, each tape with a thickness of ca. 1 nm) per fibril, and is concentration independent (at least from 0.6 to 7 mM). Scanning TEM mass per unit length measurements also supports that the fibrils are made typically of eight tapes (A.A., M.B., N.B., L.C., and D. Holmes, unpublished data). The energy required to break such a fibril, scission energy ɛsc, is ɛsc = 8 ɛtape ≈ 200 kBT (comparable to covalent bond energies!), and is much higher than that of a single ribbon ɛsc = 2 ɛtape ≈ 50 kBT. This high ɛsc results in fibrils of extraordinary predicted equilibrium average length Lfibril ≈ 108 km!, compared with Lribbon ≈ 1 μm, for c = 6 mM (Fig. 4d).
Fibril formation is readily explicable by our model of stabilization by twist (Fig. 1b). Indeed, β-sheet ribbons have an intrinsically left-handed twist, because of the l-chirality of peptides (16). The fibrils also exhibit a left-handed twist with a helix pitch hfibril of ca. 120–200 nm (Fig. 3c). From the observed geometrical characteristics of P11-II ribbons and of fibrils, our theory estimates hribbon ≈ 120–200 nm, elastic constants kbend and ktwist, and twist angle γθ = 1° for isolated P11-II ribbons and ɛ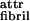 ≈ 0.015 kBT for fibrils (Table 1). The magnitude of ɛ
≈ 0.015 kBT for fibrils (Table 1). The magnitude of ɛ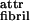 is expected to be similar both for P11-I and P11-II, because of the identity of their “white” polar sides. However, P11-I ribbons are three times more twisted than P11-II ones (compare twist angles γθ in Table 1). The higher elastic penalty ɛ
is expected to be similar both for P11-I and P11-II, because of the identity of their “white” polar sides. However, P11-I ribbons are three times more twisted than P11-II ones (compare twist angles γθ in Table 1). The higher elastic penalty ɛ , associated with untwisting P11-I ribbons compared with P11-II ones, seems to result in lower overall magnitude of ɛfibril for P11-I compared with P11-II, and thus prevents the stacking of P11-I ribbons into fibrils. This difference in the values of ɛ
, associated with untwisting P11-I ribbons compared with P11-II ones, seems to result in lower overall magnitude of ɛfibril for P11-I compared with P11-II, and thus prevents the stacking of P11-I ribbons into fibrils. This difference in the values of ɛ explains why P11-I ribbons do not combine into fibrils up to c = 25 mM, whereas P11-II ribbons form fibrils at c < 1 mM.
explains why P11-I ribbons do not combine into fibrils up to c = 25 mM, whereas P11-II ribbons form fibrils at c < 1 mM.
The value of ɛfibril for P11-II is such that the corresponding energy of attraction between ribbons per their persistence length, ɛfibril⋅l̃ribbon/b2 ≈ 20–30 kBT, where b2 is the distance between adjacent β-strands along the tape (see Fig. 1Ac), is sufficiently high to stabilize the fibrils against splitting, once the ribbons become longer than a few hundreds of peptide β-strands. It is apparent that, in such a hierarchical system of structures, the growth of the more primitive self-assembled structures up to a certain critical size, enables new (weaker) interactions to come into play, and this leads to the formation of the next hierarchical structure. Once the growth of this new structure is established, the concentration and the size of the previous structure become fixed. Indeed, Fig. 3e shows that the concentrations of peptide in monomeric and single tape states saturate at c ≈ 0.09 mM, at which point ribbons emerge. The single tape length is also seen to saturate at 3–4 peptides per tape (Fig. 4d). Similarly, the population of peptides in ribbons (Fig. 3e) and the ribbon lengths (Fig. 4d) become constant when fibrils are first formed. The transitions to higher order aggregates are thus reminiscent of (but not identical to) the formation of micelles in surfactant solutions. We can also compare the stabilization of fibrils and fibers by twist, to the stabilization of micelles by amphiphilic interactions.
The fibrils have a persistence length l̃fibril ≈ 20–70 μm, as opposed to l̃ribbon ≈ 0.7–1.5 μm, which makes the fibrils considerably more rigid than the ribbons. Indeed, a plywood-like stack made of p layers is expected to be between p and p3 times more rigid than the primary layers (p is for a loose stack, and p3 for a stack with well-glued layers). Hence, the expected persistence length of the P11-II fibrils made of p = 4 ribbons, is up to 64 times higher than the ribbon persistence length, i.e., in good agreement with our observations (Table 1).
The rigidity of the fibrils/fibers gives rise to the formation of a nematic phase at c ≥ 0.9 mM (0.001 vol/vol). The texture in the optical micrograph (Fig. 3f) and its dependence on flow, is characteristic of viscoelastic nematic fluids of semirigid polymers (17). The isotropic-to-nematic phase separation gap is narrow [0.8 mM < cI < cN < 0.9 mM (relative gap width w ≡ cN/cI − 1 < 0.13)], and is insensitive to temperature variations up to at least 60°C. Polydisperse rigid-rod solutions have much wider phase separation gaps (w ≈ 2; ref. 18). The fibrils behave more like typical semirigid (worm-like) chains with hard-core excluded volume interactions, for which w ≈ 0.09 (ref. 18). The isotropic-to-nematic transition of such chains with rectangular cross section W1 × W2 is predicted (18) to occur at volume fractions ΦIN ≈ 5.5 W/l̃fibril (where W ≈ 2W1W2/(W1 + W2), provided that L ≥ l̃fibril; this yields for P11-II, ΦIN ≈ 0.0004–0.0015 vol/vol (corresponding to cIN ≈ 0.4–1.5 mM), in agreement with our observations. l̃ribbon for P11-I is one to two orders of magnitude shorter than l̃fibril of P11-II. The isotropic-to-nematic transition of solutions of such semiflexible ribbons of P11-I is predicted to occur at ΦIN ≈ 0.015–0.05 vol/vol (corresponding to 15–50 mM). Indeed, we find that P11-I forms nematic phase at c ≈ 13 mM (Table 1).
At c ≥ 4 mM, the birefringent solution of P11-II becomes a self-supporting birefringent gel (Fig. 3g). Rheological measurements show that fibril-based gels are brittle and do not relax even after days, behavior reminiscent of permanent gels of semirigid polymers (A.A., N.B., T.C.B.M., and P. Mawer, unpublished data). In contrast, tape-based gels are more extendable (6) and relax slowly with time, behavior indicative of transient gels of semiflexible polymers. We conclude that the type of polymer (tape, ribbon, or fibril, each associated with its own characteristic flexibility, contour length, and crosslinking mechanism) determines the liquid crystalline and gelation properties of its solution.
Fibril and fiber formation is characterized by slow kinetics. Fibril formation takes up to several weeks to complete, depending on concentration, as monitored extensively by CD and TEM (data not shown). The formation of nematic phases of P11-II, after fibril formation, takes several hours for c ≈ 4 mM, and several weeks for c ≤ 1 mM. Gelation occurs even more slowly, e.g., for c = 6 mM it takes a week. Transitions from one self-assembling structure to another take even longer than their initial formation: when an aged (2 months old) P11-II nematic gel (c = 6 mM) was diluted to a concentration where ribbons are intrinsically stable (c = 0.2 mM), CD and TEM revealed that, even after standing for 4 months at the lower concentration, numerous fibrils coexist still with ribbons. The extremely slow kinetics originates from the multiplicity of molecular interactions in fibrils. The high tape scission energy ɛtape ≈ 24.5 kBT of P11-II peptide ensures that peptide dissociation from free ends of tapes, ribbons, or fibrils is a rare event: the effective dissociation rate τ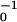 exp(−ɛtape) ≈ 1010 τ
exp(−ɛtape) ≈ 1010 τ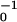 corresponds to a dissociation time τ of the order of 1 s for τ0 ≈ 100 ps (τ0 is a typical diffusion time of a peptide “rod” over a distance of 0.1 nm). The energy required to break the ribbon, ɛsc = 2ɛtape ≈ 50 kBT, gives rise to a scission (breaking) time of a ribbon of τ0 exp(2ɛtape) ≈ 104 yr. Nucleation of short ribbons in solutions of monomer peptide, in the absence of fibril seeds, is expected to be faster than nucleation of fibrils. To form fibrils, ribbons need to wind around each other, which is likely to be highly hindered in a dense system of entangled ribbons. Alternatively, they can dissociate into smaller fragments followed by reassembly; this process will be slow because of the high ɛsc. We therefore expect very slow fibril formation with defective “fresh” fibrils. Aged fibrils will be far more perfect and hence even more stable; to disintegrate they need to lose monomers at their ends. This is a very slow process because the fibrils are very long (Fig. 1d and Table 1).
corresponds to a dissociation time τ of the order of 1 s for τ0 ≈ 100 ps (τ0 is a typical diffusion time of a peptide “rod” over a distance of 0.1 nm). The energy required to break the ribbon, ɛsc = 2ɛtape ≈ 50 kBT, gives rise to a scission (breaking) time of a ribbon of τ0 exp(2ɛtape) ≈ 104 yr. Nucleation of short ribbons in solutions of monomer peptide, in the absence of fibril seeds, is expected to be faster than nucleation of fibrils. To form fibrils, ribbons need to wind around each other, which is likely to be highly hindered in a dense system of entangled ribbons. Alternatively, they can dissociate into smaller fragments followed by reassembly; this process will be slow because of the high ɛsc. We therefore expect very slow fibril formation with defective “fresh” fibrils. Aged fibrils will be far more perfect and hence even more stable; to disintegrate they need to lose monomers at their ends. This is a very slow process because the fibrils are very long (Fig. 1d and Table 1).
There are a variety of biological and designed proteins that, similarly to our model peptides, self-assemble into twisted amyloid β-sheet polymers typically 5–10 nm in diameter (7, 8, 19–22), e.g., characterizing Alzheimer's and prion diseases, and of the de novo β-sandwich protein betabellin 15D (23). The scheme (Fig. 1) provides an explanation of the factors governing the structures and stabilities of these fibrous aggregates. In the case of Alzheimer's disease, the pathological, amyloid-forming peptide β-AP (1–39/42) is cleaved from the much longer precursor protein APP, and subsequently self-assembles into amyloid fibrils (protofilaments) comprised of stacks of usually two to four twisted β-sheet tapes (Fig. 1Ae′). Three or more of theses fibrils are wound around each other in the fibers (Fig. 1Af′). A central hollow core is observed in some amyloid fibers (19) and is particularly enhanced in the fibers formed in vitro by the SH3 domain of the protein PI3-kinase (22). The size of the hole will depend on whether the fibrils in the fiber interact with their β-strands parallel to each other as in Fig. 1 Af and Af′, or at an angle determined by the interactions between segments of the proteins bordering the edges of the fibrils. Whatever the details of each specific system, there is little doubt that it is the intrinsic twist of the unifying, underlying β-sheet scaffold of the constituent β-tapes that governs the way tapes stack and ultimately the distinctive morphology of amyloid fibrils and fibers as elaborated in Fig. 1. It has been pointed out that the β-sheets in amyloid fibrils tend to be flatter than the β-sheets in globular proteins (22). This observation is in accordance with our model (Fig. 1B): the β-sheets in the tapes have to untwist to stack together in the fibrils. The energies involved in fibril formation, together with the presence of the twist in β-sheets as shown above for P11-II peptide, also explain the remarkable stability and accumulation of amyloids in vivo, as well as the slow kinetics, and the nucleation-growth mechanism of amyloid self-assembly found by in vitro studies (10, 24). In the case of the intermediate states observed in amyloid formation from more complex proteins (8), ɛtrans may be thought of as the energy difference between the ground and intermediate states of a two-state model. To complete the list of similarities, we have found that a 1.2-mM solution of fibrils of Alzheimer's β-AP (1–40) peptide, when sucked into an optical capillary, produces a banded nematic texture, similar to that observed for P11-II. We propose that systematic analysis of the self-assembly of amyloid peptides by using the scheme summarized in Fig. 1 can provide unique quantitative information on the various energies that stabilize amyloid fibrils, and thus enhance our understanding of the associated life-threatening diseases.
We are thus led to the belief that a wide variety of peptides could be designed to behave according to the scheme in Fig. 1, opening up an exciting new class of chiral tape-like and fibrillar macromolecular structures that can form liquid crystals and gels. Moreover, by appropriate peptide design, a combination of desirable properties, such as biological functionality, high stability of fibrils or fibers, and responsiveness to external triggers such as solvent polarity, temperature, pH, or ionic strength can be incorporated, making these new materials much more versatile than existing peptidic biomaterials such as collagen (25). We envisage that this unique combination of properties makes these materials ideally suited for use in biomedical and other applications, such as for the production of bioscaffolds to control the shape and alignment of cells for tissue engineering, new suture materials, templates for helical crystallization of macromolecules, and matrices for separation of chiral molecules.
The scheme (Fig. 1) is also applicable to any chiral molecule able to undergo one-dimensional self-assembly, such as the hemoglobin S protein, which forms 14-stranded twisted filaments in sickle cell anemia (26), and a variety of synthetic organic molecules (27), and possibly to non-chiral molecules that interact with chiral counterions (28). As such, it contributes to our understanding of the way in which chirality is expressed in the form of self-assembled structures. Moreover, it provides a prescription for measurement of the interaction energies governing the self-assembly process.
Supplementary Material
Acknowledgments
We thank Dr. P. McPhie for assistance with TEM. We thank the United Kingdom Engineering and Physical Sciences Research Council and Schlumberger Cambridge Research for financial support, and the Royal Society for the award of a Dorothy Hodgkin Fellowship to A.A.
References
- 1.Artsaenko O, Kettig B, Fiedler K, Conrad K, During K. Mol Breeding. 1998;4:313–319. [Google Scholar]
- 2.Urry D. Trend Biotechnol. 1999;17:249–257. doi: 10.1016/s0167-7799(99)01306-2. [DOI] [PubMed] [Google Scholar]
- 3.Panitch A, Yamaoka T, Fournier M J, Mason T L, Tirrell D A. Macromolecules. 1999;32:1701–1703. [Google Scholar]
- 4.Krejchi M T, Atkins E D T, Waddon A J, Fournier J, Mason T L, Tirrell D A. Science. 1994;265:1427–1432. doi: 10.1126/science.8073284. [DOI] [PubMed] [Google Scholar]
- 5.Zhang S G, Altman M. React Funct Polym. 1999;41:91–102. [Google Scholar]
- 6.Aggeli A, Bell M, Boden N, Keen J N, Knowles P F, McLeish T C B, Pitkeathly M, Radford S E. Nature (London) 1997;386:259–262. doi: 10.1038/386259a0. [DOI] [PubMed] [Google Scholar]
- 7.Fandrich M, Fletcher M A, Dobson C M. Nature (London) 2001;410:165–166. doi: 10.1038/35065514. [DOI] [PubMed] [Google Scholar]
- 8.Fink A L. Folding Des. 1998;3:R9–R23. doi: 10.1016/S1359-0278(98)00002-9. [DOI] [PubMed] [Google Scholar]
- 9.Aggeli A, Bell M, Boden N, Keen J N, McLeish T C B, Nyrkova I, Radford S E, Semenov A. J Mater Chem. 1997;7:1135–1145. [Google Scholar]
- 10.Terzi E, Holzemann G, Seelig J. J Mol Biol. 1995;252:633–642. doi: 10.1006/jmbi.1995.0525. [DOI] [PubMed] [Google Scholar]
- 11.Nyrkova I, Semenov A, Aggeli A, Boden N. Eur Phys J B. 2000;17:481–497. [Google Scholar]
- 12.Israelachvili J N. Intermolecular and Surface Forces. London: Academic; 1985. [Google Scholar]
- 13.Nyrkova I, Semenov A N, Aggeli A, Bell M, Boden N, McLeish T C B. Eur Phys J B. 2000;17:499–513. [Google Scholar]
- 14.Perutz M F, Johnson T, Suzuki M, Finch J T. Proc Natl Acad Sci USA. 1994;91:5355–5358. doi: 10.1073/pnas.91.12.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manning M C, Illangasekare M, Woody R W. Biophys Chem. 1988;31:77–86. doi: 10.1016/0301-4622(88)80011-5. [DOI] [PubMed] [Google Scholar]
- 16.Chothia C. J Mol Biol. 1973;75:295–302. doi: 10.1016/0022-2836(73)90022-3. [DOI] [PubMed] [Google Scholar]
- 17.Dobb M G, Johnson D J, Saville B P. J Polym Sci. 1977;15:2201–2211. [Google Scholar]
- 18.Semenov A N, Khokhlov A R. Sov. Phys. Usp. 1988. 31. [Google Scholar]
- 19.Sunde M, Blake C F. Q Rev Biophys. 1998;31:1–39. doi: 10.1017/s0033583598003400. [DOI] [PubMed] [Google Scholar]
- 20.Kirschner D A, Elliott-Bryant R, Szumowski K E, Gonnerman W A, Kindy M S, Sipe J D, Cathcart E S. J Struct Biol. 1998;124:88–98. doi: 10.1006/jsbi.1998.4047. [DOI] [PubMed] [Google Scholar]
- 21.Seilheimer B, Bohrmann B, Bondolfi L, Muller F, Stuber D, Dobeli H. J Struct Biol. 1997;119:59–71. doi: 10.1006/jsbi.1997.3859. [DOI] [PubMed] [Google Scholar]
- 22.Jimenez J, Guijarro J, Orlova E, Zurdo J, Dobson C, Sunde M, Saibil H R. EMBO J. 1999;18:815–821. doi: 10.1093/emboj/18.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lim A, Saderolm M J, Makhov A M, Kroll M, Yan Y, Perera L, Griffith J D, Erickson B W. Protein Sci. 1998;7:1545–1554. doi: 10.1002/pro.5560070708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Harper J, Lansbury P. Annu Rev Biochem. 1997;66:385–407. doi: 10.1146/annurev.biochem.66.1.385. [DOI] [PubMed] [Google Scholar]
- 25.Byrom D. Biomaterials: Novel Materials from Biological Sources. New York: Stockton; 1991. [Google Scholar]
- 26.Eaton W A, Hofrichter J. Advances in Protein Chemistry. Vol. 40. San Diego: Academic; 1990. pp. 63–262. [DOI] [PubMed] [Google Scholar]
- 27.Terech P, Weiss R G. Chem Rev. 1997;97:3133–3159. doi: 10.1021/cr9700282. [DOI] [PubMed] [Google Scholar]
- 28.Oda R, Huc I, Candau S, MacKintosh F C. Nature (London) 1999;399:566–569. doi: 10.1038/21154. [DOI] [PubMed] [Google Scholar]
- 29.Grishina I B, Woody R W. Faraday Discuss. 1994;99:245–262. doi: 10.1039/fd9949900245. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}