

Abstract
STriatal Enriched tyrosine Phosphatase (STEP) has recently been identified as a critical player in the regulation of synaptic function. It is highly restricted to neurons within the CNS and acts by down-regulating the activity of the MAP kinases, the tyrosine kinase Fyn, and NMDA receptors. By modulating these substrates, STEP acts on several parallel pathways that impact upon the progression of synaptic plasticity. Recent advances have demonstrated the importance of STEP in normal cognitive function and its possible involvement in cognitive disorders, such as Alzheimer’s disease.
Introduction
Tyrosine phosphorylation of synaptic receptors and signaling molecules regulates synaptic activity (1,2). Considerable work has characterized the kinases involved in activity-dependent synaptic plasticity, with relatively less emphasis on the participating protein tyrosine phosphatases (PTPs). The identification and characterization of tyrosine phosphatases that participate in this process has begun and a number of PTPs specifically expressed within the brain have been identified (3).
One of these PTPs was named STEP (for STriatal-Enriched tyrosine Phosphatase, also known as PTPN5), and recent evidence suggests it plays an important role in synaptic plasticity. The past decade has seen considerable advances in our understanding of the function of STEP, as well as the identification of several target proteins by which STEP controls the development of synaptic plasticity. This review concentrates on three groups of proteins that STEP regulates: the mitogen-activated protein kinases (MAPKs), the tyrosine kinase Fyn, and the NMDA receptor complex. Tyrosine phosphorylation of one member of the MAPK family, the extracellular signal regulated kinase (ERK), is necessary for the expression and maintenance of synaptic plasticity in many brain regions (4), and disruption of the ERK pathway leads to a disruption of learning and memory. Activation of the Src family of non-receptor tyrosine kinases is also regulated by tyrosine phosphorylation. One of the functions of these kinases is to phosphorylate NMDA receptors, thereby modulating their channel conductance properties and facilitating their movement to neuronal plasma membranes (2). This potentiates their activity and is required for the induction of several forms of long-term potentiation (LTP) and long-term depression (LTD) (5,6). This review discusses the properties of STEP that are necessary for its ability to regulate these three families of proteins and its role in synaptic function, learning and CNS pathology.
Molecular properties of STEP
STEP is specifically expressed within neurons of the central nervous system (7). As its name indicates, the highest expression level is within the striatum (8). However, more recent work has found that it is expressed at lower levels in multiple brain regions including the neocortex, amygdala, hippocampus, and embryonic spinal cord (9,10).
Tyrosine phosphatases are broadly divided into the receptor-like and the non-receptor, intracellular phosphatases (11,3). Of the approximately 100 tyrosine phosphatases identified in the human genome, STEP falls into a small subset of the non-receptor tyrosine phosphatases (12,13). Based on sequence homology, its closest relatives are HePTP and PTP-SL that are also expressed in a restricted fashion, with HePTP found only in leucocytes and PTP-SL enriched within the cerebellum (14–17).
STEP mRNA is alternatively spliced into two main variants (Figure 1). The protein products are termed STEP46 and STEP61 based on their observed electrophoretic mobility (7,18). STEP46 is cytosolic, while STEP61 is membrane-bound and differs from STEP46 by the presence of an extra 172 amino acids at its N-terminus (Figure 1). STEP61 is not a plasma membrane spanning protein; rather, the N-terminal sequence targets STEP61 to intracellular organelles including the endoplasmic reticulum (ER) and the postsynaptic density (9,18,19). The orientation of STEP61 on the ER is not currently known. The ability of STEP to regulate cytosolic proteins favors an orientation in which the catalytic domain faces the cytosolic compartment, although additional work is needed to clarify this point. Two additional alternatively spliced variants are expressed that lack an active phosphatase domain (20,21). The functions of these inactive variants are not known, although it is possible that in the absence of a catalytic domain, they may act as dominant-negative STEP isoforms that compete with the active isoforms for binding to substrates. In this review, we will concentrate on STEP46 and STEP61 as most of the work to date has studied the function of these two variants.
Figure 1. STEP structure.
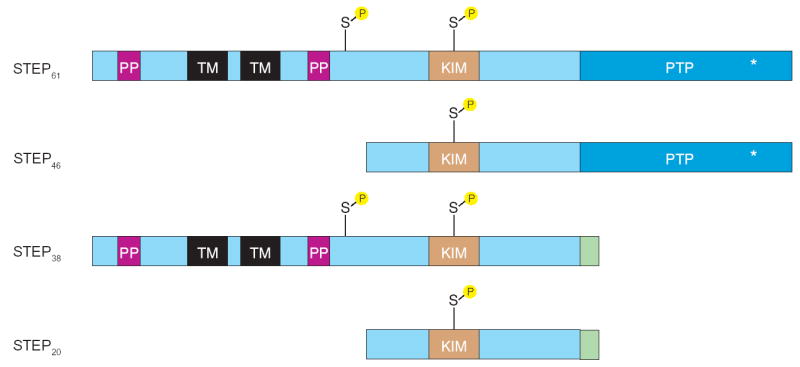
Alternative splicing produces four STEP isoforms. STEP46 and STEP61 contain the catalytic domain, which is absent from the other two isoforms STEP38 and STEP20. STEP46 is cytosolic while STEP61 is targeted to the endoplasmic reticulum and the postsynaptic density. These two isoforms differ by an additional 172 amino acids at the N-terminus of STEP61. This domain contains two transmembrane (TM) domains, two polyproline and two adjacent PEST domains (PP). The first polyproline domain interacts with Fyn, while the PEST sequences are sites of potential cleavage. Domains that are shared by STEP46 and STEP61 include the binding site for ERK, the kinase interacting motif (KIM), and the approximately 280 amino acid phosphatase domain (PTP) containing an 11 amino acid catalytic site (*). STEP61 has two serine PKA phosphorylation sites (S), whereas STEP46 contains only the one within the KIM domain. Phosphorylation within the KIM domain sterically prevents the association of ERK with STEP, and leads to enzyme inactivation. The second serine site in STEP61 is immediately adjacent to a PEST site, and is thought to facilitate proteolytic cleavage at that site. The functions of STEP38 and STEP20 are not known at present. These two inactive isoforms may function as dominant negative variants that compete with the active STEP variants for substrates and, by binding to these substrates, preserve (or prevent) their tyrosine dephosphorylation. Note that these variants also have a novel C-terminal 10 amino acid sequence (green), introduced by the alternative splicing, of unknown function.
Both STEP46 and STEP61 have a C-terminal domain of approximately 280 amino acids that contains a catalytic site with the consensus sequence (I/V)HCXAGXXR(S/T)G (Figure 1). A kinase-interaction motif (KIM) is located N-terminal to the phosphatase domain. This domain is uniquely found in STEP, HePTP and PTP-SL and is the binding site for members of the MAP kinase family. The N-terminal domain of STEP61 contains two polyproline rich regions. The first of these was shown to mediate, at least in part, the interactions of STEP61 with substrates (for example, Fyn; 22). Two hydrophobic transmembrane domains and two PEST sequences are also present. The latter sequences are potential sites for proteolytic cleavage, and two studies have shown that STEP61 is cleaved after hypoxia/anoxia in rat forebrain or excitotoxic glutamate stimulation (23,24); however, whether the PEST sites in STEP61 are the points of cleavage remains to be determined.
Baseline expression of STEP isoforms varies depending on the tissue examined. Thus, the striatum and portions of the amygdala (central nucleus) express both STEP46 and STEP61. The hippocampus, neocortex, spinal cord, and lateral aspects of the amygdala only express the larger STEP61 variant. This variation in isoform levels is reflected in the stronger immunohistochemical staining within the striatum and central nucleus compared, for example, to the hippocampus. STEP is expressed throughout the length of the neuron in a Golgi-like impregnation pattern (9). Thus, the somata, dendritic arbors and axonal processes are STEP immunoreactive. The projection targets of striatal neurons (globus pallidus and substantia nigra) have only neuritic staining with no detectable STEP immunoreactivity in cell bodies. A conclusion of this latter finding is that, although the majority of work to date has emphasized the function of STEP postsynaptically, STEP is also present presynaptically and may regulate synaptic transmission through presynaptic mechanisms. For example, ERK has been shown to regulate synapsin I by phosphorylation at key regulatory serine residues (25), and phosphorylation at these sites leads to the movement of synaptic vesicles from a reserve pool to a readily useable pool (26). The presence of STEP in this compartment would allow for the inactivation of ERK and a decrease in the pool of vesicles immediately available for fusion. This hypothesis is currently being tested.
Regulation of STEP activity
Studies on the regulation of STEP activity have focused upon the striatum where it is expressed in medium spiny neurons that make up about 90% of the neuronal cell types within this brain region (reviewed in 27). Dopaminergic inputs from the midbrain and glutamatergic afferents from the cortex converge on the spines of these neurons (28). Considerable evidence indicates that the integration of these two synaptic inputs promotes their impact on synaptic function and plasticity, although the mechanisms for this remain unclear (29). Recent findings suggest that STEP is involved in the integration of these signals. Stimulation of dopamine D1 receptors is coupled to adenylyl cyclase through Gαs leading to increased cAMP levels, which in turn activates the PKA pathway. PKA phosphorylates both STEP46 and STEP61 at a regulatory serine residue within their respective KIM domains, as well as a serine residue in the novel 172 amino acid N-terminal domain of STEP61. The effect of phosphorylation is to decrease STEP’s enzymatic activity towards MAP kinases (Figure 2). This may be due to directly affecting the catalytic activity of the phosphatase, as is suggested by in vitro studies (30), which would result in effects on any substrate. Additionally phosphorylation within the KIM domain has been demonstrated to prevent STEP from binding to ERK; however, it remains to be determined whether interactions with other substrates are also affected by phosphorylation within the KIM domain.
Figure 2. STEP regulation.
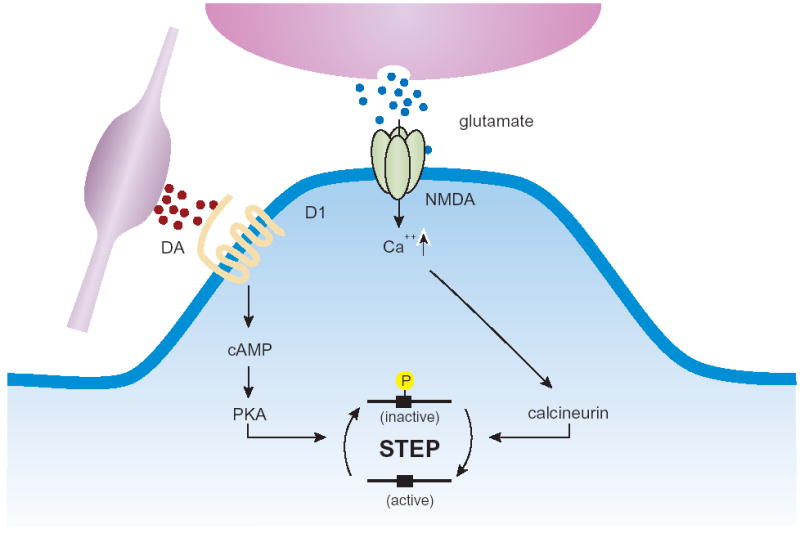
Dopamine stimulation of D1 receptors leads to cAMP synthesis, PKA activation and phosphorylation of STEP. Phosphorylation of the regulatory serine within the KIM domain prevents STEP from interacting with some substrates, such as ERK. Glutamate stimulation of NMDA receptors allows Ca2+ influx and activation of the serine phosphatase calcineurin leading to dephosphorylation of the regulatory KIM domain serine residue and thereby activation of STEP.
Phosphorylation of STEP is stimulated by D1 selective agonists, blocked by D1 receptor antagonists and not blocked by D2 receptor antagonists (31). Glutamate stimulation reverses this process and activates STEP. Stimulation of NMDA receptors, but not AMPA receptors, results in the influx of Ca2+ and activation of the serine/threonine phosphatase calcineurin. As a result, STEP is dephosphorylated at the KIM domain regulatory serine residue (30). Furthermore, it has been demonstrated that PP-1 can act to dephosphorylate the regulatory serine residue in the KIM domain of STEP (32) and the highly related HePTP (33).
STEP functions
The specificity of PTPs towards their substrates arises through amino acid modules that target PTPs to cellular compartments, while additional motifs lead to their interactions with substrate proteins. As mentioned above, STEP, along with its closest relatives HePTP and PTP-SL, contain a KIM domain that is necessary for binding to MAPK family members ERK, p38α, and JNK (33). All three of these PTPs dephosphorylate the regulatory tyrosine in the activation loop of MAPKs and thereby inactivate them (30,34–36).
The ability of STEP to regulate ERK (Figure 3) has been shown in a number of studies. In corticostriatal cultures, ERK is rapidly activated (within 2 minutes) in response to glutamate stimulation, followed by a delayed inactivation of ERK to baseline phosphorylation levels by 20–30 minutes. This delayed inactivation of ERK is mediated by STEP through its delayed dephosphorylation within the KIM domain in response to NMDA receptor-dependent activation of calcineurin (30) (Figure 2). Thus, STEP acts to regulate the temporal profile of ERK activity, and consequently helps to control its translocation to the nucleus, and subsequent downstream nuclear signaling.
Figure 3. STEP dephosphorylates ERK, Fyn and the NMDA receptor complex.
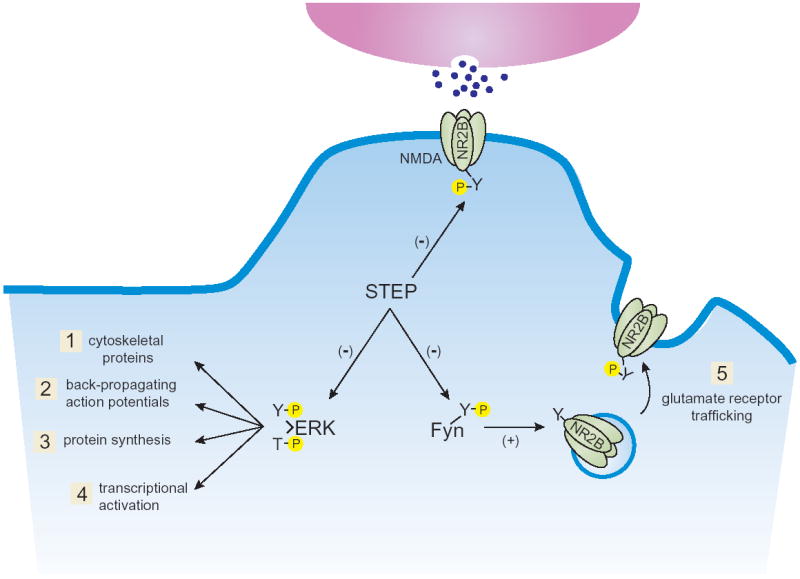
ERK, Fyn and the NR2B subunit of the NMDA receptor are potential STEP substrates. Active ERK is required for synaptic plasticity in all brain regions tested to date. In its activated state, ERK (1) phosphorylates cytoskeletal proteins, (2) regulates back-propagating action potentials, (3) stimulates protein synthesis, and (4) activates transcription. These processes work in parallel to promote synaptic plasticity. Fyn activation has also been implicated in synaptic plasticity through a variety of mechanisms including regulation of (5) glutamate receptor trafficking. Tyrosine phosphorylation of the NR2B subunit of the NMDA receptor results in exocytosis of NMDA receptor–containing endosomes.
In a second study, STEP was found to play an important role in a signal transduction cascade that mediates the effects of psychostimulant drugs on ERK activation (32). Psychostimulant drugs of abuse exert their addictive effects by increasing extracellular dopamine in the nucleus accumbens, where they likely alter the plasticity of corticostriatal glutamatergic transmission. Cocaine and amphetamine activate ERK in a subset of medium spiny neurons of the dorsal striatum and nucleus accumbens, through the combined action of NMDA and D1-dopaminergic receptors. The activation of ERK involves D1-dopamine receptor-dependent regulation of PKA, phosphorylation of the regulatory protein DARPP-32, inhibition of the serine/threonine phosphatase, PP-1, and inhibition of STEP. Thus, activation of ERK, by a protein phosphatase cascade, functions as a detector of coincidence of dopamine and glutamate signals converging on accumbens medium spiny neurons and is critical for long-lasting effects of drugs of abuse.
Recently, a series of in vivo investigations directly tested the hypothesis that STEP might be involved in regulating synaptic plasticity (37). ERK activation is required for the consolidation of many forms of long-term memory, including fear conditioning (38). Mutations of PTPs in their catalytic domain create inactive variants that may be used as substrate-trapping proteins to identify potential substrates. Inactive PTPs bind to their substrates but do not release them, as release requires dephosphorylation of the target protein (39). A substrate-trapping mutant of STEP46 was made by mutating a required cysteine in the catalytic domain to a serine. This STEP variant was made cell permeable by attaching a TAT-peptide to the N-terminus. It was infused into the lateral amygdala of rats to determine whether it would bind to ERK, disrupt ERK signaling, and thereby block consolidation of long-term memories after fear conditioning. Animals were trained on a standard protocol where a shock is paired with an acoustic cue. Short-term memory was not affected in these animals, implying that the substrate trapping TAT-STEP protein did not block the acquisition of this form of memory. However, 24 hours after fear conditioning, long-term memory was disrupted, indicating an effect on the consolidation of fear memories.
There were two striking observations in that study. The first was the rapidity of ERK activation after fear conditioning. Phosphorylated ERK (pERK) was detected in lateral amygdala neurons within five minutes of training, returned to baseline levels by 15 minutes, and then increased again by one hour. The initial activation of ERK is thought to occur through the convergence onto lateral amygdala neurons of auditory thalamic inputs in response to the conditioning stimulus (tone) and somatosensory thalamic inputs in response to the unconditioned stimulus (electrical foot shock). Both inputs are required for the establishment of LTP in the lateral amygdala and the consolidation of fear conditioning (40,41).
Activation of ERK was followed within an additional few minutes by the de novo translation of STEP (37). The translation of STEP was blocked by anisomysin, not affected by actinomycin D, and blocked by inhibitors of MAPK. Importantly, neither shock alone nor tone alone led to ERK activation or STEP translation. Within minutes after the de novo synthesis of STEP, pERK levels returned to baseline levels. These results support a feedback model by which STEP regulates the duration that ERK is active. Additional modulatory inputs are likely to be involved. For example, if a dopaminergic input arrives to these same neurons, then STEP will be phosphorylated and no longer interact with ERK, leading to a more persistent pERK signal. Additional studies are needed to determine whether the infused TAT-STEP that prevented the consolidation of fear conditioning did so through its ability to block ERK signaling only, or whether it also disrupts other components of synaptic plasticity, through the regulation of STEP substrates such as Fyn or NMDA receptors.
As was mentioned above, mutations of PTPs in their catalytic domain create inactive variants that may be used as substrate-trapping proteins. This type of inactive STEP protein was used to identify a second STEP substrate, the non-receptor tyrosine kinase Fyn (Figure 3) (22). STEP interacts with Fyn through its KIM domain, although the first polyproline sequence present in STEP61 is also involved in Fyn binding (22). Interestingly, the related tyrosine kinases, Src, Lyn and Pyk2, which are also present within the postsynaptic density, did not interact directly with STEP under the conditions used in this study (22). Two tyrosine residues are phosphorylated in the Src family of non-receptor kinases, and the enzymatic activity of these proteins depends upon which tyrosine is phosphorylated. STEP specifically catalyzes the dephosphorylation of Tyr420, leading to the inactivation of Fyn. Conversely, a second PTP (PTPα) dephosphorylates Tyr531, and dephosphorylation of this residue activates Fyn (42,43).
The NMDA receptor is a third potential STEP substrate. The NR1 subunit was initially shown to associate with STEP through co-immunoprecipitation experiments using hippocampal tissue (10) and more recently it has been shown that NMDA receptor subunits and STEP interact directly (44). STEP regulates NMDA receptor trafficking by controlling the level of tyrosine phosphorylation of the NR2B subunit (45). Tyrosine phosphorylation of NR2B at Tyr1472 by Src-family members, including Fyn, is required for the movement of NMDA receptors into membranes (46,47). Dephosphorylation of the NR2B subunit at that same residue leads to endocytosis of NMDA receptors through a clathrin- and adaptor protein-2-mediated mechanism (48). Current studies are determining whether this is through the direct dephosphorylation of the NMDA receptor by STEP, an indirect effect through its ability to reduce Fyn activity and thus decrease NMDA Tyr1472 phosphorylation levels, or whether both mechanisms work together in a cooperative fashion (Figure 4).
Figure 4. STEP activation may lead to abnormal NMDA receptor endocytosis in Alzheimer’s disease.
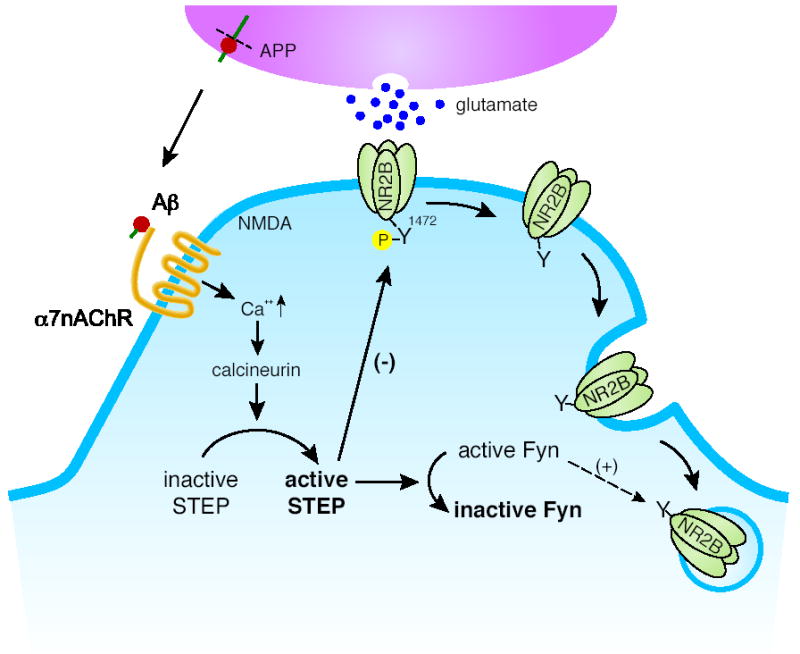
Aβ-peptide binding to the α7 nicotinic acetylcholine receptor (α7nAChR) leads to Ca2+ influx, calcineurin activation, and STEP dephosphorylation. Dephosphorylation activates STEP, which in turn inactivates Fyn. Fyn has been implicated in the phosphorylation of a regulatory tyrosine (Y1472) on the NR2B subunit of the NMDA receptor that leads to exocytosis of this receptor. In the absence of Fyn-mediated tyrosine phosphorylation, the NMDA receptor is internalized by endocytosis. Active STEP opposes trafficking to the membrane by dephosphorylating Fyn and dephosphorylating the Y1472 site on the NR2B subunit.
An initial electrophysiological study looked at the ability of STEP to regulate NMDA receptor channel properties (10). STEP affects the function of synaptic NMDA receptors in both spinal cord cultures and hippocampal CA1 pyramidal neurons. Exogenously applied STEP decreased the open probability and mean channel open time of NMDA receptors in single channel recording from excised patches of spinal cord neurons (10). Furthermore, infusing a functionally inhibitory STEP antibody increased the NMDA receptor-mediated component of synaptic responses. Because NMDA receptors are critically important for the induction of LTP, it was important to examine the role of STEP in this form of synaptic plasticity (10). Microinfusion of active STEP protein into the postsynaptic neuron blocked LTP induction at hippocampal Schaffer collateral CA1 synapses. Conversely, infusion of the functionally inhibitory antibody caused an increase in basal synaptic transmission, thereby occluding LTP induction. Thus, STEP appears to directly affect the conductance properties of NMDA receptors as well as regulating NMDA receptor trafficking, and together, these mechanisms oppose the development of synaptic plasticity.
Significance of STEP in pathological states
Recent studies have linked STEP to the cognitive decline observed in Alzheimer’s disease (45). The abnormal secretion of beta-amyloid peptide (Aβ) has been implicated in Alzheimer’s disease, and the appearance of plaques and neurofibrillary tangles have been thought to be a pathogenic cause of the disorder. A second model posits that soluble Aβ interferes with synaptic function itself and at an earlier time point (49,50).
Snyder et al. (45) directly tested the synaptic hypothesis of Alzheimer’s disease by asking whether Aβ might disrupt NMDA receptor trafficking. Aβ promoted the endocytosis of NMDA receptors in hippocampal cultures without affecting the total level of these receptors. Moreover, a similar decreased level of glutamate receptors was found on neuronal plasma membranes in a mouse model of Alzheimer’s disease that secretes high levels of Aβ. As mentioned above, exocytosis and endocytosis of NMDA receptors are regulated, in part, by tyrosine phosphorylation of the NR2B subunit. Aβ-induced endocytosis of NMDA receptors was blocked by preincubation of hippocampal cultures with the substrate-trapping TAT-STEP protein. The implication was that STEP might normally be involved in the endocytosis of glutamate receptors, and that it was being inappropriately activated by Aβ.
The study went on to determine the signaling pathway by which Aβ-induced endocytosis occurred (Figure 4). Aβ bound to the α7 nicotinic acetylcholine receptor (α7nAChR), leading to Ca2+ influx and activation of calcineurin. Calcineurin activity resulted in dephosphorylation of the regulatory serine within the KIM domain of STEP, thereby activating it. Active STEP could now dephosphorylate Fyn and/or NR2B, promoting endocytosis of NMDA receptors. A second study has also implied a role for STEP in the actions of Aβ. In a different transgenic mouse model of Alzheimer’s disease, the investigators found increased levels of α7 nAChR, decreased active Fyn, and increased STEP protein levels in the dentate gyrus (51).
It remains to be determined exactly how the substrate trapping TAT-STEP prevents NMDA receptor endocytosis. Given the fact that it acts as a substrate-binding protein, one possible model is that TAT-STEP binds to the Tyr1472 site and blocks normal dephosphorylation of that site. The increased tyrosine phosphorylation of NR2B would promote its localization at the plasma membrane. These observations support the hypothesis that one of the earliest pathological events in Alzheimer’s disease is a tyrosine dephosphorylation-mediated endocytosis of glutamate receptors, and that this process may be involved in the progressive cognitive loss in affected patients. Because STEP is an integral part of the signaling pathway between Aβ and the NMDA receptor, inhibiting STEP activity is a potential avenue for new therapeutic agents in the treatment of Alzheimer’s disease.
Conclusion
STEP regulates the activity of the MAPKs, Fyn, and NMDA receptors, and by regulating these substrates, it opposes the development of synaptic plasticity. Future work will determine whether STEP also plays a role in memory consolidation in brain regions outside of the amygdala. Additional studies should focus on STEP’s contribution to CNS disorders, because of its critical substrates and high levels of expression in the striatum. As Confucius said “A journey of a thousand miles begins with a single step.” The possibilities are only just beginning to be recognized and the path that clarifies the roles of STEP will be an exciting journey.
References
- 1.Soderling TR, Derkach VA. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000;23:75–80. doi: 10.1016/s0166-2236(99)01490-3. [DOI] [PubMed] [Google Scholar]
- 2.Salter MW, Kalia LV. Src kinases: a hub for NMDA receptor regulation. Nat Rev Neurosci. 2004;5:317–328. doi: 10.1038/nrn1368. [DOI] [PubMed] [Google Scholar]
- 3.Paul S, Lombroso PJ. Receptor and nonreceptor protein tyrosine phosphatases in the nervous system. Cell Mol Life Sci. 2003;60:2465–2482. doi: 10.1007/s00018-003-3123-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sweatt JD. Mitogen-activated protein kinases in synaptic plasticity and memory. Curr Opin Neurobiol. 2004;14:311–317. doi: 10.1016/j.conb.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 5.Nicoll RA. Expression mechanisms underlying long-term potentiation: a postsynaptic view. Philos Trans R Soc Lond B Biol Sci. 2003;358:721–726. doi: 10.1098/rstb.2002.1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Collingridge GL, Isaac JT, Wang YT. Receptor trafficking and synaptic plasticity. Nat Rev Neurosci. 2004;5:952–962. doi: 10.1038/nrn1556. [DOI] [PubMed] [Google Scholar]
- 7.Lombroso PJ, Murdoch G, Lerner M. Molecular characterization of a protein tyrosine phosphatase enriched in striatum. PNAS USA. 1991;88:7242–7246. doi: 10.1073/pnas.88.16.7242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lombroso PJ, Naegele JR, Sharma E, Lerner M. A protein tyrosine phosphatase expressed within dopaminoceptive neurons of the basal ganglia and related structures. J Neuroscience. 1993;13:3064–3074. doi: 10.1523/JNEUROSCI.13-07-03064.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boulanger LM, Lombroso PJ, Raghunathan A, During MJ, Wahle P, Naegele JR. Cellular and molecular characterization of a brain-enriched protein tyrosine phosphatase. J Neuroscience. 1995;15:1532–1544. doi: 10.1523/JNEUROSCI.15-02-01532.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pelkey K, Askalan R, Paul S, Kalia LV, Nguyen T-H, Pitcher GM, Salter MW, Lombroso PJ. Tyrosine phosphatase STEP is a tonic brake on induction of long-term potentiation. Neuron. 2002;34:127–138. doi: 10.1016/s0896-6273(02)00633-5. [DOI] [PubMed] [Google Scholar]
- 11.Stoker AW. Protein tyrosine phosphatases and signalling. J Endocrinol. 2005;185:19–33. doi: 10.1677/joe.1.06069. [DOI] [PubMed] [Google Scholar]
- 12.Andersen JN, Jansen PG, Echwald SM, Mortensen OH, Fukada T, Del Vecchio R, Tonks NK, Moller NP. A genomic perspective on protein tyrosine phosphatases: gene structure, pseudogenes, and genetic disease linkage. FASEB J. 2004;18:8–30. doi: 10.1096/fj.02-1212rev. [DOI] [PubMed] [Google Scholar]
- 13.Alonso A, Sasin J, Bottini N, Friedberg I, Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J, Mustelin T. Protein tyrosine phosphatases in the human genome. Cell. 2004;117:699–711. doi: 10.1016/j.cell.2004.05.018. [DOI] [PubMed] [Google Scholar]
- 14.Zanke B, Suzuki H, Kishihara K, Mizzen L, Minden M, Pawson A, Mak TW. Cloning and expression of an inducible lymphoid-specific, protein tyrosine phosphatase (HePTPase) Eur J Immunol. 1992;22:235–239. doi: 10.1002/eji.1830220134. [DOI] [PubMed] [Google Scholar]
- 15.Sharma E, Lombroso PJ. A neuronal protein tyrosine phosphatase induced by nerve growth factor. JBC. 1995;270:49–53. doi: 10.1074/jbc.270.1.49. [DOI] [PubMed] [Google Scholar]
- 16.Hendriks W, Schepens J, Brugman C, Zeeuwen P, Wieringa B. A novel receptor-type protein tyrosine phosphatase with a single catalytic domain is specifically expressed in mouse brain. Biochem J. 1995;305:499–504. doi: 10.1042/bj3050499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chirivi RG, Dilaver G, van de Vorstenbosch R, Wanschers B, Schepens J, Croes H, Fransen J, Hendriks W. Characterization of multiple transcripts and isoforms derived from the mouse protein tyrosine phosphatase gene Ptprr. Genes Cells 2004. 2004;9:919–933. doi: 10.1111/j.1365-2443.2004.00773.x. [DOI] [PubMed] [Google Scholar]
- 18.Bult A, Zhao F, Dirkx R, Sharma E, Lukacsi E, Solimena M, Naegele JR, Lombroso PJ. STEP61: A new member of a family of brain-enriched PTPs is localized to the ER. J Neuroscience. 1996;16:7821–7831. doi: 10.1523/JNEUROSCI.16-24-07821.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Oyama T, Goto S, Nishi T, Sato K, Yamada K, Yoshikawa M, Ushio Y. Immunocytochemical localization of the striatal enriched protein tyrosine phosphatase in the rat striatum: EM study. Neuroscience. 1995;69:869–880. doi: 10.1016/0306-4522(95)00278-q. [DOI] [PubMed] [Google Scholar]
- 20.Bult A, Zhao F, Dirkx R, Raghunathan A, Solimena M, Lombroso PJ. STEP: A family of brain enriched PTPs: Alternative splicing produces transmembrane, cytosolic and truncated isoforms. European Journal of Cell Biology. 1997;72:337–344. [PubMed] [Google Scholar]
- 21.Sharma E, Zhao F, Bult A, Lombroso PJ. Identification of two alternatively spliced transcripts of STEP: a subfamily of brain-enriched protein tyrosine phosphatases. Molecular Brain Research. 1995;32:87–93. doi: 10.1016/0169-328x(95)00066-2. [DOI] [PubMed] [Google Scholar]
- 22.Nguyen TH, Liu J, Lombroso PJ. Striatal enriched phosphatase 61 (STEP61) dephosphorylates Fyn at phosphotyrosine 420. JBC. 2002;277:24274–24279. doi: 10.1074/jbc.M111683200. [DOI] [PubMed] [Google Scholar]
- 23.Gurd J, Bissoon N, Nguyen TH, Lombroso PJ, Beesley PW, Rider CC, Vannucci SJ. Hypoxia-ischemia in perinatal rat brain induces the formation of a low molecular weight isoform of the protein tyrosine phosphatase, STEP. J Neurochemistry. 1999;73:1990–1995. [PubMed] [Google Scholar]
- 24.Nguyen TH, Xu Y, Gurd J, Lombroso PJ. Ca++-dependent cleavage of striatal enriched tyrosine phosphatase (STEP) J Neurochemistry. 1999;73:1995–2001. [PubMed] [Google Scholar]
- 25.Jovanovic JN, Benfenati F, Siow YL, Sihra TS, Sanghera JS, Pelech SL, Greengard P, Czernik AJ. Neurotrophins stimulate phosphorylation of synapsin I by MAP kinase and regulate synapsin I-actin interactions. PNAS. 1996;93:3679–3683. doi: 10.1073/pnas.93.8.3679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kushner SA, Elgersma Y, Murphy GG, Jaarsma D, van Woerden GM, Hojjati MR, Cui Y, LeBoutillier JC, Marrone DF, Choi ES, De Zeeuw CI, Petit TL, Pozzo-Miller L, Silva AJ. Modulation of presynaptic plasticity and learning by the H-ras/extracellular signal-regulated kinase/synapsin I signaling pathway. J Neurosci. 2005;25:9721–9734. doi: 10.1523/JNEUROSCI.2836-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Packard MG, Knowlton BJ. Learning and memory functions of the Basal Ganglia. Annu Rev Neurosci. 2002;25:563–593. doi: 10.1146/annurev.neuro.25.112701.142937. [DOI] [PubMed] [Google Scholar]
- 28.Kotter R. Postsynaptic integration of glutamatergic and dopaminergic signals in the striatum. Prog Neurobio. 1994;44:163–196. doi: 10.1016/0301-0082(94)90037-x. [DOI] [PubMed] [Google Scholar]
- 29.Cepeda C, Levine MS. Dopamine and N-methyl-D-aspartate receptor interactions in the neostriatum. Dev Neurosci. 1998;20:1–18. doi: 10.1159/000017294. [DOI] [PubMed] [Google Scholar]
- 30.Paul S, Nairn A, Wang P, Lombroso PJ. NMDA-mediated activation of the protein tyrosine phosphatase, STEP, regulates the duration of ERK signaling. Nature Neuroscience. 2003;6:34–42. doi: 10.1038/nn989. [DOI] [PubMed] [Google Scholar]
- 31.Paul S, Hisayuki Y, Snider G, Picciotto M, Nairn A, Lombroso PJ. Dopamine/D1 receptor mediates the phosphorylation and inactivation of the protein tyrosine phosphatase, STEP, through a PKA-mediated pathway. J Neuroscience. 2000;20:5630–5638. doi: 10.1523/JNEUROSCI.20-15-05630.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Valjent E, Pascoli V, Svenningsson P, Paul S, Enslen H, Corvol J-C, Stipanovich A, Caboche J, Lombroso PJ, Nairn AC, Greengard P, Hervé D, Girault J-A. Regulation of a protein phosphatase cascade allows convergent dopamine and glutamate signals to activate ERK in the striatum. PNAS. 2004;102:491–496. doi: 10.1073/pnas.0408305102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nika K, Huynh H, Williams S, Paul S, Bottini N, Tasken K, Lombroso PJ, Mustelin Hematopoietic protein tyrosine phosphatase (HePTP) phosphorylation by cAMP-dependent protein kinase in T cells: dynamics and subcellular location. Biochem J. 2004;378:335–342. doi: 10.1042/BJ20031244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pulido R, Zuniga A, Ullrich A. PTP-SL and STEP protein tyrosine phosphatases regulate the activation of the extracellular signal-regulated kinases ERK1 and ERK2 by association through a kinase interaction motif. EMBO J. 1998;17:7337–50. doi: 10.1093/emboj/17.24.7337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Saxena M, Williams S, Brockdorff J, Gilman J, Mustelin T. Inhibition of T cell signaling by mitogen-activated protein kinase-targeted hematopoietic tyrosine phosphatase (HePTP) JBC. 1999;274:11693–11700. doi: 10.1074/jbc.274.17.11693. [DOI] [PubMed] [Google Scholar]
- 36.Zuniga A, Torres J, Ubeda J, Pulido R. Interaction of mitogen-activated protein kinases with the kinase interaction motif of the tyrosine phosphatase PTP-SL provides specificity and retains ERK2 in the cytoplasm. JBC. 1999;274:21900–21907. doi: 10.1074/jbc.274.31.21900. [DOI] [PubMed] [Google Scholar]
- 37.Paul S, Olausson P, Moran TD, Ruchkina I, Tronson N, Salter MW, Taylor J, Lombroso PJ. Disruption of Fear Conditioning By STEP: A Striatal Enriched Phosphatase, Through the Regulation of MAP Kinase. Society for Neuroscience. 2004 Abstracts 632.11. [Google Scholar]
- 38.Schafe GE, Atkins CM, Swank MW, Bauer EP, Sweatt JD, LeDoux JE. Activation of ERK/MAP kinase in the amygdala is required for memory consolidation of Pavlovian fear conditioning. J Neurosci. 2000;20:8177–8187. doi: 10.1523/JNEUROSCI.20-21-08177.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Flint A, Tganis T, Barford D, Tonks N. Development of substrate trapping mutants to identify physiological substrates of PTPs. PNAS. 1997;94:1680–1685. doi: 10.1073/pnas.94.5.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schafe GE, LeDoux JE. Memory consolidation of auditory pavlovian fear conditioning requires protein synthesis and protein kinase A in the amygdala. J Neurosci. 2000;20:RC96. doi: 10.1523/JNEUROSCI.20-18-j0003.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Blair HT, Schafe GE, Bauer EP, Rodrigues SM, LeDoux JE. Synaptic plasticity in the lateral amygdala: a cellular hypothesis of fear conditioning. Learn. Mem. 2001;8:229–242. doi: 10.1101/lm.30901. [DOI] [PubMed] [Google Scholar]
- 42.Zheng XM, Wang Y, Pallen CJ. Cell transformation and activation of pp60c-src by overexpression of a protein tyrosine phosphatase. Nature. 1992;359:336–339. doi: 10.1038/359336a0. [DOI] [PubMed] [Google Scholar]
- 43.Bhandari V, Lim KL, Pallen CJ. Physical and functional interactions between receptor-like protein-tyrosine phosphatase alpha and p59fyn. JBC. 1998;273:8691–8698. doi: 10.1074/jbc.273.15.8691. [DOI] [PubMed] [Google Scholar]
- 44.Braithwaite SP, Adkisson M, Leung J, Nikolich K, Urfer R, Oksenberg D. Striatal Enriched Tyrosine Phosphatase Regulates NMDA Receptor Trafficking. Society for Neuroscience. 2005 doi: 10.1111/j.1460-9568.2006.04837.x. Abstracts 842.15. [DOI] [PubMed] [Google Scholar]
- 45.Snyder EM, Nong Y, Almeida CG, Paul S, Moran T, Choi EY, Nairn AC, Salter MW, Lombroso PJ, Gouras GK, Greengard P. Regulation of NMDA receptor trafficking by beta-amyloid. Nature Neuroscience. 2005;8:1051–1058. doi: 10.1038/nn1503. [DOI] [PubMed] [Google Scholar]
- 46.Dunah AW, Sirianni AC, Fienberg AA, Bastia E, Schwarzschild MA, Standaert DG. Dopamine D1-dependent trafficking of striatal N-methyl-D-aspartate glutamate receptors requires Fyn protein tyrosine kinase but not DARPP-32. Mol Pharmacol. 2004;65:121–129. doi: 10.1124/mol.65.1.121. [DOI] [PubMed] [Google Scholar]
- 47.Cheung HH, Gurd JW. Tyrosine phosphorylation of the N-methyl-D-aspartate receptor by exogenous and postsynaptic density-associated Src-family kinases. J Neurochem. 2001;78:524–534. doi: 10.1046/j.1471-4159.2001.00433.x. [DOI] [PubMed] [Google Scholar]
- 48.Lavezzari G, McCallum J, Lee R, Roche KW. Differential binding of the AP-2 adaptor complex and PSD-95 to the C-terminus of the NMDA receptor subunit NR2B regulates surface expression. Neuropharmacology. 2003;45:729–737. doi: 10.1016/s0028-3908(03)00308-3. [DOI] [PubMed] [Google Scholar]
- 49.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–9. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 50.Kamenetz F, Tomita T, Hsieh H, Seabrook G, Borchelt D, Iwatsubo T, Sisodia S, Malinow R. APP processing and synaptic function. Neuron. 2003;37:925–37. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- 51.Chin J, Palop JJ, Puolivali J, Massaro C, Bien-Ly N, Gerstein H, Scearce-Levie K, Masliah E, Mucke L. Fyn kinase induces synaptic and cognitive impairments in a transgenic mouse model of Alzheimer's disease. J Neurosci. 2005;25:9694–9703. doi: 10.1523/JNEUROSCI.2980-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]