

Abstract
Glioblastoma is the most common primary malignant brain tumor of adults and one of the most lethal of all cancers. Patients with this disease have a median survival of 15 months from the time of diagnosis despite surgery, radiation, and chemotherapy. New treatment approaches are needed. Recent works suggest that glioblastoma patients may benefit from molecularly targeted therapies. Here, we address the compelling need for identification of new molecular targets. Leveraging global gene expression data from two independent sets of clinical tumor samples (n = 55 and n = 65), we identify a gene coexpression module in glioblastoma that is also present in breast cancer and significantly overlaps with the “metasignature” for undifferentiated cancer. Studies in an isogenic model system demonstrate that this module is downstream of the mutant epidermal growth factor receptor, EGFRvIII, and that it can be inhibited by the epidermal growth factor receptor tyrosine kinase inhibitor Erlotinib. We identify ASPM (abnormal spindle-like microcephaly associated) as a key gene within this module and demonstrate its overexpression in glioblastoma relative to normal brain (or body tissues). Finally, we show that ASPM inhibition by siRNA-mediated knockdown inhibits tumor cell proliferation and neural stem cell proliferation, supporting ASPM as a potential molecular target in glioblastoma. Our weighted gene coexpression network analysis provides a blueprint for leveraging genomic data to identify key control networks and molecular targets for glioblastoma, and the principle eluted from our work can be applied to other cancers.
Keywords: epidermal growth factor receptor vIII, Glioblastoma, modules, weighted gene coexpression network analysis, network-based screening
Molecularly targeted therapies are transforming the treatment of cancer (1). Small molecule inhibitors that target key enzymes on which cancer cells depend, raise the possibility of rational approaches to cancer therapy. We have demonstrated that targeted inhibition of the epidermal growth factor receptor (EGFR), a receptor tyrosine kinase commonly amplified, overexpressed, or mutated in glioblastoma, promotes significant clinical response in a subset of glioblastoma patients, and we have identified the molecular determinants of this response (2). This provides a proof-of-principle for the potential efficacy of molecularly targeted therapy for glioblastoma; identifying new drug targets is a critical next step.
The wealth of molecular information provided by genomic technologies provides a remarkable opportunity for new target discovery (3, 4). Gene expression data can provide a key first step toward constructing a systems level view of the perturbed networks in cancer cells, thus potentially identifying key genes, networks, or pathways that can be therapeutically targeted (5). However, the identification of key molecular targets still remains a challenge. Recent work highlights the potential for uncovering oncogenic pathways and molecular targets, when genomic data are analyzed at the level of gene coexpression modules or metagenes or when aggregated gene sets are used to assess modules enriched for key biological processes (6–8). Integrating this type of data with studies in model systems in which modules can be studied in response to relevant molecular perturbations (e.g., oncogene overexpression or pharmacological inhibition) may further facilitate the identification and validation of novel molecular targets (9–11). Here, we adopt an unbiased strategy to detect an oncogenic module in glioblastoma and integrate this with studies in isogenic cell systems to identify and validate ASPM (abnormal spindle-like microcephaly associated) as a previously undescribed glioblastoma target.
Results
Identification of genes with expression levels that are highly correlated may help shed light on shared biological processes or common regulatory mechanisms that could potentially be targeted. Therefore, we performed global gene expression profiling on RNA from 120 glioblastoma patient samples (data set 1, n = 55 (12); and data set 2, n = 65). To facilitate the identification of gene modules (groups of highly coexpressed genes), we constructed a weighted gene coexpression network based on pairwise Pearson correlations between the expression profiles. Unsupervised hierarchical clustering was used to detect groups, or modules, of highly coexpressed genes (13). To facilitate reproducibility of this analysis, the complete gene expression data, module composition, and statistical software code are available upon request.
Five gene coexpression modules were detected in glioblastoma data set 1 (Fig. 1a). These modules were significantly enriched for genes with the following specific ontologic classes: (i) mitosis/cell cycle (185 genes, P = 7.2 × 10−42); (ii) immune response (606 genes, P = 2.4 × 10−36); (iii) neurogenesis (143 genes, P = 4.0 × 10−4); (iv) cytoplasm (1,112 genes P = 1.1 × 10−12); and (v) metabolism (136 genes, P = 1.8 × 10−2) (EASE software: http://david.niaid.nih.gov/david/ease1.htm) (Fig. 1d). The fact that unsupervised clustering based on a coexpression measure resulted in modules enriched for biologically important processes, including cancer-related themes, suggests that these modules are a robust feature of the molecular architecture of glioblastoma.
Fig. 1.

Detection of gene coexpression modules in glioblastoma and breast cancer. (a) In glioblastoma data set 1, five gene coexpression modules were detected. (b) The network genes derived from data set 1 were mapped to data set 2. The genes maintain the same color coding for module association from data set 1, facilitating visual inspection of module conservation. The same module association in both data sets was found for 87.4% of genes (P = 2.2 × 10−16). (c) Two of the five glioblastoma modules (brown, cell cycle mitosis; blue, immune response) were detected in a set of 77 breast cancer samples. (d) The green/red expression diagram shows the relative expression of the module across 353 samples, including 180 glioblastomas, 66 meningiomas, 64 normal adult body tissues of different types, 18 adult normal brain tissue samples, 11 fetal brain tissue samples, and 14 fetal nonbrain tissue samples. The MCM is highly expressed in only a subset of glioblastomas (type 2A) (12), and its expression pattern is similar to a fetal proliferation signature. (e) Mean expression of ASPM in the 353 clinical samples, including glioblastomas, meningiomas, normal brain, normal body tissues, fetal brain, and fetal body tissues.
To determine whether these modules were reproducible, we used the genes from data set 1 to construct a weighted gene coexpression network in data set 2. We found the same five gene coexpression modules as in data set 1: 87.4% of the genes in data set 2 were embedded within the same module in data set 1 (Pearson χ2 test, P = 2.2 × 10−16, Rand index measure of agreement = 0.9) (Fig. 1b). Thus, gene coexpression modules are highly preserved in both glioblastoma data sets.
To determine whether these modules were detectable in another cancer type, we analyzed a publicly available breast cancer data set (14). This data set was sufficiently large and contained gene expression data from a different microarray platform, allowing for array platform independent conclusions. Probe sets that were common to both array platforms were mapped, and a weighted gene coexpression network was constructed based for the breast cancer data set. To determine whether glioblastoma modules were present in breast cancer, we assigned glioblastoma module colors to the genes in the hierarchical clustering tree of the breast cancer data. This revealed that two glioblastoma modules (the cell cycle/mitosis module and the immune response module) were highly preserved (Fig. 1c) (data available upon request). This suggested that the cell cycle/mitosis module may be involved in biological processes that are shared by both cancer types.
To further assess whether the mitosis/cell cycle module (MCM) is present in other cancers, we determined whether the genes contained within the module are part of the “metasignatures” of cancer that have been derived from metaanalyses of a large numbers of samples of different cancer types (15). Two metasignatures (MS) of cancer have been identified by large-scale metaanalyses across multiple cancer types, including a MS of “undifferentiated cancer” (15). Of the 48 genes of the “undifferentiated cancer” MS that were present in the glioblastoma data set, 33 (69%) were present in the MCM (P = 2.7 × 10−31).
To correlate individual expression profiles with the entire module, we summarized the expression profile of the module genes by the first module eigengene, which is defined by using the singular value decomposition of the expression data (16). To determine whether this MCM is a proliferation cluster, we correlated the module eigengene with Ki67 and PCNA (two clinically used markers of cancer cell proliferation and members of the module) (17). The module eigengene was highly correlated with both Ki67 and PCNA (Ki67: r = 0.74; P < 6.2 × 10−7 for data set 1; and r = 0.81; P < 1 × 10−20 for data set 2; PCNA: r = 0.79 P < 1 × 10−20 for data set 1; and r = 0.80; P < 1 × 10−20 for data set 2) (Fig. 5 a–d, which is published as supporting information on the PNAS web site). We next examined the expression pattern of these module genes across 353 samples including glioblastoma and other tissues (both tumor and normal). Expression of MCM genes goes up or down together across a wide range of tissue types, including glioblastoma, meningioma, normal brain, fetal brain, a range of normal nonbrain tissues, and a range of fetal nonbrain tissues (Fig. 1e). The pattern of expression of this module in a subset of glioblastomas is quite similar to that of fetal tissues (including fetal brain), and quite unlike that of normal mature brain or body tissues. Further, this module is highly expressed in only a subset of glioblastomas, the type 2A pattern, which we have shown to be associated with poor prognosis (12). The module is not highly expressed in some other subsets of glioblastoma, including the type 2B pattern that we have shown to be a highly aggressive tumor type as well (12). This raises the possibility that this proliferation module is specific to a subset of glioblastomas and that it shares similarity with a fetal proliferation signature.
Highly connected “hub” genes are thought to play an important role in organizing the behavior of biological modules (18–21). Therefore, we set out to identify the MCM hubs (22). We defined a connectivity measure (K) for each gene based on its Pearson correlation with all of the other genes in the module as described in Methods (13). Because highly connected hub genes are far more likely than nonhub genes to be essential for survival in lower organisms (18–20), we hypothesized that intramodular hub genes may be associated with survival in cancer. To define a measure of prognostic significance, we used a univariate Cox proportional hazards regression model to regress patient survival on the individual gene expression profiles. The resulting univariate Cox-regression p-values were used to define a measure of prognostic significance as follows: GS = –log10(Cox P value), i.e., this measure of gene significance is proportional to the number of zeroes in the P value. In the MCM, intramodular connectivity K and prognostic significance GS, were significantly correlated in both glioblastoma data sets (r = 0.59, P = 7.1 × 10−19 in data set 1, and r = 0.59, P = 6.5 × 10−19 in data set 2) (Fig. 2a and b).
Fig. 2.

Clinical association between hub gene status and outcome in glioblastoma and breast cancer. (a) Scatterplot between connectivity K in the MCM (x axis) and gene significance defined as GS = –log10(Cox P value) (y axis) for glioblastoma data set 1. (b) Analogous scatterplot for glioblastoma data set 2. (c) Scatterplot between intramodular connectivity K in the breast cancer network (x axis) and K in the glioblastoma network (y axis). (d) Scatterplot between intramodular connectivity K and prognostic gene significance in breast cancer (14).
Highly connected hub genes of the glioblastoma MCM tend to be highly connected in the breast cancer network as well: the correlation between the respective connectivity measures was highly significant (r = 0.62; P < 1.9 × 10−20) (Fig. 2c). Next, we determined whether the highly connected hub genes identified in glioblastoma are also related to a clinical outcome in breast cancer. Because the overall survival data were not available for that data set, we considered cancer recurrence time as the clinical outcome of interest. For each gene, we defined a measure of prognostic significance by correlating individual gene expression profiles with recurrence time. Specifically, we defined a prognostic gene significance measure as minus the logarithm of the Spearman correlation test P value GS = −log(P value). Consistent with the association between K and outcome in glioblastoma, intramodular connectivity K was also significantly associated with prognostic significance for recurrence in breast cancer (r = 0.70; P < 3.4 × 10−20) (Fig. 2d). Thus, the hub genes identified in glioblastoma were also predictive of breast cancer recurrence.
Some of the most highly connected genes within the MCM already have been identified as potential cancer targets (topoisomerase IIα, ARKB, PTTG1/sercuin, Survivin, and EZH2) (23–27). To identify a potential novel gene target, we looked for the most highly connected genes that have not been extensively studied as cancer targets. This led us to study the ASPM gene, because it had the highest K value in both glioblastoma data sets of any gene that has not been previously recognized as a cancer target. ASPM is the human ortholog of a Drosophila mitotic spindle protein, encoding the protein microcephalin (28–30). ASPM is thought to regulate neuroblast proliferation (29), and it has recently been shown to be a key regulator of brain size through evolution (31–33). Mutations within this gene are associated with primary human microcephaly (29, 30). A recent study demonstrated increased ASPM in ovarian and uterine cancers, suggesting that it may play a role in other cancer types (34), although it was not part of the MS of undifferentiated cancer (15), suggesting that it may have specificity for only a few tumor types including glioblastoma. Because ASPM is expressed at a very low level in normal brain (and normal body tissues) relative to glioblastoma (Fig. 1f), we reasoned that it could present a compelling molecular target.
Strikingly, the traditional proliferation markers Ki67 (Cox regression P = 0.13) and PCNA (P = 0.021) were less associated with glioblastoma survival than 9 of the top 10 most connected hub genes, including ASPM. Specifically, for the combined glioblastoma data set, the P values in the univariate Cox model for these nine hub genes were as follows: TOP2A (P = 0.00088), RACGAP1 (P = 0.0022), KIF4A (P = 0.0030), TPX2 (P = 0.0021), CDC2 (P = 0.0072), EZH2 (P = 0.024), CDC20 (P = 0.0029), KIF14 (P = 0.0020), RAMP (P = 0.015), and ASPM (P = 0.0059). These results suggest that the hub genes, including ASPM, may be more predictive of clinical outcome than the traditional markers of proliferation, PCNA and Ki67. Further, we found that the top third most connected genes encode proteins that are known to interact physically and/or functionally to regulate metaphase to anaphase transition (Fig. 6, which is published as supporting information on the PNAS web site).
To identify a potential molecular mechanism underlying regulation of the MCM, we used a series of isogenic U87 glioblastoma cells engineered to express EGFR, EGFRvIII, and PTEN in relevant combinations (2) (Fig. 3). These molecular alterations are common in glioblastoma (35–37), and they play a critical role in determining response to EGFR kinase inhibitor therapy (2). We performed global transcriptional profiling from RNA that was extracted from duplicate cultures of each of the isogenic U87 lines and analyzed the expression of the MCM. MCM genes were significantly up-regulated in the EGFRvIII overexpressing cells (Fig. 3a). Of note, these are also the most proliferative of the cells. These data suggested that the module is potentially downstream of EGFRvIII signaling; possibly via the PI3K pathway signaling because PTEN coexpression inhibited up-regulation of this module. We therefore analyzed expression of a series of these genes in response to the EGFR inhibitor erlotinib. Expression of each of six representative hub genes tested (ASPM, PRC1, ARKB, MELK, PTTG1, and TOP2α) was increased by EGFRvIII, which was abrogated by treatment with the EGFR inhibitor erlotinib (Fig. 3b). Thus, up-regulation of the MCM is regulated by EGFRvIII signaling in glioblastoma cells, likely via its ability to confer a proliferative advantage to these cells.
Fig. 3.

Modeling of the MCM in isogenic glioblastoma cells with varying levels of EGFRvIII, EGFR, and PTEN expression, and effect of EGFRvIII inhibition on the MCM module. (a) Gene expression profiling was performed in two independent samples for each cell line from RNA extracted from serum starved isogenic U87 glioblastoma cells (unlabeled) or U87 cells expressing the EGFRvIII oncogene, WT EGFR, and the PTEN tumor suppressor protein alone or in combination as labeled. Biochemical analyses of these cell lines has been performed in ref. 2. The expression pattern of the cell cycle/mitosis module genes demonstrated that this module is downstream of EGFRvIII, and is inhibited by coexpression of PTEN. ASPM (red arrow) is identified as one of the hub genes within this module. (b) Erlotinib treatment inhibits expression a representative subset of cell cycle/mitisos/undifferentiated cancer MS genes U87vIII overexpressing/PTEN deficient glioblastoma cells.
To validate the biological significance of ASPM, we used siRNA to stably knock down ASPM in U87 cells expressing EGFRvIII and in low passage explant culture from a glioblastoma patient. Low-passage primary patient-derived glioblastoma cells treated with two different ASPM siRNAs showed specific and dramatic inhibition of proliferation (Fig. 4a), as did five independent clones of U87-EGFRvIII cells with stably expressing ASPM siRNA (Fig. 4b). These results suggest that ASPM is a potential molecular target in glioblastoma.
Fig. 4.

Inhibition of ASPM by RNAi inhibits glioblastoma cell growth and neural stem cell self renewal. (a) Expression of two different ASPM siRNAs dramatically inhibits the proliferation of low-passage primary patient-derived glioblastoma cell line. (b) Stable infection with ASPM siRNA inhibits the growth of five independent stable clones of U87-EGFRvIII-ASPM siRNA expressing cells. This is mediated by a GI arrest (data not shown). (c) Differentiation of mouse embryonic neurospheres by basic fibroblast growth factor or EGF diminishes ASPM expression. (d) siRNA-mediated knockdown of ASPM in mouse embryonic neurospheres inhibits neurosphere formation. The inset shows ASPM expression after treatment with control (lane 1), 25 (lane 2), 50 (lane 3), or 100 nM (lane 4) ASPM. The graph shows numbers of secondary neurospheres generated from progenitors treated with 100 nM siRNA.
Brain tumor stem cells can be found in glioblastoma and have been hypothesized to play a role in their pathogenesis (38–41). Because this could potentially result from overexpression of genes that promote self renewal and because our data suggested that the module shares some similarities with fetal brain, we tested the functional role of ASPM in fetal murine neural stem/progenitor cells. These cells form floating neurospheres when cultured in the presence of basic fibroblast growth factor and/or EGF. Withdrawal of mitogen from neurosphere induces differentiation into neurons, astrocytes, and oligodendrocytes. ASPM was highly expressed in murine neurospheres and expression dramatically declined during differentiation (Fig. 4c). When murine neurosphere cultures were incubated in the presence of ASPM siRNA (Fig. 4d) and then recultured at clonal density as spheres, there was a marked loss in the production of secondary spheres, suggesting that ASPM regulates self-renewal neural stem/progenitor cells. Thus, ASPM promotes neural stem cell self-proliferation, further implicating it as a potential molecular target in glioblastoma.
Discussion
We used WGCNA to identify gene coexpression modules and therapeutic targets in glioblastoma. Several computational methods for incorporating biological pathway information and gene sets into microarray data analysis have been proposed. For example, gene set enrichment analysis (GSEA) determines whether an a priori defined set of genes shows statistically significant, concordant differences between two biological states (6). Although WGCNA shares the philosophy of GSEA of focusing on gene sets as opposed to individual genes, it does not make use of a priori defined gene sets. Instead, gene sets (modules) are constructed from the expression data by using unsupervised clustering. Although it is advisable to relate the resulting modules to gene ontology information for assessing their biological plausibility, it is not required.
WGCNA also alleviates the multiple testing problem inherent in microarray data analysis. Instead of relating thousands of genes to the clinical outcome, it focuses on the relationship between a few (here 5) modules and the clinical trait. It is worth repeating that the modules were constructed in an unsupervised manner, i.e., without regard to the clinical outcome. Because the modules may correspond to biological pathways, focusing the analysis on module eigengenes (and equivalently intramodular hub genes) amounts to a biologically motivated data reduction scheme. WGCNA starts from the level of thousands of genes, identifies clinically interesting gene modules, and finally uses intramodular connectivity to suggest suitable targets. Because the expression profiles of intramodular hub genes inside an interesting module are highly correlated (in our data, r > 0.90) typically dozens of targets result. Although these targets are statistically equivalent, they may differ in terms of biological plausibility or clinical utility. In many applications, the list of module hub genes may be further winnowed down based on (i) biological plausibility based on external gene (ontology) information, (ii) the availability of protein biomarkers for further validation, (iii) the availability of suitable mouse models for further validation, and/or, (iv) the druggability, i.e., the opportunity for therapeutic intervention.
Understanding how broad cancer-related modules interact with specific molecular lesions in an individual patient is a critical step in finding new molecular targets. Our finding that the MCM is downstream of EGFRvIII signaling suggests a potentially important link by which this process is switched on by an upstream molecular lesion. It is not surprising that the EGFRvIII expressing, PTEN-deficient glioblastoma cells also were the most proliferative. Many of the hub genes identified here (ASPM, BUB1, HEK, STK6, NEK2, PTTG1, PRC1, KNSL2, CCNB1, CDC2, and CDC20) also have been shown to be downstream of other key molecular lesions such as BRCA1 in breast cancer (42) or the human papilloma virus proteins E6/E7 in patients with cervival cancer (for which a “proliferation cluster” of 123 genes associated with HPV E6/E7 in clinical samples strikingly overlapped with the glioblastoma mitosis/cell cycle module; P = 2.2 × 10−16) (43). In addition, there was a highly significant overlap with genes that have been shown to be highly overexpressed in high grade breast cancer (P = 3.6 × 10−75) (44). These data indicate that the MCM may be up-regulated by a number of key molecular lesions that confer a proliferation advantage, thus raising the possibility that common therapies targeting this module may be useful in patients with different types of aggressive cancer.
ASPM had the highest connectivity index in both glioblastoma data sets for any gene not already known to be a cancer target, and it is expressed at very low level in normal brain (and normal body tissues) relative to glioblastoma. ASPM also has been recognized as a critical regulator of brain size, likely via its role in promoting neuroblast proliferation and symmetric division (28–30, 45). Our data showing that neural stem cell differentiation results in loss of ASPM expression and that siRNA-mediated knockdown of ASPM specifically inhibits neural stem cell self renewal and glioblastoma growth suggests the possibility that this gene may be involved in glioblastoma pathogenesis by promoting a stem cell phenotype. Further studies will be necessary to examine the suitability of targeting ASPM in glioblastomas and to determine whether it mediates its effects on glioblastoma by promoting cancer stem cell self-renewal. In summary, this study provides a blueprint for using genomic data to identify key control networks and molecular targets for glioblastoma and, potentially, for other cancers.
Methods
Microarray Data.
Glioblastoma gene expression profiling with Affymetrix high-density oligonucleotide microarrays was performed and analyzed as described in ref. 12. Quantification was performed by using model-based expression and the perfect match minus mismatch method implemented in dCHIP. We used the breast cancer microarray data (14) (Agilent) to find prognostic genes for breast cancer recurrence. The Ingenuity Pathways Knowledge Base (Ingenuity Systems, Redwood City, CA) was used to identify to subnetwork of potential interactions (46).
Genomic and Functional Analysis in Glioblastoma Cell Lines.
The isogenic U87MG expressing PTEN, EGFR, and EGFRvIII in varying combinations have been reported in ref. 2. In brief, cell lines were grown in duplicate cultures under serum free conditions for 48 h, and RNA was isolated by using the Qiagen (Valencia, CA) RNeasy Mini Kit Gene. Expression analysis by using Affymetrix HG-U133A arrays was performed and analyzed, as described above.
EGFR Inhibitor Treatment and siRNA Studies.
The EGFR tyrosine kinase inhibitor Erlotinib (Tarceva, OSI-774) was kindly provided by Genentech (South San Francisco, CA). U87MG and U87-EGFRvIII cells (1 × 105) were seeded, respectively, in 100-mm culture dishes and maintained in DMEM supplemented with 10% FBS. Cells were incubated in 5% CO2, 95% humidity incubator for 3 days to reach 50–70% confluency. Then all cells were switched to serum-free medium. The next day U87-EGFRvIII cells were treated by 5 μM OSI-774, whereas U87MG and U87-EGFRvIII control group received the equivalent vehicle. Twenty-four hours later, cell total RNA was isolated by Qiagen RNeasy Mini Kit. RT-PCR analysis of expression of selected genes after treatment is described in the Supporting Methods, which is published as supporting information on the PNAS web site. The specific methods for siRNA studies are available in Supporting Methods. For proliferation assays, 1,500 cells per well in eight replicates were seeded into 96-well plates. Cells were fixed and stained by 0.25% crystal violet in methanol every day or every other day. Stained plates were densitometry scored by AlphaImager 2200 software and plot in Microsoft (Redmond, WA) Excel.
Neurosphere Cell Culture and Transfection.
Cerebral cortex was isolated from embryonic day 12 mice. Cells were dissociated and cultured at 50,000 cells/ml in neurosphere formation medium [Neural Basal medium (Invitrogen, Carlsbad, CA) with B27 (GIBCO BRL, Carlsbad, CA), basic fibroblast growth factor (Peprotech, Rocky Hill, NJ), EGF (Chemicon, Temecula, CA), heparin (Sigma-Aldrich, St. Louis, MO) and penicillin-streptomycin (Gemini Bioproducts, West Sacramento, CA)] for a week. Growth factors were added every 3 days. Neurospheres were dissociated and plated onto polyl-ornithine (Sigma)/fibronectin-coated six-well plates in neural basal medium with 2% FBS (GIBCO BRL). Six hours later, the serum medium was removed and replaced with neurosphere formation medium without heparin and penicillin-streptomycin. Twenty-four hours later, cells were transfected with 100 nM siRNA targeting ASPM and 100 nM control siRNA targeting firefly luciferase by using lipofectAMINE 2000 (Invitrogen). The cells were incubated with reagents for 6 h and passaged for secondary neurosphere formation assay.
Secondary Neurosphere Formation Assay.
Cells were lifted off the plate with TriplExpress (GIBCO BRL) and then placed into neurosphere formation medium at 1,000 cells/ml and 100 cells/ml. Neurospheres were propagated for 1 week, and the number and the size of the secondary neurospheres formed were measured by using Microcomputer Imaging Device program.
A comprehensive materials and methods section is available upon request. We also provide the entire statistical code, the data, and a weighted gene coexpression network analysis tutorial so that the reader can reproduce all of our findings.
Supplementary Material
Acknowledgments
This work was supported by National Institute of Neurological Disorders and Stroke Grants NS050151 and NS43137, National Cancer Institute (NCI) Grant CA119347 and CA108633, the Brain Tumor Funders' Collaborative, and an Accelerate Brain Cancer Cure Award (to P.S.M.). This work also was supported by the Harry Allgauer Foundation through The Doris R. Ullman Fund for Brain Tumor Research Technologies, a Henry E. Singleton Brain Tumor Fellowship (to P.S.M.), and a generous donation from the Ziering Family Foundation in memory of Sigi Ziering. A.C.S. was supported by NCI Grant CA25956 and The Barrow Neurological Foundation. M.C. was supported by University of California, Los Angeles (UCLA) Tumor Cell Biology Training Grant 5T32CA09056 funded by NCI. Microarray studies were supported by the National Institutes of Health Neuroscience Microarray consortium, UCLA site (U24HS052108). P.G.F. is a Damon Runyon-Lilly Clinical Investigator supported by the Damon Runyon Cancer Research Foundation.
Abbreviations
- EGFR
epidermal growth factor receptor
- MCM
mitosis/cell cycle module
- MS
metasignatures.
Footnotes
The authors declare no conflict of interest.
References
- 1.Sawyers C. Nature. 2004;432:294–297. doi: 10.1038/nature03095. [DOI] [PubMed] [Google Scholar]
- 2.Mellinghoff IK, Wang MY, Vivanco I, Haas-Kogan DA, Zhu S, Dia EQ, Lu KV, Yoshimoto K, Huang JH, Chute DJ, et al. N Engl J Med. 2005;353:2012–2024. doi: 10.1056/NEJMoa051918. [DOI] [PubMed] [Google Scholar]
- 3.Liu ET, Karuturi KR. N Engl J Med. 2004;350:1595–1597. doi: 10.1056/NEJMp048050. [DOI] [PubMed] [Google Scholar]
- 4.Ebert BL, Golub TR. Blood. 2004;104:923–932. doi: 10.1182/blood-2004-01-0274. [DOI] [PubMed] [Google Scholar]
- 5.Hood L, Heath JR, Phelps ME, Lin B. Science. 2004;306:640–643. doi: 10.1126/science.1104635. [DOI] [PubMed] [Google Scholar]
- 6.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Proc Natl Acad Sci USA. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Segal E, Friedman N, Koller D, Regev A. Nat Genet. 2004;36:1090–1098. doi: 10.1038/ng1434. [DOI] [PubMed] [Google Scholar]
- 8.Mootha VK, Lindgren CM, Eriksson KF, Subramanian A, Sihag S, Lehar J, Puigserver P, Carlsson E, Ridderstrale M, Laurila E, et al. Nat Genet. 2003;34:267–273. doi: 10.1038/ng1180. [DOI] [PubMed] [Google Scholar]
- 9.Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, et al. Nature. 2006;439:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- 10.Lamb J, Ramaswamy S, Ford HL, Contreras B, Martinez RV, Kittrell FS, Zahnow CA, Patterson N, Golub TR, Ewen ME. Cell. 2003;114:323–334. doi: 10.1016/s0092-8674(03)00570-1. [DOI] [PubMed] [Google Scholar]
- 11.Kang Y. Expert Rev Mol Diagn. 2005;5:385–395. doi: 10.1586/14737159.5.3.385. [DOI] [PubMed] [Google Scholar]
- 12.Freije WA, Castro-Vargas FE, Fang Z, Horvath S, Cloughesy T, Liau LM, Mischel PS, Nelson SF. Cancer Res. 2004;64:6503–6510. doi: 10.1158/0008-5472.CAN-04-0452. [DOI] [PubMed] [Google Scholar]
- 13.Zhang B, Horvath S. Stat Appl Genet Mol Biol. 2005;4:17. doi: 10.2202/1544-6115.1128. [DOI] [PubMed] [Google Scholar]
- 14.van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 15.Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. Proc Natl Acad Sci USA. 2004;101:9309–9314. doi: 10.1073/pnas.0401994101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alter O, Brown PO, Botstein D. Proc Natl Acad Sci USA. 2000;97:10101–10106. doi: 10.1073/pnas.97.18.10101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Quinones-Hinojosa A, Sanai N, Smith JS, McDermott MW. J Neurooncol. 2005;74:19–30. doi: 10.1007/s11060-004-5758-0. [DOI] [PubMed] [Google Scholar]
- 18.Han JD, Bertin N, Hao T, Goldberg DS, Berriz GF, Zhang LV, Dupuy D, Walhout AJ, Cusick ME, Roth FP, Vidal M. Nature. 2004;430:88–93. doi: 10.1038/nature02555. [DOI] [PubMed] [Google Scholar]
- 19.Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 20.Carlson MR, Zhang B, Fang Z, Mischel PS, Horvath S, Nelson SF. BMC Genomics. 2006;7:40. doi: 10.1186/1471-2164-7-40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barabasi AL, Albert R. Science. 1999;286:509–512. doi: 10.1126/science.286.5439.509. [DOI] [PubMed] [Google Scholar]
- 22.Barabasi AL, Oltvai ZN. Nat Rev Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 23.Hamid T, Malik MT, Kakar SS. Mol Cancer. 2005;4:3. doi: 10.1186/1476-4598-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Larsen AK, Gobert C. Pathol Oncol Res. 1999;5:171–178. doi: 10.1053/paor.1999.0209. [DOI] [PubMed] [Google Scholar]
- 25.Sellers WR, Loda M. Cancer Cell. 2002;2:349–350. doi: 10.1016/s1535-6108(02)00187-3. [DOI] [PubMed] [Google Scholar]
- 26.Andrews PD. Oncogene. 2005;24:5005–5015. doi: 10.1038/sj.onc.1208752. [DOI] [PubMed] [Google Scholar]
- 27.Altieri DC. Oncogene. 2003;22:8581–8589. doi: 10.1038/sj.onc.1207113. [DOI] [PubMed] [Google Scholar]
- 28.Bond J, Roberts E, Mochida GH, Hampshire DJ, Scott S, Askham JM, Springell K, Mahadevan M, Crow YJ, Markham AF, et al. Nat Genet. 2002;32:316–320. doi: 10.1038/ng995. [DOI] [PubMed] [Google Scholar]
- 29.Bond J, Scott S, Hampshire DJ, Springell K, Corry P, Abramowicz MJ, Mochida GH, Hennekam RC, Maher ER, Fryns JP, et al. Am J Hum Genet. 2003;73:1170–1177. doi: 10.1086/379085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woods CG, Bond J, Enard W. Am J Hum Genet. 2005;76:717–728. doi: 10.1086/429930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mekel-Bobrov N, Gilbert SL, Evans PD, Vallender EJ, Anderson JR, Hudson RR, Tishkoff SA, Lahn BT. Science. 2005;309:1720–1722. doi: 10.1126/science.1116815. [DOI] [PubMed] [Google Scholar]
- 32.Ponting C, Jackson AP. Curr Opin Genet Dev. 2005;15:241–248. doi: 10.1016/j.gde.2005.04.009. [DOI] [PubMed] [Google Scholar]
- 33.Kouprina N, Pavlicek A, Mochida GH, Solomon G, Gersch W, Yoon YH, Collura R, Ruvolo M, Barrett JC, Woods CG, et al. PLoS Biol. 2004;2:E126. doi: 10.1371/journal.pbio.0020126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kouprina N, Pavlicek A, Collins NK, Nakano M, Noskov VN, Ohzeki J, Mochida GH, Risinger JI, Goldsmith P, Gunsior M, et al. Hum Mol Genet. 2005;14:2155–2165. doi: 10.1093/hmg/ddi220. [DOI] [PubMed] [Google Scholar]
- 35.Choe G, Horvath S, Cloughesy TF, Crosby K, Seligson D, Palotie A, Inge L, Smith BL, Sawyers CL, Mischel PS. Cancer Res. 2003;63:2742–2746. [PubMed] [Google Scholar]
- 36.Ermoian RP, Furniss CS, Lamborn KR, Basila D, Berger MS, Gottschalk AR, Nicholas MK, Stokoe D, Haas-Kogan DA. Clin Cancer Res. 2002;8:1100–1106. [PubMed] [Google Scholar]
- 37.Smith JS, Tachibana I, Passe SM, Huntley BK, Borell TJ, Iturria N, O'Fallon JR, Schaefer PL, Scheithauer BW, James CD, et al. J Natl Cancer Inst. 2001;93:1246–1256. doi: 10.1093/jnci/93.16.1246. [DOI] [PubMed] [Google Scholar]
- 38.Hemmati HD, Nakano I, Lazareff JA, Masterman-Smith M, Geschwind DH, Bronner-Fraser M, Kornblum HI. Proc Natl Acad Sci USA. 2003;100:15178–15183. doi: 10.1073/pnas.2036535100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Cancer Res. 2003;63:5821–5828. [PubMed] [Google Scholar]
- 40.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 41.Galli R, Binda E, Orfanelli U, Cipelletti B, Gritti A, De Vitis S, Fiocco R, Foroni C, Dimeco F, Vescovi A. Cancer Res. 2004;64:7011–7021. doi: 10.1158/0008-5472.CAN-04-1364. [DOI] [PubMed] [Google Scholar]
- 42.Bae I, Rih JK, Kim HJ, Kang HJ, Haddad B, Kirilyuk A, Fan S, Avantaggiati ML, Rosen EM. Cell Cycle. 2005;4:1641–1666. doi: 10.4161/cc.4.11.2152. [DOI] [PubMed] [Google Scholar]
- 43.Rosty C, Sheffer M, Tsafrir D, Stransky N, Tsafrir I, Peter M, de Cremoux P, de La Rochefordiereqq A, Salmon R, Dorval T, et al. Oncogene. 2005;24:7094–7104. doi: 10.1038/sj.onc.1208854. [DOI] [PubMed] [Google Scholar]
- 44.Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, Nordgren H, Farmer P, Praz V, Haibe-Kains B, et al. J Natl Cancer Inst. 2006;98:262–272. doi: 10.1093/jnci/djj052. [DOI] [PubMed] [Google Scholar]
- 45.Fish JL, Kosodo Y, Enard W, Paabo S, Huttner WB. Proc Natl Acad Sci USA. 2006;103:10438–10443. doi: 10.1073/pnas.0604066103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calvano SE, Xiao W, Richards DR, Felciano RM, Baker HV, Cho RJ, Chen RO, Brownstein BH, Cobb JP, Tschoeke SK, et al. Nature. 2005;437:1032–1037. doi: 10.1038/nature03985. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
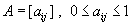{kind=link}
{kind=link}