

Abstract
Alzheimer's disease is the most common cause of dementia and is widely believed to be due to the accumulation of β-amyloid peptides (Aβ) and their interaction with the cell membrane. Aβs are hydrophobic peptides derived from the amyloid precursor proteins by proteolytic cleavage. After cleavage, these peptides are involved in a self-assembly-triggered conformational change. They are transformed into structures that bind to the cell membrane, causing cellular degeneration. However, it is not clear how these peptide assemblages disrupt the structural and functional integrity of the membrane. Membrane fluidity is one of the important parameters involved in pathophysiology of disease-affected cells. Probing the Aβ aggregate-lipid interactions will help us understand these processes with structural detail. Here we show that a fluid lipid monolayer develop immobile domains upon interaction with Aβ aggregates. Atomic force microscopy and transmission electron microscopy data indicate that peptide fibrils are fragmented into smaller nano-assemblages when interacting with the membrane lipids. Our findings could initiate reappraisal of the interactions between lipid assemblages and Aβ aggregates involved in Alzheimer's disease.
INTRODUCTION
The excessive generation and accumulation of the Aβ peptide, a peptide 40–42 amino-acids long, in the vulnerable brain regions of patients with Alzheimer's disease (AD), is believed to be the major cause for the disease. Human Aβ peptide is expressed intracellularly, however, if the peptide concentration is too high, the peptides aggregate, and are further secreted out of the cell. These peptides are produced by a proteolytic processing of large APPs by an aspartyl protease (BACE-1) and a multiprotein complex mainly consisting of γ-secretase (1). Aβ is expressed in both soluble and assembled forms, and is observed in many cellular compartments, such as the endoplasmic reticulum, the Golgi, the Golgi intermediate compartment, and the trans-Golgi network (2). The BACE-1 cleavage is believed to occur in the trans-Golgi and endocytic compartments, and the γ-secretase cleavage is mediated by the activity of presenilins normally localized to the endoplasmic reticulum and pre-Golgi compartments.
Though the assemblage kinetics of the Aβ aggregates is well known, exactly how this diverse molecular species, residing in multiple subcellular sites, elicits cellular toxicity is not well understood. There are a number of different points of view as to the toxicity of Aβ. Early work has shown that Aβ accumulation in APP mutant neurons inhibits the activities of the proteasome (3). Amyloid peptide toxicity may be due to its interaction with metals such as copper, and proteins like acetylcholinesterase (4). Neuronal signaling is affected by amyloid peptide interaction with transducers of the Wnt/β-catenin signaling pathway, including β-catenin and glycogen synthase kinase 3β resulting in toxicity (4). There are several membrane binding proteins known to be associated with amyloid-triggered toxicity (5). Others believe it to be either pore formation or the formation of nanoaggregates that is responsible for disrupting membrane functions (6–9). Although this is the interest of this study, as we see significant lipid association with amyloid peptides and associated changes in the lipid and amyloid organization, it should be noted that the amyloid toxicity is not exclusively correlated to the lipid binding alone. As membrane lipids are intimately associated with these peptides in both soluble and assembled form, it is crucial to know how the different assemblage states of the peptides affect membrane dynamics and functionality. This is particularly important as there is compelling evidence indicating that the toxicity of these aggregates lies in the soluble oligomers and not with the matured fibrils (polymers) (10), since for the same number of monomers we have many more soluble oligomers than fibrils. It is not known whether the toxicity is due to the size of the soluble oligomers or to the number of them present in the cells, but the interaction of the membrane lipids with the peptides, as well as the change in aggregate size, is important. These structurally transformed aggregates are immobile and known to interact with cells in their immediate proximity (11,12). While these processes are well established, there are some pertinent questions that remain unanswered.
What is the relationship between the fibrils and soluble oligomers with respect to membrane malfunction? How stable are the fibrils when in contact with the cell membrane? Is it possible to decompose the toxic nano-aggregates into smaller, nontoxic aggregates in the membrane micro environments or vice versa? There is a growing consensus suggesting that amyloidogenic processing occurs in domains in the membrane enriched with secretases BACE-1 and γ-secretase. Recent efforts in the quest for a vaccine for AD suggest that the matured Aβ fibrils produced by BACE-1 and γ-secretase are dynamic entities that can be broken down by antibodies in transgenic animals and AD patients' brains (13). However it should not be ruled out that this could be due to a mass action of the antibodies, or via stimulation of cellular clearance mechanisms (14). Grimm et al. (15) recently proposed that Aβ1–42 promotes breakdown of the lipid sphingomyelin as part of its physiological function, whereas Aβ1–40 reduces the cholesterol production. Hence understanding the Aβ fibril assemblage modulation could be very important to discern these vital processes. Neuronal cells are highly sensitive to microrheological changes because of their highly polarized morphology and large number of specialized microdomains.
In this study we have investigated the interaction of fibrils and soluble oligomers with cell membrane model systems. Previous studies with model monolayers have demonstrated that mixtures of lipids mimicking the composition of the outer leaflet of plasma membranes exhibit a similar kind of domain structure and stability to that of live cells (16). However, even though these studies were performed using a lipid mixture, they still do not represent the native condition of cells. This is because of the lack of actin, cytoskeleton, membrane proteins, and the inner lipid leaflet. Actin alone has been shown to act as a barrier that hinders the diffusion of membrane lipids (17–19). Even in regard to this, the lipid monolayer is a good model system for studying the interactions between lipids and peptides. This system was used in the single-particle tracking (SPT) experiments and the atomic force microscopy (AFM) experiments as well as in part of the transmission electron microscopy (TEM) study. Another model system for the cell membrane is represented by giant vesicles. These vesicles have a diameter ∼1–10 μm and the interior of these vesicles consists of freely moving smaller nested vesicles. This is a minimal system with great relevance to membrane protein interactions where both intervesicular transport as well as lipid-peptide interactions can be visualized. The giant-unilamellar vesicle (GUV) system was used in the fluorescence resonance energy transfer (FRET) experiments as well as in part of the transmission electron microscopy (TEM) study. This system is far from the cellular bilayer where the lipids are distributed asymmetrically over the inner and outer leaflets. Both exofacial and cytofacial leaflets of the cell membrane bilayer are different in lipid composition and microfluidity. Furthermore, our experimental condition does not provide the micro environment of the cytoskeleton interior that is present in vivo (20). These caveats are kept in mind when interpreting the current data. Despite all these shortcomings, this system is easy to replicate in vivo with different membrane components. Specifically it is easy to add amyloid peptide in different aggregates and study their interaction. Using these model systems we observe that the amyloid aggregates have an affinity for the lipid bilayer in giant vesicles. In lipid monolayers we observe drastic interfacial rheological changes associated with peptide aggregate binding. We also observe fragmentation of matured fibrils, as well as fragmentation of the protofibrils, on association with lipids in our model systems. In this work, we show that Aβ peptide, whatever the form of aggregate, might be involved in the generation of immobile micro domains which will have drastic cellular implications particularly in neuronal cells.
MATERIALS AND METHODS
Lipids
1-Palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG); 1,2-dioleoyl-sn-glycero-3-phosphate (DOPA); 1,2-dioleoyl-sn-glycero-3-{[N (-5-amino-1-carboxypentyl)imino-di-acetic acid] succinyl} (Nickel Salt); and egg yolk phosphatidylcholine were obtained from Avanti Polar Lipids (Birmingham, AL). 1,2-Dihexadecanoyl-sn-glycero-3-phosphoethanolamine, triethylammonium salt, and Texas Red (TR) was obtained from Molecular Probes (Eugene, OR). All lipids were used without further purification.
Peptides
Aβ1–42 and Aβ1–40 were prepared by Fmoc-solid phase peptide synthesized in W.M. Keck Facility at Yale University (New Haven, CT). The peptides were purified using RP-HPLC, analyzed by matrix-assisted laser desorption ionization time of flight mass spectrometry and lyophilized at the PAN facility Stanford University. FITC-Aβ1–42 and Aβ42–1 were obtained from AnaSpec (San Jose, CA).
Aβ peptide preparation
The Aβ1–40 and Aβ1–42 were suspended in 90:10% hexafluoroisopropanol/trifluoroacetic acid for pretreatment. After complete dissolution, the solvent was removed using dry Argon gas. The remaining film was placed in a vacuum chamber. The film was redissolved in a 1 mM NaOH solution, and the pH was adjusted to 7.4 using a 20 mM NaOH solution to obtain Aβ peptide in monomeric form. This solution was aliquoted and lyophilized again. The lyophilized powder was dissolved in phosphate-buffered saline (PBS) at pH = 7.4 and 155 mM NaCl to a working concentration of 200 μM. To prepare the soluble oligomer fractions of the peptides, the Aβ1–42 sample was placed at 4°C for 24 h and the Aβ1–40 was incubated for 72 h at room temperature. Mature fibrils were prepared by incubating the lyophilized peptide dissolved in PBS along with 5% preformed seed at 37°C for 15–30 days. The monomers were thereafter separated from the fibrils by centrifuging the sample at 5000 rpm for 10 min, thereby pelleting the matured fibril, leaving the monomer in the supernatant which is removed. When used, the working concentration of the peptides was 200 nM.
Miniature Langmuir trough
In studying the Aβ-lipid monolayer interactions, a need arose for a miniature Langmuir trough. The reasons include reactant amounts and a need to improve single particle tracking (SPT) measurements. There have been a few attempts at making miniature Langmuir troughs, the goal usually being aimed at improving diffusion measurements at the air-water interface and reducing contamination. Lipid monolayer tracking experiments at the air-water interface have inherent problems not found in supported bilayer experiments, due to the influence of air currents and convective flows in the subphase. There are a few different approaches to address these problems; one uses a circular ring residing in the subphase right below the interface in a Langmuir trough, which acts as a barrier to subphase flow (21). Another group (22) encapsulated their setup under glass and reduced the trough size to suppress air flow and reduce contamination. The resulting trough was small and light, allowing it to fit on a microscope stage. Our approach is along the same lines, reducing the size; the “trough” that we constructed consists of a sessile drop sitting on a polydimethylsiloxane stage with an inlet and an outlet (Fig. 1). Surrounding the sessile drop are glass sides to reduce air flow, evaporation, and contamination; however, the setup is not entirely enclosed. The setup contains three droplet stages, to facilitate multiple experiments at the same time. The area per molecule is adjusted by either injecting more subphase liquid into the interior of the drop or by removing some of the subphase. Reduced subphase volume is of importance when, in our case using hybridized oligonucleotides as linkers between monolayer lipids and fluorescently tagged subphase vesicles as probes, this system is described in Single Particle Tracking (SPT). A reduced subphase volume allows for shorter hybridization time and smaller amounts of analyte. This setup has a subphase volume of ∼40 μL, which is approximately two orders-of-magnitude less than the subphase of a conventional Langmuir trough. With this setup we see a great reduction in convective flow and also a much shorter experiment turnover time. Formation of a drop is accomplished by injecting subphase liquid through the inlet channel; the size of the drop can be further adjusted by either injecting more liquid through the inlet or by aspirating the subphase through the outlet channel. A monolayer can then be spread on top of the drop; all lipid monolayers are spread using a chloroform solution to ensure quick solvent evaporation and immiscibility, depositing the lipids at the interface only. The Aβ peptides in PBS solution are introduced through the inlet channel, and simultaneously, the same amount of subphase is withdrawn using the outlet, so that the volume of the drop is maintained constant. To remove excess peptides or other material that has been introduced into the system, clean subphase liquid can be injected at the same time as the subphase with excess material is aspirated. The drawback with this smaller trough is that there are some problems when air bubbles form and rise to the interface; the experiment then needs to be aborted, and we do not yet have a way of measuring the surface pressure. The miniature Langmuir trough was used for the SPT, AFM, and part of the TEM experiments.
FIGURE 1.

Novel micro Langmuir trough. With a subphase volume of ∼40 μL, this “trough” is much smaller than a conventional Langmuir trough. The area per molecule can be varied by adjusting the volume of the drop. The monolayer is spread on top of the drop. Peptides and vesicles are injected into the interior through the inlet. As peptides and vesicles are injected, subphase fluid is removed at the same rate through the outlet channel, such that constant volume is maintained. The radius of the drop is ∼3 mm. We do not yet have a functional way for this system to control the surface pressure if we want to have a ability to inject substances into the subphase.
Atomic force microscopy (AFM)
AFM analysis was performed using a Digital Instruments Multimode NanoScope IIIa SPM (Veeco Metrology Group, Santa Barbara, CA). Silicon AFM tips (model PPP-NCH) for tapping mode were purchased from Molecular Imaging (Tempe, AZ) and was used for imaging the sample at a rate of 0.5–2 Hz depending on the scan size. AFM is widely used to investigate the aggregate structure of amyloid-forming peptides (23–26). The samples to be investigated were prepared using the micro Langmuir trough. Experiments were conducted using pairs of drops; both drops' subphase consisted of 40 μL PBS, one drop having a lipid monolayer, the other one not. The monolayer, which consisted of POPG and DOPA lipids, was spread on the surface of the drop. Two microliters of Aβ1–40 is then injected into the interior of the drop. Once introduced, the peptide quickly absorbed to the drop-air, drop-lipid interface. The drop-air interface is the control system, allowing us to know what the peptide looks like before interacting with the monolayer lipids. The monolayers were thereafter transferred to a silicon wafer using a Langmuir-Schaffer horizontal transfer technique where the wafer was brought into contact with the drop interface. Once the monolayer had been transferred, the wafer was carefully washed in Millipore water to remove salts from the buffer solution that would otherwise interfere with the imaging. The films were then investigated by AFM to gain information on their structural features at the submicrometer level.
Single particle tracking (SPT)
To deduce the interfacial rheological properties of the monolayers and the effect of the Aβ peptides, SPT was implemented (27,28). Similarly to AFM experiments already described, experiments were conducted using pairs of drops, but this time, the reference system was that of a lipid monolayer alone (POPG/DOPA) spread at the air-water interface, and the other system was that of a lipid monolayer, spread first, after which peptide aggregates were introduced into the interior of the drop. Tracking of the lipids was facilitated using a technique developed by Yoshina-Ishii and Boxer (29). This technique allows for “smart tethering” of fluorescently labeled vesicles to lipids in a monolayer or bilayer. The smart linker is a modified lipid, where a oligomer 16-bases-long is attached covalently to the lipid headgroup. Using this type of linker you can select for the vesicle that has the complementary lipid attached to it. In this study we labeled egg-yolk phosphatidylcholine vesicles with 1.5% Texas Red (TR) lipids. Enough DNA-lipids were added to allow for a maximum of one DNA-lipid per vesicle. This way we reduce the likelihood of getting more than one tether per vesicle. However, if we have perfect insertion of the DNA-lipid into the vesicle, we probably have a number of vesicles that do not have the DNA-lipid in it, so they will not attach to the monolayer and can be flushed away. A monolayer with 1:10,000 DNA-lipids is spread on top of the drop on the “trough”, and once the chloroform from the spreading solution evaporated, TR-labeled vesicles with the complimentary lipids are injected into the subphase using the injection channel in the stage. Once the complimentary oligonucleotides in the TR-labeled vesicles have hybridized with the DNA-lipids in the monolayer, these vesicles are constrained to the two-dimensional air-water interface. After hybridization, the subphase is flushed with clean buffer to remove any excess fluorescent vesicles not attached to the monolayer. If Aβ aggregates are used in the system, these are injected into the subphase after the monolayer is spread, but before the injection of the fluorescent vesicles. After incubation for 10 min, the subphase is flushed to remove any peptide not yet interacting with the monolayer to avoid interactions between the peptide and the vesicles. A Nikon Microphot-SA fluorescence microscope (Nikon, Marunouchi, Tokyo) equipped with a Princeton Instruments PentaMAX intensified CCD camera (Princeton Instruments, Trenton, NJ) was used to image the SPT experiments. Images were captured at a rate of 11 frames per second and analyzed using Metamorph v.6.3 from Molecular Devices (Downingtown, PA). Further analysis was conducted using Microsoft Excel (Microsoft, Redmond, WA).
Confocal microscopy, FRET, and giant vesicle preparation
Confocal microscopy
The membrane-Aβ peptide interaction in giant vesicles was studied using confocal microscopy utilizing the fluorescence resonance energy transfer (FRET) technique. To facilitate these studies, a five-well sample viewer was prepared for viewing the samples. Using a diamond-tipped drill, 10 holes were drilled in a microscopy slide. Six strips of double-sided tape were used to form five channels, each channel with a hole at both ends. A coverslip functionalized with a His-tag protein was then attached, using double-sided tape and epoxy glue to form five enclosed channels, each with an inlet and an outlet. The His-tag protein allowed us to fix the vesicles to the coverslip. The sample is viewed through the No. 1.5 coverslip, covering the channels. An inverted confocal microscope, Leica SP2 AOBS, was used to capture the images (Leica, Wetzlar, Germany). The excitation wavelengths were 488 nm (FITC-Aβ) and 543 nm (TR).
Giant vesicle preparation
Phospholipid stock solutions were dissolved in chloroform to yield a concentration of 0.2 mg/ml. Giant unilamellar vesicles (GUVs) with POPG, DOPA, Ni lipids, and TR-lipids (1:1:0.02:0.01 molar ratio) were prepared using the rotary evaporation method (30). Aliquots of giant vesicles suspended in PBS at pH = 7.4 were used in the FRET experiments.
Fluorescence resonance energy transfer (FRET)
Fluorescence resonance energy transfer (FRET) is a robust and sensitive technique widely used to study the distance between two different fluorophores and is only possible when two fluorophores are in close proximity (31). This technique has been used for similar systems where the interactions between peptides and membrane lipids have been studied (32,33). To accomplish FRET, a donor fluorophore is excited by an incoming photon. If an acceptor fluorophore is in close proximity, the acquired energy can be transferred nonradiatively between the fluorophores, and a photon of lower energy is emitted from the acceptor fluorophore. The efficiency of FRET is dependent on the inverse sixth power of the distance between the molecules, making this a very useful method for visualizing and studying the incorporation of peptides into a bilayer. Other techniques for studying the incorporation of the peptide into bilayers could be accomplished by incubating the peptide with vesicles followed by centrifugation and spectroscopic analysis. The major disadvantage with that technique is that it is harder to separate larger aggregates from the vesicles, and also requires two steps—whereas the FRET technique used here is a one-step technique in which incorporation is easily seen visually. The increase in FRET intensity of the acceptor or reduction of the donor fluorescence intensity gives a direct measure of proximity information between the donor-acceptor carrying moieties.
Transmission electron microscopy (TEM)
Transmission electron microscopy (TEM) was facilitated using a JEOL 1230 electron microscope (JEOL, Peabody, MA) at the Cell Sciences Imaging Facility at Stanford University. All the TEM work was conducted with the subphase consisting of PBS. A drop of PBS is formed on the same sample stage described earlier; the Aβ peptide aggregate is injected into the interior of the drop. If a lipid monolayer is desired for the experiment, this is spread before injection of the peptide. The system is allowed to equilibrate for 15 min; at that time a TEM grid is brought to the air-water interface and the aggregates are transferred horizontally. After the peptides have been fixed to the grid, the uranyl acetate (UA) stain is applied with a drop of 2% UA solution on the back side of the grid. The drop is allowed to engulf the grid and after 5 min, the grid is washed three times by immersion in a rinse made of the same solution, but without the UA. Since the peptides and lipids are already fixed to the surface when the stain is applied, there should be minimal effect of the stain on the peptide structures.
RESULTS AND DISCUSSION
FRET
To study the membrane-Aβ peptide interaction, TR-lipids were incorporated in the bilayer of giant vesicles and the Aβ peptide was labeled with FITC. Ten microliters of GUVs in PBS buffer were injected into the sample holder described earlier. The vesicles were allowed to attach and were imaged in the absence of peptides (data not shown). FITC-Aβ1–42 soluble oligomer/fibrillar mixture was introduced into the sample chamber and imaged. Fig. 2, A–D, is a confocal fluorescence image of a giant vesicle using different excitation and emission wavelengths. Fig. 2 A depicts the vesicle exciting and imaging the TR-lipids. Fig. 2 B depicts the FRET after peptide has been added to the system. Significant FRET is observed, which implies that FITC-Aβ is localized in close proximity with the membrane TR-lipids. Imaging the FITC channel after peptide addition (Fig. 2 C), a weak fluorescence is observed everywhere except for where the vesicle is located, where very little or no fluorescence at all is observed—indicating energy transfer between the FITC-labeled bound peptide and the TR lipids in the membrane. The reduction in fluorescence seen in Fig. 2 C can, in addition to the FRET, be attributed to the metal quenching by the Ni-lipids in the vesicles (34). However, FRET can be seen in Fig. 2 B, which would not be seen if all the quenching seen in Fig. 2 C would be attributed to the Ni-lipids. This quenching should be seen in all imaged channels, not just in the FITC channel; therefore, we attribute the quenching seen in Fig. 2 C partly to FRET.
FIGURE 2.

Giant vesicles in confocal microscope. Scale bar represents 1 μm. (A) Exciting Texas Red directly; imaging Texas Red emission. (B) The same vesicle under FRET, exciting FITC and imaging Texas Red, we see a weaker signal than direct excitation, as expected. (C): Exciting the FITC-labeled peptide reveals energy transfer to the TR-labeled membrane lipid as there is no increase in FITC signal along the GUV perimeter. (D) FRET of nested vesicle moving inside giant vesicle, suggesting that peptides can pass through the vesicle membrane.
The Förster radius for the FITC/TR donor-acceptor pair is R0–50 Å (35). Hence, the observation of FRET reveals that the peptide aggregate must reside within 100 Å of the membrane TR lipid. The incorporation of the peptide in the bilayer was near-complete during the time of our sample preparation (5 min). This indicates that the binding of Aβ aggregates to the membrane is essentially instantaneous, since we see no increase in FRET with time. Green fluorescent protein was used as a control to check the fluorescence signal bleed in the instrument. The FRET intensity for the same amount of the protein added was minimal (data not shown), indicating the observed FRET is consistent with the membrane Aβ peptide interaction. The Aβ aggregates can move through the giant vesicle membrane to its interior; this observation was made through the visible FRET in small vesicles nested inside the giant vesicles. Brownian motion of the nested vesicles was observed, suggesting that they are not invaginations of the bigger vesicles, but freely moving entities (Fig. 2 D). These vesicles are seen inside the GUVs before the peptide is added, so the vesicles are not invaginated because of the peptide. The peptide is transferred to the internal vesicles since FRET is seen in these nested vesicles, indicating that Aβ can rapidly cross the giant vesicle bilayer and incorporates with the bilayer of nested vesicles. The mechanism for this transfer is not known, but we speculate that the peptide that is incorporated into the outer membrane is transferred as the nested vesicles collide with the encapsulating membrane. If pore formation is the mechanism that rules this system, then pores could be formed and the peptides transfer through the pores; however, we have seen no evidence for pore formation in our system. No experiments were conducted on unlabeled Aβ, so it is not known if this membrane penetration is due to the FITC label. We see the same results for both Aβ1–40 and Aβ1–42. It should be mentioned that passage of the soluble oligomers through the membrane is noteworthy, as these species are implicated in the neuronal toxicity (36). The soluble oligomers of Aβ (37,38), IAAP (39), and α-synuclein (40) have previously been shown to be localized to the cell membrane, inducing membrane leakage and, as a result, an ion imbalance. To the best of our knowledge, this is the first time that it has been shown that a mixture of matured fibrils and protofibrils has generated more soluble oligomers (an activity mediated by giant vesicles). This observation has biological implications because it has been shown that introducing the peptides to the cell either by microinjections of Aβ1–42 or by a cDNA-expressing cytosolic-Aβ1–42 rapidly induces cell death of primary human neurons, both in fibrillar and nonfibrillar forms. Concentrations as low as 1 pM or 1500 molecules/cell are known to be neurotoxic (41). The present results show that assembled Aβ preparations have an ability to form toxic nanoaggregates when in contact with membrane lipids. A closer look at the GUV suggests that the fluorescence is granular, not uniform. This indicates that the peptides binding to the membrane result in a surface excess, which appears as granular aggregates. This may be due to the peptides binding to the membrane and changing the lipid domains from ordered liquid crystalline lipid domains to partially ordered domains. Consequently this leads to a curvature imbalance of the vesicular membrane (42). Further, the defects will also lead to diffusion of the soluble oligomers into the membrane, a mechanism that has previously been observed in the case of α-synuclein membrane interactions (43).
Aggregate structures as observed by AFM
To further explore the nature of Aβ1–40 aggregates in the membrane environment, Aβ1–40 assemblages were incorporated into lipid monolayers and studied using AFM. The lipids used in this system have a negative charge, making them more representative of the inner leaflet of the cell, where the peptides are formed. Fig. 3 A depicts an AFM image of a DOPA/POPG (1:1) monolayer without the peptide. The lipid monolayer alone forms a smooth film, with wavelike features that are attributed to the separation of the lipids into smaller domains when the system is dehydrating. The lighter regions in the height mode AFM images represent higher topography. In systems with lipids, the darker regions are assigned to the monolayer absorbed to the silicon wafer, and in the systems without lipids, the darker regions are assigned to the silicon wafer itself (23,24). Injecting the soluble oligomers alone and letting them absorb to the air-water interface results in polydisperse flat discoid aggregates (Fig. 3 B), these objects not only have a height difference (3–25 nm taller than the silicon wafer), but also a difference in viscoelastic response, which is apparent in the phase mode image (data not shown). The soluble oligomers adsorbed at the air-water interface form domains that are between 3- and 25-nm high, which is higher than the fibrils or the soluble oligomers of Aβ1–42 on graphite or mica substrates (25). This suggests that the air-water interface induces lateral aggregation of the Aβ1–40 peptides on the water surface. These domains are flat, with a maximum height of 21 Å. Solid-state NMR studies suggest a parallel β-helical arrangement, where two adjacent strands of Aβ1–42 pack with a Cα-Cα distance of 4.8 ± 0.5 Å (44). The smallest observed peptide peaks are high enough to accommodate four units of Aβ1–40 peptide monomers, indicating that soluble oligomers consist of Aβ1–40, arranged one above the other. A lateral assembly of several units will result in a discoid of 25 nm in diameter as observed in the AFM (25). Previous studies indicate that monolayers can condense upon transfer to a solid support and form solid phase islands consisting of lateral aggregates (45). A striking difference in aggregate distribution is observed when the peptides are injected into a drop with a lipid monolayer at the air-water interface (Fig. 3 C). When the introduced peptide aggregates incorporate into the lipid monolayer, the average diameter of the disklike soluble oligomers is reduced from 25 nm to 16 nm. The lipids further reduces the average height of the aggregates from 13.3 Å to 9.5 Å. Fig. 3 D displays the change in aggregate size in the two different systems. We see a reduction in aggregate size and also a change in the size distribution. The peptide at the air-water interface is fairly polydisperse, whereas, when the same system has interacted with lipids, the size distribution is much narrower. This indicates that the lipids induce fragmentation of the peptide assemblages. An aggregate height of ∼13.3 Å would represent a trimeric or dimeric peptide aggregate, whereas a height of 9.5 Å would represent a dimer. We find a reduction in aggregate number from three to two soluble oligomer units. Our AFM data suggest that the soluble oligomers at the air-water interface, or soluble oligomers incorporated in a lipid monolayer, accommodate smaller peptide units compared to the findings of previous studies (25,46). This could be due to our method of preparation of the sample.
FIGURE 3.

AFM height mode images. (A) Lipid monolayer 0.5 μm × 0.5 μm 10-nm height scale reveals very small height undulations, indicating a fairly uniform monolayer. (B) Aβ1–40 without lipid monolayer reveals many high peaks. (C) Introducing the peptides into a lipid monolayer reduces the peak height and also peak width, in comparison to the system in panel B. (D) Aggregate size and size distribution, peptide with lipids, and peptide alone, the system with lipids induce a breakup of larger aggregates.
TEM analysis visualizing the Aβ aggregate-lipid interaction
The results so far presented suggest that lipophilic Aβ aggregates interact with the cellular lipid membrane leading to structural changes of the aggregates. We used TEM to image matured fibrils and soluble oligomers of the Aβ1–40 and Aβ1–42 peptides incorporated in lipid monolayers and bilayers of GUVs. We used both systems and will hereafter in this section refer to the peptides as Aβ. We investigated whether these aggregates sever the cell membrane integrity upon incorporation. Previous studies of incorporation of Aβ peptides in lipid membranes, utilizing AFM to investigate vesicles, suggested such possibilities (39,47). Fig. 4 A is a TEM image of the soluble oligomeric fraction of the Aβ peptide at the air-water interface. The image shows small aggregates with a diameter of ∼6–8 nm. However, when the same fraction is introduced into a drop with a lipid monolayer at the air-water interface, the soluble oligomers of Aβ are almost nonexistent in the TEM images. We presume that the soluble oligomers are divided into smaller fragments as seen with the AFM, and that these aggregates are of such a small dimension that their contrast is too low to be visible (Fig. 4 B). Matured Aβ fibrils absorbed to the air-water interface and incubated for 1 h appear as a fibrous mesh (Fig. 4 C), with the individual fibers having a diameter of ∼6–8 nm and variable length. When these fibrils are incubated in a drop covered by a lipid monolayer for 1 h, they fragment. The fragments appear as a pattern of globular structures of soluble oligomers of ∼3–4 nm in diameter. Fig. 4 D shows a fragmentation pattern with generation of globular, short fibrils of 25–75 nm in length. This indicates that matured Aβ fibrils can generate soluble oligomers in the presence of lipids. In connection with this observation, it is worth mentioning that there is evidence suggesting that, in specific micro environments such as inflammatory conditions, the fibrils also elicit toxicity like that of soluble oligomers (48). In the light of the present discoveries, this could be due to the generation of smaller aggregates from the matured fibrils upon interaction with lipids. We propose that when fibrils are in contact with the lipid domains, it is not necessary that the whole fibril is in contact with the membrane lipids. Instead, we suggest that since different parts of long fibrils can be in contact with membrane domains, they can be in contact with different subdomains. A fibril that is firmly anchored into different subdomains, which might be moving independently in different directions, is slowly teased apart (Fig. 5). The disruption of fibrillar structures into oligomeric structures has also been observed in tubulin-lipid complexes (49). Our observation is also consistent with the previous work of Tashima et al. (50), who reported that there was no fibril formation in the liposomal preparation without cholesterol. The Aβ aggregates' effect on giant vesicles is indicative of amyloid dissolution in the vesicle membrane. The incorporation of aggregates into the membrane leads to both ordered and disordered domains in the membrane. The disordered domains appear as dark regions in the TEM images, where more of the UA molecules have been incorporated. The UA incorporates in and around the peptide aggregates, which are more prevalent in the disordered domains (Fig. 6 A). An image at higher magnification of the ordered and disordered regions is shown in Fig. 6 B. The total conversion of mature fibrils to soluble oligomers is visible in Fig. 6 C. Examination of the lipid membrane at a higher magnification (Fig. 6 D) reveals that the peptide aggregates are globular and localized to the disordered domains, adjacent to the ordered areas of the membrane. The Triton-X micelle is also known to produce such disordered domains when interacting with the lipids in the membrane (51). This induced disorder is in turn compensated by the formation of larger-ordered domains as well. The soluble oligomeric forms of several amyloid assemblages are known to dramatically increase the ionic permeability of planar lipid bilayers. Micropipette aspiration studies indicate that the Aβ aggregates induce a strong membrane destabilization and leakage of the entrapped solutes in GUVs (38). Studies of the Aβ peptide interaction with plasma, endosomal, and lysosomal membrane compartments in the brain of an AD patient have indicated a decrease in fluidity of the membrane (52,53). This corroborates with our findings in the monolayer studies, where peptide incorporation reduces total diffusion in the system.
FIGURE 4.

TEM images of soluble oligomers and fibrils Aβ1–40. The scale bar represents 100 nm. (A) Soluble oligomers at the air-water interface. (B) Soluble oligomers in a lipid monolayer, structures are too small to see, but the stain is visible, indicating peptide presence. (C) Fibrils at the air-water interface, large three-dimensional networks are present. (D) With a lipid monolayer present, the mature fibrils are broken down into soluble oligomeric components.
FIGURE 5.

Proposed model for the fractionation of Aβ fibrils. The fibril consists of soluble oligomeric substructures; these come together to form a fibril because of hydrophobic interactions. The hydrophobic peptide segments preferentially incorporate into the hydrophobic tails of the monolayer lipids. Micro domains in the monolayer moving in different directions can tease the fibril apart, decomposing it into smaller fragments.
FIGURE 6.

Giant vesicles with Aβ1–40 peptide incorporation. The scale bar represents 100 nm. The uranyl acetate incorporates in and around the peptide aggregates. (A) Protofibrillar peptide aggregates are more incorporated in the disordered domain, the disordered domain being outside the GUVs. (B) Higher magnification image, revealing ordered and disordered regions, arrows pointing out the peptide in the disordered regions. (C) The total conversion of mature fibrils to soluble oligomers is visible; soluble oligomers can be seen, but no fibrils. The interior of the GUVs have less visible oligomers than the surroundings, but is darker, indicating more of the smaller peptides in those regions. (D) Looking at the lipid membrane at a higher magnification reveals that the peptide aggregates are globular, localized to the disordered domains, whereas where the ordered domains are, no oligomers can be seen.
Peptide influence on monolayer fluidity
To investigate the influence of Aβ1–40 aggregates on the lipid membrane fluidity, Aβ1–40 soluble oligomers were incorporated into lipid monolayers and studied using SPT. The lipid monolayer alone is fluid, with no solid domains (54). The “micro trough” was developed for these experiments and a great reduction in convective flow was seen, compared to conventional Langmuir troughs. However, some flow still exists and will have to be taken into account. The motion of the vesicles can be divided up into a convective part and a Brownian motion part. Subtracting the average displacement from the total displacement of each vesicle leaves random movement (see Eq. 1). This scheme allows for tracking of individual vesicles, and based on their positions in each image and the time-lapse between each image, we can calculate the diffusion coefficient for the lipids. This position is analyzed using the SPT plug-in of the Metamorph software. The output from the program is particle-coordinates in each image frame and the elapsed time from the first image:
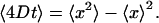 |
(1) |
Using this relationship, where x is the direction vector, the random walk diffusion can be calculated and the diffusivity, D, can be calculated. In the system without peptide, there are no apparent domains, where the vesicle trajectories display a random walk (Fig. 7 A). The diffusion coefficient for the plain POPG/DOPA monolayer is 0.2 μm2/s (Fig. 7 B). When peptides are introduced, solid domains appear, and in these regions the vesicles become motionless (Fig. 7 C). The whole monolayer is not immobile, and fluid domains can be found, but for the most part the monolayer motion is retarded. The reduction in mobility is not only seen in the trajectories but in the diffusion coefficient—which, for the solid domain, is reduced to less than half of that with the plain monolayer (Fig. 7 D).
FIGURE 7.

Lipid diffusion experiments investigating fluidity of membrane. (A) Trajectories of tracked lipids in a monolayer without peptide. (B) Diffusion coefficient histogram for the lipid monolayer, the y axis representing the fractional distribution with the total number total number of tracked vesicles being 123. (C) Trajectories of tracked lipids in a monolayer with incorporated peptide. (D) Diffusion coefficient histogram for lipid monolayer with incorporated peptide, the y axis representing the fractional distribution with the total number of tracked vesicles being 144.
CONCLUSIONS
Aβ peptide-membrane interactions have implications not only for the production of Aβ but also for the onset of cell death. Our FRET studies on cell membrane model systems suggest that Aβ inserts itself into the bilayers. FRET is observed in GUVs, indicating incorporation of the peptide in the membrane. Upon excitation with blue light and imaging the green channel (which is the Aβ fluorophore color), we see that there is peptide around the GUV. However, at the location of the GUV, there is no green fluorescence, which suggests that all the fluorescence in this location is transferred to the TR lipids in the membrane. Since the Aβ peptides are hydrophobic, the peptides are believed to insert into the carbon chain part of the phospholipids. This assumption is supported by the AFM studies that demonstrate that the peptides are incorporated into the lipid monolayer. Using visible FRET in nested vesicles inside the GUVs, the Aβ aggregates were also found to move through the giant vesicle membrane to its interior. Nested vesicles were observed to display rapid motion, suggesting that they are not invaginations of the bigger vesicles, but freely moving entities. The nested vesicles displayed substantial FRET, indicating that FITC-Aβ can rapidly cross the GUV bilayer and incorporate in the bilayer of nested vesicles (Fig. 2 D). This is a very important observation as these peptide aggregates are responsible for cell death. The SPT experiments further confirm that the Aβs are incorporated into the lipid monolayer as revealed though a large reduction in monolayer fluidity. Our results are consistent with previous reports demonstrating that the incorporation of Aβ peptides into a lipid bilayer decreases the membrane fluidity (52). We see a formation of solid domains when the peptide is introduced; the mobility in these domains is very low, but there are still fluid regions, and in these regions the lipid monolayer exhibits the same mobility as before the introduction of the peptides. Our TEM and AFM results demonstrate the degeneration of mature Aβ fibrils into soluble oligomers. The ability of Aβ to insert into the plasma membrane has many implications for both cell survival and cell-driven fibril degeneration. Although we have examined Aβ-lipid interactions in different model systems, we have not taken into account the effect of other cellular proteins residing in the membranes of cells. Integral membrane proteins will also have an effect on Aβ-membrane interactions whether as competitors for Aβ binding or as modulators of bilayer properties. Our results demonstrate the consequences of Aβ-lipid interactions, which may play a role not only in the normal cellular processing and turnover of Aβ but in the progression of disease processes in Alzheimer's disease.
Acknowledgments
This work was supported by the Center on Polymer Interfaces and Macromolecular Assemblies, a National Science Foundation sponsored partnership.
References
- 1.Sisodia, S. S., and P. H. S. George-Hyslop. 2002. Gamma-secretase, notch, Aβ and Alzheimer's disease: where do the presenilins fit in? Nat. Rev. Neurosci. 3:281–290. [DOI] [PubMed] [Google Scholar]
- 2.Greenfield, J. P., J. Tsai, G. K. Gouras, B. Hai, G. Thinakaran, F. Checler, S. S. Sisodia, P. Greengard, and H. X. Xu. 1999. Endoplasmic reticulum and trans-Golgi network generate distinct populations of Alzheimer β-amyloid peptides. Proc. Natl. Acad. Sci. USA. 96:742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Almeida, C., R. Takahashi, and G. Gouras. 2006. Beta-amyloid accumulation impairs multivesicular body sorting by inhibiting the ubiquitin-proteasome system. J. Neurosci. 26:4277–4288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fuentealba, R., G. Farias, J. Scheu, M. Bronfman, M. Marzolo, and N. Inestrosa. 2004. Signal transduction during amyloid-[β]-peptide neurotoxicity: role in Alzheimer disease. Brain Res. Rev. 476:275–289. [DOI] [PubMed] [Google Scholar]
- 5.Verdier, Y., M. Zarándi, and B. Penke. 2004. Amyloid-β-peptide interactions with neuronal and glial cell plasma membrane: binding sites and implications for Alzheimer's disease. J. Pept. Sci. 10:229–248. [DOI] [PubMed] [Google Scholar]
- 6.Selkoe, D. J. 2003. Aging, amyloid, and Alzheimer's disease: a perspective in honor of Carl Cotman. Neurochem. Res. 28:1705–1713. [DOI] [PubMed] [Google Scholar]
- 7.Lashuel, H. 2005. Membrane permeabilization: a common mechanism in protein-misfolding diseases. Sci. Aging Knowledge Env. 38:pe28. [DOI] [PubMed] [Google Scholar]
- 8.Tanzi, R. E. 2005. The synaptic Aβ hypothesis of Alzheimer disease. Nat. Neurosci. 8:977–979. [DOI] [PubMed] [Google Scholar]
- 9.Bitan, G., M. D. Kirkitadze, A. Lomakin, S. S. Vollers, G. B. Benedek, and D. B. Teplow. 2003. Amyloid beta protein (Aβ) assembly: Aβ40 and Aβ42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA. 100:330–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lacor, P. N., M. C. Buniel, L. Chang, S. J. Fernandez, Y. S. Gong, K. L. Viola, M. P. Lambert, P. T. Velasco, E. H. Bigio, C. E. Finch, G. A. Krafft, and W. L. Klein. 2004. Synaptic targeting by Alzheimer's-related amyloid β-oligomers. J. Neurosci. 24:10191–10200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vetrivel, K. S., H. P. Cheng, W. Lin, T. Sakurai, T. Li, N. Nukina, P. C. Wong, H. A. Xu, and G. Thinakaran. 2004. Association of γ-secretase with lipid rafts in post-Golgi and endosome membranes. J. Biol. Chem. 279:44945–44954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ravi, L. B., S. Poosala, D. Ahn, F. J. Chrest, E. L. Spangler, R. Jayakumar, E. Nagababu, J. G. Mohanty, M. Talan, D. K. Ingram, and J. M. Rifkind. 2005. Red cell interactions with amyloid-β (1–40) fibrils in a murine model. Neurobiol. Dis. 19:28–37. [DOI] [PubMed] [Google Scholar]
- 13.Gelinas, D. S., K. DaSilva, D. Fenili, P. S. George-Hyslop, and J. McLaurin. 2004. Immunotherapy for Alzheimer's disease. Proc. Natl. Acad. Sci. USA. 101:14657–14662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lombardo, J., E. Stern, M. McLellan, S. Kajdasz, G. Hickey, B. Bacskai, and B. Hyman. 2003. Amyloid-β antibody treatment leads to rapid normalization of plaque-induced neuritic alterations. J. Neurosci. 23:10879–10883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Grimm, M. O. W., H. S. Grimm, A. J. Patzold, E. G. Zinser, R. Halonen, M. Duering, J. A. Tschape, B. D. Strooper, U. Muller, J. Shen, and T. Hartmann. 2005. Regulation of cholesterol and sphingomyelin metabolism by amyloid-β and presenilin. Nat. Cell Biol. 7:1118–1123. [DOI] [PubMed] [Google Scholar]
- 16.Devaux, P., and R. Morris. 2004. Transmembrane asymmetry and lateral domains in biological membranes. Traffic. 5:241–246. [DOI] [PubMed] [Google Scholar]
- 17.Wawrezinieck, L., H. R. D. Marguet, and P.-F. Lenne. 2005. Fluorescence correlation spectroscopy diffusion laws to probe the submicron cell membrane organization. Biophys. J. 89:4029–4042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fujiwara, T., K. Ritchie, H. Murakoshi, K. Jacobson, and A. Kusumi. 2002. Phospholipids undergo hop diffusion in compartmentalized cell membrane. J. Cell Biol. 157:1071–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lommerse, P. H., H. P. Spaink, and T. Schmidt. 2004. In vivo plasma membrane organization: results of biophysical approaches. Biochim. Biophys. Acta Biomembr. 1664:119–131. [DOI] [PubMed] [Google Scholar]
- 20.Sheetz, M., J. Sable, and H.-G. Döbereiner. 2006. Continuous membrane-cytoskeleton adhesion requires continuous accommodation to lipid and cytoskeleton dynamics. Annu. Rev. Biophys. Biomol. Struct. 35:417–434. [DOI] [PubMed] [Google Scholar]
- 21.Forstner, M. B., D. S. Martin, A. M. Navar, and J. A. Kas. 2003. Simultaneous single-particle tracking and visualization of domain structure on lipid monolayers. Langmuir. 19:4876–4879. [Google Scholar]
- 22.Hu, Y. F., K. Y. C. Lee, and J. Israelachvili. 2003. Sealed minitrough for microscopy and long-term stability studies of Langmuir monolayers. Langmuir. 19:100–104. [Google Scholar]
- 23.Ekelund, K., E. Sparr, J. Engblom, H. Wennerstrom, and S. Engstrom. 1999. AFM study of lipid monolayers. 1. Pressure-induced phase behavior of single and mixed fatty acids. Langmuir. 15:6946–6949. [Google Scholar]
- 24.Stolz, M., D. Stoffler, U. Aebi, and C. Goldsbury. 2000. Monitoring biomolecular interactions by time-lapse atomic force microscopy. J. Struct. Biol. 131:171–180. [DOI] [PubMed] [Google Scholar]
- 25.Arimon, M., I. Diez-Perez, M. J. Kogan, N. Durany, E. Giralt, F. Sanz, and X. Fernandez-Busquets. 2005. Fine structure study of a β(1–42) fibrillogenesis with atomic force microscopy. FASEB J. 19:1344–1346. [DOI] [PubMed] [Google Scholar]
- 26.Kowalewski, T., and D. M. Holtzman. 1999. In situ atomic force microscopy study of Alzheimer's β-amyloid peptide on different substrates: new insights into mechanism of β-sheet formation. Proc. Natl. Acad. Sci. USA. 96:3688–3693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hong, Q. A., M. P. Sheetz, and E. L. Elson. 1991. Single-particle tracking—analysis of diffusion and flow in two-dimensional systems. Biophys. J. 60:910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saxton, M. J., and K. Jacobson. 1997. Single-particle tracking: applications to membrane dynamics. Annu. Rev. Biophys. Biomol. Struct. 26:373–399. [DOI] [PubMed] [Google Scholar]
- 29.Yoshina-Ishii, C., and S. G. Boxer. 2003. Arrays of mobile tethered vesicles on supported lipid bilayers. J. Am. Chem. Soc. 125:3696–3697. [DOI] [PubMed] [Google Scholar]
- 30.Moscho, A., O. Orwar, D. T. Chiu, B. P. Modi, and R. N. Zare. 1996. Rapid preparation of giant unilamellar vesicles. Proc. Natl. Acad. Sci. USA. 93:11443–11447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy, 2nd Ed. Kluwer Academic, New York.
- 32.Marine, S., E. Zamiara, S. Smith, E. Stec, J. McGarvey, O. Kornienko, G. Jiang, K. Wong, J. Stack, B. Zhang, M. Ferrer, and B. Strulovici. 2006. A miniaturized cell-based fluorescence resonance energy transfer assay for insulin-receptor activation. Anal. Biochem. In press. [DOI] [PubMed]
- 33.Andra, J., H. Cock, P. Garidel, J. Howe, and K. Brandenburg. 2005. Investigation into the interaction of the phosphoporin PhoE with outer membrane lipids: physicochemical characterization and biological activity. Med. Chem. 1:537–546. [DOI] [PubMed] [Google Scholar]
- 34.Birch, D., K. Suhling, A. Holmes, T. Salthammer, and R. Imhof. 1993. Metal ion quenching of perylene fluorescence in lipid bilayers. Pure Appl. Chem. 65:1687–1692. [Google Scholar]
- 35.Patel, R. C., U. Kumar, D. C. Lamb, J. S. Eid, M. Rocheville, M. Grant, A. Rani, T. Hazlett, S. C. Patel, E. Gratton, and Y. C. Patel. 2002. Ligand binding to somatostatin receptors induces receptor-specific oligomer formation in live cells. Proc. Natl. Acad. Sci. USA. 99:3294–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lambert, M. P., A. K. Barlow, B. A. Chromy, C. Edwards, R. Freed, M. Liosatos, T. E. Morgan, I. Rozovsky, B. Trommer, K. L. Viola, P. Wals, C. Zhang, C. E. Finch, G. A. Krafft, and W. L. Klein. 1998. Diffusible, nonfibrillar ligands derived from Aβ 1–42 are potent central nervous system neurotoxins. Proc. Natl. Acad. Sci. USA. 95:6448–6453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Michikawa, M., J. S. Gong, Q. W. Fan, N. Sawamura, and K. Yanagisawa. 2001. A novel action of Alzheimer's amyloid β-protein (Aβ): oligomeric Aβ promotes lipid release. J. Neurosci. 21:7226–7235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ambroggio, E. E., D. H. Kim, F. Separovic, C. J. Barrow, C. J. Barrow, K. J. Barnham, L. A. Bagatolli, and G. D. Fidelio. 2005. Surface behavior and lipid interaction of Alzheimer β-amyloid peptide 1–42: a membrane-disrupting peptide. Biophys. J. 88:2706–2713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anguiano, M., R. J. Nowak, and P. T. Lansbury. 2002. Protofibrillar islet amyloid polypeptide permeabilizes synthetic vesicles by a pore-like mechanism that may be relevant to type II diabetes. Biochemistry. 41:11338–11343. [DOI] [PubMed] [Google Scholar]
- 40.Volles, M. J., and P. T. Lansbury. 2002. Vesicle permeabilization by protofibrillar α-synuclein is sensitive to Parkinson's disease-linked mutations and occurs by a pore-like mechanism. Biochemistry. 41:4595–4602. [DOI] [PubMed] [Google Scholar]
- 41.Zhang, Y., R. McLaughlin, C. Goodyer, and A. LeBlanc. 2002. Selective cytotoxicity of intracellular amyloid β-peptide (1–42) through p53 and Bax in cultured primary human neurons. J. Cell Biol. 156:519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bagatolli, L. A., and E. Gratton. 1999. Two-photon fluorescence microscopy observation of shape changes at the phase transition in phospholipid giant unilamellar vesicles. Biophys. J. 77:2090–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nuscher, B., F. Kamp, T. Mehnert, S. Odoy, C. Haass, P. J. Kahle, and K. Beyer. 2004. Alpha-synuclein has a high affinity for packing defects in a bilayer membrane: a thermodynamics study. J. Biol. Chem. 279:21966–21975. [DOI] [PubMed] [Google Scholar]
- 44.Petkova, A. T., Y. Ishii, J. J. Balbach, O. N. Antzutkin, R. D. Leapman, F. Delaglio, and R. Tycko. 2002. A structural model for Alzheimer's β-amyloid fibrils based on experimental constraints from solid state NMR. Proc. Natl. Acad. Sci. USA. 99:16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Johnson, S. J., T. M. Bayerl, D. C. McDermott, G. W. Adam, A. R. Rennie, R. K. Thomas, and E. Sackmann. 1991. Structure of an adsorbed dimyristoylphosphatidylcholine bilayer measured with specular reflection of neutrons. Biophys. J. 59:289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harper, J. D., S. S. Wong, C. M. Lieber, and P. T. Lansbury. 1999. Assembly of a β-amyloid protofibrils: an in vitro model for a possible early event in Alzheimer's disease. Biochemistry. 38:8972–8980. [DOI] [PubMed] [Google Scholar]
- 47.Lin, H., R. Bhatia, and R. Lal. 2001. Amyloid β-protein forms ion channels: implications for Alzheimer's disease pathophysiology. FASEB J. 15:2433–2444. [DOI] [PubMed] [Google Scholar]
- 48.Weninger, S. C., and B. A. Yankner. 2001. Inflammation and Alzheimer disease: the good, the bad, and the ugly. Nat. Med. 7:527–528. [DOI] [PubMed] [Google Scholar]
- 49.Raviv, U., D. J. Needleman, Y. L. Li, H. P. Miller, L. Wilson, and C. R. Safinya. 2005. Cationic liposome-microtubule complexes: pathways to the formation of two-state lipid-protein nanotubes with open or closed ends. Proc. Natl. Acad. Sci. USA. 102:11167–11172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tashima, Y., R. Oe, S. Lee, G. Sugihara, E. J. Chambers, M. Takahashi, and T. Yamada. 2004. The effect of cholesterol and monosialoganglioside (GM1) on the release and aggregation of amyloid β-peptide from liposomes prepared from brain membrane-like lipids. J. Biol. Chem. 279:17587–17595. [DOI] [PubMed] [Google Scholar]
- 51.Heerklotz, H. 2002. Triton promotes domain formation in lipid raft mixtures. Biophys. J. 83:2693–2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Waschuk, S. A., E. A. Elton, A. A. Darabie, P. E. Fraser, and J. McLaurin. 2001. Cellular membrane composition defines Aβ-lipid interactions. J. Biol. Chem. 276:33561–33568. [DOI] [PubMed] [Google Scholar]
- 53.Roher, A. E., M. O. Chaney, Y. M. Kuo, S. D. Webster, W. B. Stine, L. J. Haverkamp, A. S. Woods, R. J. Cotter, J. M. Tuohy, G. A. Krafft, B. S. Bonnell, and M. R. Emmerling. 1996. Morphology and toxicity of Aβ (1–42) dimer derived from neuritic and vascular amyloid deposits of Alzheimer's disease. J. Biol. Chem. 271:20631–20635. [DOI] [PubMed] [Google Scholar]
- 54.Jacobson, K., E. D. Sheets, and R. Simson. 1995. Revisiting the fluid mosaic model of membranes. Science. 268:1441–1442. [DOI] [PubMed] [Google Scholar]