

Abstract
We investigate the potential of dual-color photon counting histogram (PCH) analysis to resolve fluorescent protein mixtures directly inside cells. Because of their small spectral overlap, we have chosen to look at the fluorescent proteins EGFP and mRFP1. We experimentally demonstrate that dual-color PCH quantitatively resolves a mixture of EGFP and mRFP1 in cells from a single measurement. To mimic the effect of protein association, we constructed a fusion protein of EGFP and mRFP1 (denoted EGFP-mRFP1). Fluorescence resonant energy transfer within the fusion protein alters the dual-channel brightness of the fluorophores. We describe a model for fluorescence resonant energy transfer effects on the brightness and incorporate it into dual-color PCH analysis. The model is verified using fluorescence lifetime measurements. Dual-color PCH analysis demonstrated that not all of the expressed EGFP-mRFP1 fusion proteins contained a fluorescent mRFP1 molecule. Fluorescence lifetime and emission spectra measurements confirmed this surprising result. Additional experiments show that the missing fluorescent fraction of mRFP1 is consistent with a dark state population of mRFP1. We successfully resolved this mixture of fusion proteins with a single dual-color PCH measurement. These results highlight the potential of dual-color PCH to directly detect and quantify protein mixtures in living cells.
INTRODUCTION
Proteins are complex biomolecules that perform important and essential cellular functions such as transporting molecules, regulating cellular processes, assembling structures, and communicating with neighboring cells. These functions are carried out through interactions between proteins. Quantitative and noninvasive observation of these protein-protein interactions in living cells is an important first step for ultimately piecing together the inner workings of the cellular machinery.
One technique capable of quantitatively studying protein association and dissociation in living cells is dual-color fluorescence fluctuation spectroscopy (FFS) (1,2,3,4,5,6). In dual-color or dual-channel FFS, two spectrally distinct fluorophores (i.e., “red” and “green”) are used to label the proteins of interest (i.e., X and Y). The emission of the two fluorophores is separated into two detectors by a dichroic mirror. Coincident signals in the “red” and “green” channels indicate association of proteins X and Y, whereas noncoincident signals indicate that the proteins are dissociated. Various statistical tools, such as cross-correlation analysis (7), dual-color photon counting histogram (PCH) analysis (8), or two-dimensional fluorescence intensity distribution analysis (9), are used to extract information about the protein interactions from the fluctuations in the two detection channels.
We previously showed that single-channel PCH detects protein association inside cells and recently expanded the technique to quantify the simultaneous presence of homo- and heterodimers (10,11). Dual-color PCH offers more sensitivity for resolving a mixture of species than single-channel PCH. For example, we showed that a mixture of cyan and yellow fluorescent protein (CFP and YFP), which have significant spectral overlap, are resolved by a single dual-color PCH experiment in vitro (8,12). In this article, we focus on expanding the use of dual-color PCH to the intracellular environment. The original theory for dual-color PCH was developed assuming that the two detectors were ideal. This assumption is not valid for the conditions found in typical cellular experiments. We recently modified dual-color PCH theory to include detector effects such as dead-time and after pulses (12). For dual-color measurements in cells, we also need to identify a suitable pair of fluorescent proteins. CFP and YFP are widely used in many fluorescence applications, and we initially worked with this pair, but quickly realized that the large spectral overlap makes the pair very challenging to resolve with dual-color PCH. Thus we chose an alternate pair of fluorescent proteins, EGFP and mRFP1, which exhibit significantly less spectral overlap. We show that this pair is experimentally resolvable from a single dual-color PCH measurement in a cell. The sensitivity of dual-color PCH is highest at low concentrations. We characterized and corrected for the contribution of cellular autofluorescence in dual-color PCH analysis at very low concentrations. An EGFP-mRFP1 fusion protein was created to serve as a model heterodimer. This fusion protein exhibits fluorescence resonance energy transfer (FRET). We describe a theory that takes the effect of FRET upon the dual-channel brightnesses into account. Dual-channel PCH analysis of cells reveals that the expressed fusion protein is best described as a mixture of species, since not all fusion proteins contain a fluorescent mRFP1. Fluorescence lifetime and emission spectra measurements of the fusion protein are in agreement with the dual-color PCH result. The existence of mRFP1 in a spectroscopic dark and bright state best describes the experimental data. We also describe a procedure that takes into account the depletion of fluorophores during a PCH measurement. This allowed us to directly resolve the mixture of the fusion protein with a single dual-color PCH measurement.
THEORY
Dual-color PCH
Dual-color PCH analysis utilizes the joint probability of observing 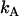 photon counts in channel A and
photon counts in channel A and 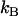 photon counts in channel B (throughout the rest of the article, we refer to the two detection channels as A and B rather than “red” and “green”). The theory of dual-color PCH was developed by Chen et al. (8) for the case of ideal detectors. This theory was later modified to include nonideal detector effects (12). The theoretical dual-color PCH for a single species, denoted as
photon counts in channel B (throughout the rest of the article, we refer to the two detection channels as A and B rather than “red” and “green”). The theory of dual-color PCH was developed by Chen et al. (8) for the case of ideal detectors. This theory was later modified to include nonideal detector effects (12). The theoretical dual-color PCH for a single species, denoted as 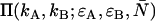 , is described by three parameters: i),
, is described by three parameters: i), 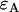 the molecular brightness in channel A, ii),
the molecular brightness in channel A, ii), 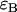 the molecular brightness in channel B, and iii),
the molecular brightness in channel B, and iii), 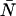 the average number of molecules in the observation volume. For multiple species, the theoretical dual-color PCH is obtained by successive convolutions of each individual species' theoretical PCH function (8). We refer to a species' combination of
the average number of molecules in the observation volume. For multiple species, the theoretical dual-color PCH is obtained by successive convolutions of each individual species' theoretical PCH function (8). We refer to a species' combination of 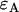 and
and 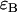 as its “brightness signature”. Since the dichroic mirror splits each fluorophore's emission differently, multiple species are resolved through differences in their brightness signatures.
as its “brightness signature”. Since the dichroic mirror splits each fluorophore's emission differently, multiple species are resolved through differences in their brightness signatures.
FRET effects on the dual-channel brightness
The association of labeled proteins may bring the two fluorophores in close enough proximity to each other that FRET occurs. In FRET, the excited donor fluorophore transfers energy to the acceptor fluorophore via a dipole-dipole interaction. The efficiency of this transfer, denoted E, depends on the orientation and distance between the donor and acceptor. FRET changes the fluorescence properties of the donor and acceptor molecules. Specifically, it changes the fluorescence lifetime of the donor (13,14,15), and the molecular brightness of the donor and acceptor (10). In the following expressions, we assume that each donor molecule is associated with an acceptor. We denote the donor and acceptor brightnesses for a single detection channel in the absence of FRET as 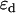 and
and 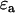 . For the EGFP/mRFP1 pair, EGFP acts as the donor fluorophore and mRFP1 as the acceptor.
. For the EGFP/mRFP1 pair, EGFP acts as the donor fluorophore and mRFP1 as the acceptor.
The expressions for the single-channel brightness of the donor 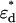 , acceptor
, acceptor 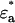 , and donor-acceptor complex
, and donor-acceptor complex 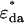 in the presence of FRET are (10)
in the presence of FRET are (10)
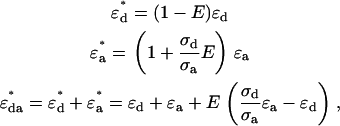 |
(1) |
where 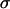 denotes the absorption cross section at the specific excitation wavelength
denotes the absorption cross section at the specific excitation wavelength 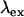 . Note that Eq. 1 has been formulated for coexcitation of donor and acceptor. The extension of Eq. 1 to two detection channels is straightforward, and only requires the replacement of the single-channel brightness by the appropriate brightness in channel A or B,
. Note that Eq. 1 has been formulated for coexcitation of donor and acceptor. The extension of Eq. 1 to two detection channels is straightforward, and only requires the replacement of the single-channel brightness by the appropriate brightness in channel A or B,
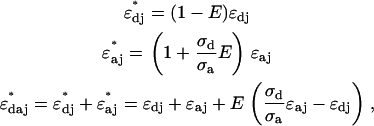 |
(2) |
where 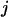 denotes the detection channel.
denotes the detection channel.
A special case worth noting is when the donor and acceptor brightnesses are identical and both fluorophores have comparable cross sections (as in the case of CFP and YFP excited at  ); in this case the third term in Eq. 1 cancels and the heterodimer's brightness in single-channel measurements is always the same regardless of how much FRET occurs. This fact was exploited in an intracellular study of heterodimerization using single-channel PCH (10). In dual-channel measurements, the brightness values of the donor and acceptor differ in each channel. Thus for dual-color experiments, it is generally not possible to find excitation conditions where the influence of FRET on the heterodimer's brightness vanishes. In other words, FRET must be accounted for in dual-color brightness analysis.
); in this case the third term in Eq. 1 cancels and the heterodimer's brightness in single-channel measurements is always the same regardless of how much FRET occurs. This fact was exploited in an intracellular study of heterodimerization using single-channel PCH (10). In dual-channel measurements, the brightness values of the donor and acceptor differ in each channel. Thus for dual-color experiments, it is generally not possible to find excitation conditions where the influence of FRET on the heterodimer's brightness vanishes. In other words, FRET must be accounted for in dual-color brightness analysis.
Equation 2 predicts the brightness of a heterodimer in the presence of FRET, provided that the donor's and acceptor's brightnesses can be measured independently and provided that the FRET efficiency is known. The FRET efficiency is determined from measurements of the donor's lifetime in the presence and absence of the acceptor,
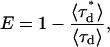 |
(3) |
where 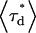 is the intensity-based average lifetime of the donor in the presence of FRET and
is the intensity-based average lifetime of the donor in the presence of FRET and 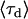 is the intensity-based average lifetime of the donor in the absence of FRET. The intensity-based average lifetime
is the intensity-based average lifetime of the donor in the absence of FRET. The intensity-based average lifetime 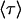 of a multi-exponential decay
of a multi-exponential decay 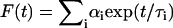 is defined as
is defined as 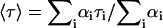 .
.
Apparent FRET efficiency and apparent brightness
We note that Eq. 3 is valid only if all donors are associated with an acceptor. If there are more donors than acceptors, as would be for a mixture of EGFP and EGFP-mRFP1, then the lifetime obtained from the sample would be a composite lifetime described by
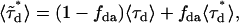 |
(4) |
where 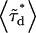 is the average lifetime of the mixture and
is the average lifetime of the mixture and 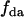 is the fraction of donor-acceptor complexes,
is the fraction of donor-acceptor complexes, 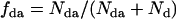 , where
, where 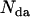 is the number of acceptor-donor complexes and
is the number of acceptor-donor complexes and 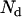 is the number of free donors. Mixtures of donor and donor-acceptor complexes are best described by an apparent FRET efficiency
is the number of free donors. Mixtures of donor and donor-acceptor complexes are best described by an apparent FRET efficiency 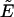 (10), which is defined as
(10), which is defined as
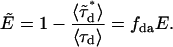 |
(5) |
Note that for a mixture of acceptor monomers and donor-acceptor dimers, all donors are associated with an acceptor (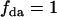 ) and Eq. 5 simplifies to Eq. 3.
) and Eq. 5 simplifies to Eq. 3.
If a single-channel histogram of a mixture is fit to a single-species model, then the brightness returned is an apparent brightness 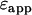 that represents the average contribution of each individual species' brightness values. Similarly, a single-species description of a mixture also leads to an apparent number of molecules
that represents the average contribution of each individual species' brightness values. Similarly, a single-species description of a mixture also leads to an apparent number of molecules 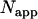 (16). The apparent brightness is useful for characterizing single-channel measurements with insufficient statistics to resolve the two species. A single-channel measurement of a two-species mixture with brightness values of
(16). The apparent brightness is useful for characterizing single-channel measurements with insufficient statistics to resolve the two species. A single-channel measurement of a two-species mixture with brightness values of 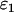 and
and 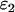 for the two species yields an apparent brightness
for the two species yields an apparent brightness 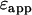 described by (11)
described by (11)
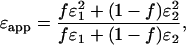 |
(6) |
where 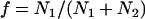 . For the mixtures, we are concerned with (EGFP and EGFP-mRFP1 or mRFP1 and EGFP-mRFP1), f is the fraction of EGFP-mRFP1 (i.e.,
. For the mixtures, we are concerned with (EGFP and EGFP-mRFP1 or mRFP1 and EGFP-mRFP1), f is the fraction of EGFP-mRFP1 (i.e., 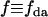 ),
), 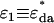 is the brightness of the fusion protein (given by Eq. 1), and
is the brightness of the fusion protein (given by Eq. 1), and 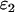 is the brightness of either EGFP or mRFP1. Equation 6 is only valid for the single-channel case. An expression for the apparent brightness in dual-channel experiments can be obtained from cumulants (12,17,18), but the resulting expressions are lengthy and not reported here. Instead, we determine the apparent brightness by fitting the dual-color PCH to a single-species model.
is the brightness of either EGFP or mRFP1. Equation 6 is only valid for the single-channel case. An expression for the apparent brightness in dual-channel experiments can be obtained from cumulants (12,17,18), but the resulting expressions are lengthy and not reported here. Instead, we determine the apparent brightness by fitting the dual-color PCH to a single-species model.
Brightness spectrum
In Chen et al. (8), we introduced the brightness spectrum 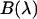 of a fluorophore,
of a fluorophore,
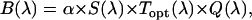 |
(7) |
where 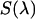 is fluorescence emission spectrum;
is fluorescence emission spectrum; 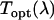 characterizes the transmission function of our microscope and includes the optical properties of the objective, the two-photon dichroic and all other optical elements;
characterizes the transmission function of our microscope and includes the optical properties of the objective, the two-photon dichroic and all other optical elements; 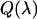 is the detection efficiency of the photon avalanche diode (APD); and
is the detection efficiency of the photon avalanche diode (APD); and 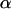 is a proportionality constant. The value of
is a proportionality constant. The value of 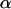 is determined from the total brightness
is determined from the total brightness 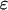 , which corresponds to the area under the
, which corresponds to the area under the 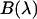 curve (8),
curve (8),
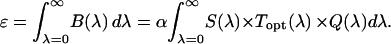 |
(8) |
The brightness spectrum of a donor-acceptor complex 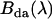 in the presence of FRET is related to the corresponding donor's and acceptor's brightness spectra (
in the presence of FRET is related to the corresponding donor's and acceptor's brightness spectra (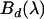 and
and 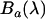 respectively) as
respectively) as
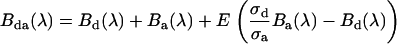 |
(9) |
Equation 9 allows us to model our fusion protein's spectrum using its constituent monomers' spectra. We define the apparent brightness spectrum 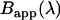 of a mixture as
of a mixture as
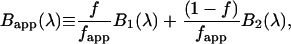 |
(10) |
where 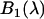 and
and 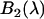 are the brightness spectra of each species, and the apparent fraction
are the brightness spectra of each species, and the apparent fraction 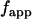 is given by
is given by
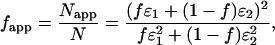 |
(11) |
with 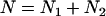 and the apparent number of molecules
and the apparent number of molecules 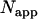 . Note that the definition of the apparent brightness spectrum in Eq. 10 reproduces the correct apparent brightness (Eq. 6),
. Note that the definition of the apparent brightness spectrum in Eq. 10 reproduces the correct apparent brightness (Eq. 6), 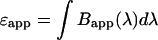 . Later we will associate
. Later we will associate 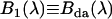 with the EGFP-mRFP1 spectrum,
with the EGFP-mRFP1 spectrum, 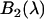 with either the EGFP or the mRFP1 spectrum, and
with either the EGFP or the mRFP1 spectrum, and 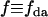 with the fraction of EGFP-mRFP1.
with the fraction of EGFP-mRFP1.
MATERIALS AND METHODS
FFS measurements
The instrumentation for dual-channel fluorescence fluctuation experiments has been described in Chen et al. (8). EGFP and mRFP1 were excited at 995 nm with an average power after the objective of 2.0 mW. The fluorescence emission is separated into two different detection channels using a 570-nm dichroic (Chroma Technology, Rockingham, VT). This dichroic optimizes the signal statistics for the EGFP/mRFP1 pair as determined according to the procedure in Chen et al. (8). The sampling frequency was 20 kHz for cellular measurements; no undersampling occurs at this frequency. Data were acquired for ∼0.5 min for most measurements. Experimental single- and dual-channel histograms were fit to theoretical PCH functions as described in Hillesheim and Mueller (12,19). Brightness values are reported with respect to a normalized Gaussian-Lorentzian point spread function. Nonideal detector effects were included in the fitting procedure. Modeling was performed by calculating PCH functions for chosen parameters and fitting them to specific models as previously explained (12).
Next we describe the alignment procedure for dual-channel measurements using dual-color PCH analysis. The two detectors must be aligned properly to ensure that both channels observe photons from the same volume. We use a low concentration solution (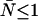 ) of a dye whose emission is approximately split by the dichroic to be used in subsequent measurements. Additionally, the dye must be easy to excite at the specified wavelength. Data are taken at a sampling frequency of 20 kHz to increase the brightness in each channel and to reduce dead-time effects. Although this frequency introduces undersampling, the effect is the same in both channels and thus it can be ignored for calibration purposes. We require the brightness in each channel to be ≥1 cpm (counts per time bin per molecule). This requirement, along with the use of a low concentration sample and longer sampling period, ensures that after-pulse and dead-time effects are small. For measurements with the 570-nm dichroic (
) of a dye whose emission is approximately split by the dichroic to be used in subsequent measurements. Additionally, the dye must be easy to excite at the specified wavelength. Data are taken at a sampling frequency of 20 kHz to increase the brightness in each channel and to reduce dead-time effects. Although this frequency introduces undersampling, the effect is the same in both channels and thus it can be ignored for calibration purposes. We require the brightness in each channel to be ≥1 cpm (counts per time bin per molecule). This requirement, along with the use of a low concentration sample and longer sampling period, ensures that after-pulse and dead-time effects are small. For measurements with the 570-nm dichroic (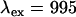 nm) we used rhodamine 6G (emission maximum:
nm) we used rhodamine 6G (emission maximum: 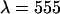 nm) in water. Rhodamine 6G was obtained from Acros Organics (Geel, Belgium). To align the two channels, we first optimize the position of both APDs to get the maximum intensity in each channel and then adjust channel B's position along the optical axis until we get agreement in the single-channel and dual-channel PCH parameters. Single-channel PCH analysis of channel A returns values for
nm) in water. Rhodamine 6G was obtained from Acros Organics (Geel, Belgium). To align the two channels, we first optimize the position of both APDs to get the maximum intensity in each channel and then adjust channel B's position along the optical axis until we get agreement in the single-channel and dual-channel PCH parameters. Single-channel PCH analysis of channel A returns values for 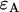 and
and 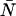 , whereas single-channel analysis of channel B returns values for
, whereas single-channel analysis of channel B returns values for 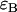 and
and 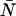 . Dual-channel analysis of the two-dimensional histogram returns values for
. Dual-channel analysis of the two-dimensional histogram returns values for 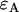 ,
, 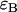 , and
, and 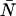 . If the three results are the same within errors, then we consider the channels to be aligned. We note that although the alignment was conducted under conditions in which nonideal detector effects are minimized, we accounted for any dead-time and/or after-pulse induced biases by including them in all fits as a precaution.
. If the three results are the same within errors, then we consider the channels to be aligned. We note that although the alignment was conducted under conditions in which nonideal detector effects are minimized, we accounted for any dead-time and/or after-pulse induced biases by including them in all fits as a precaution.
Autofluorescence and background light
Some of the cellular PCH experiments are performed at fluorescent protein concentrations where autofluorescence needs to be accounted for in the analysis. The most straightforward method for determining the autofluorescence contribution is to measure a population of untransfected or mock-transfected cells, and use their average fluorescence to correct the fluorescence fluctuation experiments of the transfected cells. However, this approach assumes that the autofluorescence of the untransfected control is identical to that of the transfected cells. We developed an alternative approach described in Hillesheim (20) that uses the intensity ratio of transfected cells. Using this procedure on EGFP and mRFP1 cells excited at 995 nm, we found the background/autofluorescence intensities to be 500 cps in channel A and 370 cps in channel B. These intensity values were consistent with those obtained from mock-transfected cells.
The autofluorescent species' 2PE cross section is ∼10–100 times smaller at excitation wavelengths of 900–1000 nm than those of the fluorescent proteins (e.g., EGFP) (21). We therefore expect the autofluorescent species' brightness to be ∼0.001–0.01 cpm given that the typical brightness of a fluorescent protein under our measurement conditions is ∼0.1 cpm. The background light is uncorrelated, and although the autofluorescence is correlated, its brightness is very low and the number of molecules high. Thus both components can be treated as a single Poisson distribution. To account for background/autofluorescence effects in the dual-color PCH fit, we simply include a fixed dual-color Poisson distribution with the means equal to the total background counts in each channel.
Lifetime measurements
A removable mirror in the emission path was used to direct the fluorescence onto a photomultiplier tube (H7421-40, Hamamatsu, Hamamatsu City, Japan) for lifetime measurements. A polarizer set at magic-angle conditions (14) and a 150 μm pinhole were placed in front of the photon multiplier tube. The photon multiplier tube output was connected to a time-correlated single-photon counting module (TimeHarp 200, Picoquant, Berlin, Germany). A photodiode (DET210, Thorlabs, Newton, NJ) detected the laser pulse for timing purposes. A 505–535 nm bandpass filter was placed before the pinhole to select for the EGFP emission, while a 605–645 nm filter was used to select for mRFP1 emission. Both filters were obtained from Semrock (Rochester, NY). Instrument response functions were obtained using Urea crystals (ICN Biomedical, Aurora, OH) via second harmonic generation (22). Fluorescence lifetime data were analyzed using Globals Unlimited (Urbana, IL). A standard solution of Alexa 488 was measured before commencing lifetime measurements in cells for calibration purposes. The single-exponential lifetime obtained from the sample (4.0 ns) was in good agreement with the lifetime reported in the literature (4.1 ns) (23).
Spectral measurements
We directly measure the modified emission spectrum, 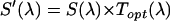 , with a charge-coupled device (CCD) spectrograph (SpectraPro-2150i spectrograph from Acton Research, Acton, MA; Ixon DV 887 CCD camera, Andor Technology, South Windsor, CT) mounted on the side port of the microscope. Spectra are first corrected for the CCD spectrograph's response. Each spectrum was then multiplied by the APD's detection efficiency
, with a charge-coupled device (CCD) spectrograph (SpectraPro-2150i spectrograph from Acton Research, Acton, MA; Ixon DV 887 CCD camera, Andor Technology, South Windsor, CT) mounted on the side port of the microscope. Spectra are first corrected for the CCD spectrograph's response. Each spectrum was then multiplied by the APD's detection efficiency 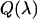 . For simplicity, we denote the quantity
. For simplicity, we denote the quantity 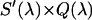 as
as 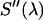 . The microscope allows us to redirect the fluorescence from the side port to the bottom port, where the fluorescence emission is detected by the APD for FFS measurements. Because the FFS and spectral measurements are performed under identical conditions, the brightness spectrum
. The microscope allows us to redirect the fluorescence from the side port to the bottom port, where the fluorescence emission is detected by the APD for FFS measurements. Because the FFS and spectral measurements are performed under identical conditions, the brightness spectrum 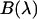 is determined from the experimentally measured brightness and the calculated spectrum
is determined from the experimentally measured brightness and the calculated spectrum 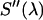 by
by 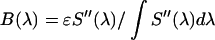 . The brightness ɛ in each cell was obtained via single-channel PCH analysis. The brightness spectra obtained from several cells were averaged together to obtain an averaged brightness spectrum for each fluorescent protein. In the case of a spectrum obtained from a mixture, the apparent brightness (Eq. 6) of the mixture is used to determine the apparent brightness spectrum.
. The brightness ɛ in each cell was obtained via single-channel PCH analysis. The brightness spectra obtained from several cells were averaged together to obtain an averaged brightness spectrum for each fluorescent protein. In the case of a spectrum obtained from a mixture, the apparent brightness (Eq. 6) of the mixture is used to determine the apparent brightness spectrum.
The change in the acceptor's brightness depends on the ratio of the donor's cross section to the acceptor's (Eqs. 1 and 2). Two-photon cross sections are generally quite difficult to measure, so we devised a way to determine the cross-sectional ratio using the brightness ratio between the donor and acceptor and their emission spectra. The two-photon absorption rate 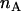 is given by
is given by 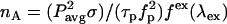 , where
, where 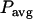 is the average laser power,
is the average laser power, 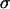 is the cross section,
is the cross section, 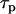 is the laser pulse duration,
is the laser pulse duration, 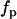 is the repetition frequency, and
is the repetition frequency, and 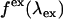 is a geometrical factor that depends on the excitation wavelength and the numerical aperture of the objective (24). The rate of fluorescence emission
is a geometrical factor that depends on the excitation wavelength and the numerical aperture of the objective (24). The rate of fluorescence emission 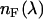 at wavelength
at wavelength 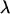 is determined from
is determined from 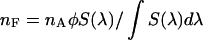 , where
, where 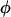 is the quantum yield and
is the quantum yield and 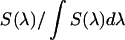 is the normalized emission spectrum. The probability that fluorescence at a wavelength λ is detected is described by the factor
is the normalized emission spectrum. The probability that fluorescence at a wavelength λ is detected is described by the factor 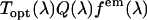 , where
, where 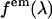 is a geometrical factor that describes the geometrical collection efficiency at wavelength λ. The factor
is a geometrical factor that describes the geometrical collection efficiency at wavelength λ. The factor 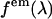 is approximately constant over the emission wavelengths of interest (
is approximately constant over the emission wavelengths of interest (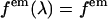 ) and thus the brightness spectrum is given by
) and thus the brightness spectrum is given by 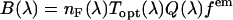 . Integrating over the brightness spectrum determines the brightness of the fluorophore,
. Integrating over the brightness spectrum determines the brightness of the fluorophore, 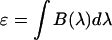 . If the donor and acceptor are measured under the same experimental conditions, then
. If the donor and acceptor are measured under the same experimental conditions, then 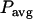 ,
, 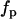 ,
, 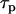 ,
, 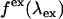 , and
, and 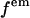 are identical. This leads to the following expression for the brightness ratio of the donor and acceptor,
are identical. This leads to the following expression for the brightness ratio of the donor and acceptor,
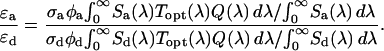 |
(12) |
The cross-sectional ratio of the donor and acceptor is then determined by
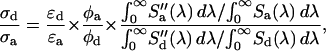 |
(13) |
where 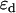 and
and 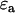 are the single-channel brightness values of the donor and acceptor (in the absence of FRET), respectively;
are the single-channel brightness values of the donor and acceptor (in the absence of FRET), respectively; 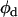 and
and 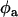 are the donor and acceptor quantum yields; and
are the donor and acceptor quantum yields; and 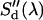 and
and 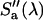 are the modified emission spectra of the donor and acceptor. The emission spectrum
are the modified emission spectra of the donor and acceptor. The emission spectrum 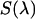 is determined by dividing the measured spectrum
is determined by dividing the measured spectrum 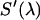 by
by 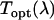 . The instrument's transmission curve
. The instrument's transmission curve 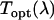 was constructed from wavelength-dependent transmission curves of the individual optical elements.
was constructed from wavelength-dependent transmission curves of the individual optical elements.
The spectra from four to six cells expressing a given fluorescent protein were measured. The spectra were normalized according to Eq. 13 and averaged together. The corresponding brightness values for each protein were obtained via single-channel PCH analysis of the cells. Using the average spectrum and brightness value, we determined a cross-sectional ratio of 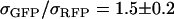 for EGFP and mRFP1 at
for EGFP and mRFP1 at 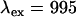 nm. Published values for the quantum yields (
nm. Published values for the quantum yields (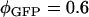 ,
, 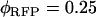 ) were used in the calculation (25,26). Using this method we also determined the cross-sectional ratio of CFP to YFP to be
) were used in the calculation (25,26). Using this method we also determined the cross-sectional ratio of CFP to YFP to be 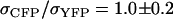 at
at 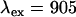 nm, which is in agreement with published values (27).
nm, which is in agreement with published values (27).
Sample preparation
pEGFP-C1 plasmid was obtained from Clontech (Moutainview, CA). A synthetic linker that encodes 12 amino acids (GHGTGSTGSGSS) was cloned into the pEGFP-C1 plasmid at BspEI and XhoI sites to generate pEGFP-C1′. pEGFP-C1′ was digested with NheI and BspEI restriction sites and gel purified to generate the backbone of the vector. The mRFP1 pRSET B plasmid was a kind gift from Dr. R. Y. Tsien (University of California, San Diego). mRFP1 was polymerase chain reaction (PCR) amplified with a 5′ primer that encodes a NheI restriction site and a 3′ primer that encodes a BspEI site. mRFP was ligated into the backbone vector of pEGFP-C1′ to generate the mRFP1-C1 vector for mammalian expression. EGFP-mRFP1 was constructed by inserting the PCR-amplified mRFP1 into the pEGFP-C1′ at EcoRI and BamHI sites. For EGFP-mRFP1 fusion protein, the total number of amino acids between the proteins is 24; there are an additional 12 amino acids due to the cloning site used. mRFP1-EGFP and mRFP1-mRFP1 were generated by ligating PCR amplified EGFP and mRFP1 into the mRFP1-C1 vector. All sequences were verified by automatic sequencing. The purity of plasmids was checked by gel electrophoresis.
COS cells were obtained from ATCC (Manassas, VA) and maintained in 10% fetal bovine serum (Hyclone Laboratories, Logan, UT) and Dulbecco's modified Eagle's medium. Cells were subcultured into eight-well coverglass chamber slides (Naglenunc International, Rochester, NY) and then transiently transfected using polyfect (Qiagen, Valencia, CA) according to manufacturer's instructions. Before conducting measurements, the growth media was removed and replaced with phosphate-buffered saline. All measurements were performed in the cell nucleus.
RESULTS
EGFP and mRFP1: independent species
The area-normalized emission spectra 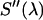 of EGFP and mRFP1 as well as the dichroic used to separate them are shown in Fig. 1 A. There is significantly less cross talk with this pair compared to CFP and YFP (Fig. 1 B); however, mRFP1's single-channel brightness is ∼4 times smaller than EGFP's and this worsens the signal statistics. We have already shown EGFP and mRFP1 to remain monomeric as a function of concentration (12) and note that the dual-channel brightnesses sum to the single-channel brightness (data not shown).
of EGFP and mRFP1 as well as the dichroic used to separate them are shown in Fig. 1 A. There is significantly less cross talk with this pair compared to CFP and YFP (Fig. 1 B); however, mRFP1's single-channel brightness is ∼4 times smaller than EGFP's and this worsens the signal statistics. We have already shown EGFP and mRFP1 to remain monomeric as a function of concentration (12) and note that the dual-channel brightnesses sum to the single-channel brightness (data not shown).
FIGURE 1.

Fluorescence emission spectra of (A) the EGFP and mRFP1 pair and (B) the CFP and YFP pair along with their respective dichroic mirror. The cross talk for each pair is indicated by the shaded region. Photons with wavelengths to the left of the dichroic curve are reflected into channel B, whereas photons with wavelengths to the right of the dichroic curve are transmitted into channel A.
To determine the limits of the dual-color PCH in resolving EGFP from mRFP1, we modeled mixtures of EGFP and mRFP1 in cells using brightness values obtained from independent measurements of EGFP alone and mRFP1 alone in COS cells. We created two-species dual-color PCH functions with varying amounts of EGFP and mRFP1 assuming a data acquisition time of 2 min. The modeled PCH functions were then fit to a single-species model. The reduced 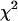 returned by the single-species fit is a measure of dual-color PCH's ability to distinguish between one- and two-species systems. A contour plot of the reduced
returned by the single-species fit is a measure of dual-color PCH's ability to distinguish between one- and two-species systems. A contour plot of the reduced 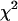 -values is shown in Fig. 2. Contours with reduced
-values is shown in Fig. 2. Contours with reduced 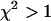 mark regions in which EGFP and mRFP1 can be resolved with a data acquisition time of 2 min. Contours with reduced
mark regions in which EGFP and mRFP1 can be resolved with a data acquisition time of 2 min. Contours with reduced 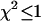 indicate regions in which EGFP and mRFP1 cannot be resolved with a data acquisition time of 2 min. To resolve the two species in these regions, data must be acquired for longer periods. Modeling predicts that the acquisition time needed to resolve EGFP and mRFP1 from a single histogram is 0.1 min for a concentration of
indicate regions in which EGFP and mRFP1 cannot be resolved with a data acquisition time of 2 min. To resolve the two species in these regions, data must be acquired for longer periods. Modeling predicts that the acquisition time needed to resolve EGFP and mRFP1 from a single histogram is 0.1 min for a concentration of 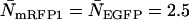 , whereas it is 0.8 min for a concentration of
, whereas it is 0.8 min for a concentration of 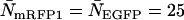 . For cells with
. For cells with 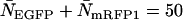 (a typical total concentration for our cellular measurements), resolving an EGFP/mRFP1 mixture from a single histogram requires a data acquisition time
(a typical total concentration for our cellular measurements), resolving an EGFP/mRFP1 mixture from a single histogram requires a data acquisition time 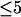 min as long as
min as long as 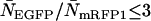 .
.
FIGURE 2.

Contour plot of the reduced 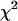 due to the misfit between a two-species dual-color PCH and a single-species model as function of
due to the misfit between a two-species dual-color PCH and a single-species model as function of 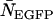 and
and 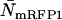 . Contours with
. Contours with 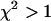 indicate regions in which EGFP and mRFP1 can be resolved, whereas contours with
indicate regions in which EGFP and mRFP1 can be resolved, whereas contours with 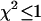 indicate regions in which they cannot. Favorable conditions for resolving the two species include low concentrations and more mRFP1 than EGFP. The number of data points used in the modeling was 2.6 × 106, corresponding to an acquisition time ∼2 min at a sampling frequency of 20 kHz.
indicate regions in which they cannot. Favorable conditions for resolving the two species include low concentrations and more mRFP1 than EGFP. The number of data points used in the modeling was 2.6 × 106, corresponding to an acquisition time ∼2 min at a sampling frequency of 20 kHz.
This modeling suggests that dual-color PCH analysis can successfully resolve this protein pair from a single histogram under a wide range of conditions. To experimentally confirm the modeling results, we coexpressed EGFP and mRFP1 in cells and performed dual-color PCH analysis. We found that we had to use twice more mRFP1 plasmid than EGFP during transfection to obtain similar expression levels of mRFP1 and EGFP. A cell expressing EGFP and mRFP1 was measured for ∼0.5 min and its fitted histogram is shown in Fig. 3 A. A fit to a single-species model returned a reduced 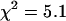 , whereas a two-species fit returned a reduced
, whereas a two-species fit returned a reduced 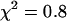 . The dual-color brightness values of the two species returned from the fit are shown in Fig. 3 B. The figure also shows the dual-color brightness values of cellular EGFP and mRFP1 measured individually as a control. Note that the fitted brightness values match the brightness values of EGFP and mRFP1 as expected. The average numbers of mRFP1 and EGFP molecules determined by the fit are
. The dual-color brightness values of the two species returned from the fit are shown in Fig. 3 B. The figure also shows the dual-color brightness values of cellular EGFP and mRFP1 measured individually as a control. Note that the fitted brightness values match the brightness values of EGFP and mRFP1 as expected. The average numbers of mRFP1 and EGFP molecules determined by the fit are 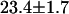 and
and 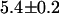 , respectively. We were able to resolve the two species in other cells using a single histogram. To our knowledge, only dual-color PCH and time-integrated fluorescence cumulant analysis (28) have successfully resolved a protein mixture from a single cellular measurement without any additional information.
, respectively. We were able to resolve the two species in other cells using a single histogram. To our knowledge, only dual-color PCH and time-integrated fluorescence cumulant analysis (28) have successfully resolved a protein mixture from a single cellular measurement without any additional information.
FIGURE 3.

(A) Experimental histogram from a cell coexpressing mRFP1 and EGFP and its fit to a two-species dual-color PCH model. (B) The brightness signatures returned from the fit match those obtained from cells expressing only mRFP1 or only EGFP. The number of molecules was 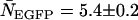 and
and 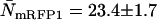 and the reduced
and the reduced 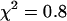 . A single-species model failed to describe the data (
. A single-species model failed to describe the data (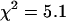 ). The data acquisition time was ∼0.5 min.
). The data acquisition time was ∼0.5 min.
EGFP-mRFP1 fusion protein
To model a heterodimer, we constructed a fusion protein of EGFP and mRFP1 in which the two fluorophores were joined by a 12-amino acid chain. We used lifetime, single, and dual-color brightness, and spectral analyses to characterize the fusion protein. We began with dual-channel FFS experiments on the EGFP-mRFP1 fusion protein and globally analyzed five cells expressing the EGFP-mRFP1 protein with a single-species model. In global PCH analysis, the brightness of each channel is linked across different data sets and the number of molecules is allowed to vary. To our surprise, the single-species model failed to describe the experimental dual-color PCHs (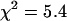 for five cells).
for five cells).
The dual-color PCH result suggests that the cells contain a mixture instead of a single species. We refit the five histograms globally to a two-species model with the brightness values of each species linked across the different data sets. The absolute value of the brightness and the number of molecules were allowed to vary. This model described the data within experimental error, and one of the brightness signatures returned by the fit was consistent with EGFP's brightness signature. This result led us to consider the following model: The cells contain a mixture of functional EGFP-mRFP1 together with a second-species EGFP-mRFP1†, where mRFP1† denotes a nonfluorescent mRFP1 protein. The brightness signature of the second species would be identical to that of EGFP. There are several possible explanations for the nonfluorescent mRFP1, including that the mRFP1 exists in dark and bright conformational states, that the construction of fusion proteins influences the fluorescence intensity of mRFP1 (29), or that the mRFP1 was photobleached in the cell selection process. We will explore these possibilities later in the article. For the moment, we will simply use a model in which some of the mRFP1 in the fusion protein is nonfluorescent without specifying the reasons for this behavior.
Using this model, we refit the histograms using a two-species model where one of the species' brightness signatures was fixed to EGFP and the other species' brightness signature was fixed to the FRET model in Eq. 2. The only inputs to the FRET model were the brightnesses of EGFP and mRFP1 in each channel. The FRET efficiency was fixed to 0.52 (see below), whereas the number of molecules of each species were allowed to vary. A global fit of the five cells returned an average fraction of fully functional fusion protein of 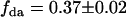 across the five cells. For simplicity, we will refer to the fraction of EGFP-mRFP1† as EGFP. A larger selection of cells measured on different days produced an average fraction of EGFP-mRFP1 of
across the five cells. For simplicity, we will refer to the fraction of EGFP-mRFP1† as EGFP. A larger selection of cells measured on different days produced an average fraction of EGFP-mRFP1 of 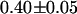 when the FRET efficiency was fixed to 0.52. Reanalysis of the data while allowing the FRET efficiency to vary resulted in an efficiency of 0.44.
when the FRET efficiency was fixed to 0.52. Reanalysis of the data while allowing the FRET efficiency to vary resulted in an efficiency of 0.44.
Next, we used fluorescence lifetime measurements to characterize cells expressing the fusion protein. A 505–535-nm bandpass filter was used to ensure that only the EGFP fluorescence emission is collected. We found that EGFP alone exhibited a monoexponential fluorescence decay with a lifetime of 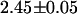 ns. The EGFP fluorescence from cells expressing the fusion protein exhibits a biexponential decay with an average lifetime of
ns. The EGFP fluorescence from cells expressing the fusion protein exhibits a biexponential decay with an average lifetime of 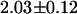 ns. Based upon the value for the average lifetime, we conclude that the apparent FRET efficiency between the EGFP and mRFP1 is 17%. However, one of the lifetimes in the fusion protein is very close to that of EGFP alone (
ns. Based upon the value for the average lifetime, we conclude that the apparent FRET efficiency between the EGFP and mRFP1 is 17%. However, one of the lifetimes in the fusion protein is very close to that of EGFP alone (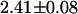 ns). We refit the decay curves, this time fixing one of the lifetimes to that obtained from EGFP alone (this led to an ∼5% increase in the reduced
ns). We refit the decay curves, this time fixing one of the lifetimes to that obtained from EGFP alone (this led to an ∼5% increase in the reduced 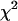 -values relative to the unrestrained fit), and obtained
-values relative to the unrestrained fit), and obtained 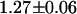 ns for the second lifetime. This result indicates that a fraction of the fusion proteins do not undergo FRET because some of the mRFP1 in the fusion proteins is not able to act as an acceptor. This is consistent with a model where only fluorescent mRFP1 acts as an acceptor. The EGFP lifetime in the fusion proteins with mRFP1† would remain unchanged, whereas those with mRFP1 would undergo FRET and exhibit a reduced lifetime. If we associate the second lifetime with EGFP-mRFP1, then the construct has a FRET efficiency of
ns for the second lifetime. This result indicates that a fraction of the fusion proteins do not undergo FRET because some of the mRFP1 in the fusion proteins is not able to act as an acceptor. This is consistent with a model where only fluorescent mRFP1 acts as an acceptor. The EGFP lifetime in the fusion proteins with mRFP1† would remain unchanged, whereas those with mRFP1 would undergo FRET and exhibit a reduced lifetime. If we associate the second lifetime with EGFP-mRFP1, then the construct has a FRET efficiency of 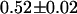 . Note that in our analysis, the preexponential amplitudes in the lifetime fit describe each species' fraction of the total population. Based upon the amplitudes of the two lifetime components, the fraction of fusion proteins with a functional mRFP1 is
. Note that in our analysis, the preexponential amplitudes in the lifetime fit describe each species' fraction of the total population. Based upon the amplitudes of the two lifetime components, the fraction of fusion proteins with a functional mRFP1 is 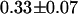 across 15 cells (Table 1). This is in excellent agreement with the value obtained via dual-color PCH analysis.
across 15 cells (Table 1). This is in excellent agreement with the value obtained via dual-color PCH analysis.
TABLE 1.
Summary of FRET measurements on the EGFP-mRFP1 fusion protein in cells
| Lifetime | Dual-color PCH | Spectra | |
|---|---|---|---|
| FRET efficiency | 0.52 ± 0.02 | 0.54 ± 0.05 | 0.52 |
| Fraction of EGFP-mRFP1 | 0.33 ± 0.07 | 0.40 ± 0.05 | 0.35 |
All three measurements agree within errors on the FRET efficiency of the fusion protein and on the fraction of EGFP-mRFP1 with fluorescent mRFP1; ∼60% of the fusion proteins did not show fluorescence of mRFP1.
Because of the large spectral separation of the fluorescence emission of EGFP and mRFP1, it is possible to selectively monitor just the EGFP or just the mRFP1 fluorescence of the fusion protein. This allows us to directly test whether our theory correctly describes the FRET-induced brightness changes of the donor and the acceptor (Eq. 1). We first investigated the donor EGFP using the same 505–535 nm bandpass filter employed in the fluorescence lifetime measurements to completely block mRFP1 emission. As a control, we measured the brightness of cells expressing EGFP alone (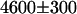 cpsm (counts per second per molecule)). Based upon our model for the EGFP-mRFP1 fusion protein (i.e., 60% EGFP-mRFP1† and 40% EGFP-mRFP1 with a FRET efficiency of E = 0.52), we expect an apparent brightness of EGFP in the mixture of
cpsm (counts per second per molecule)). Based upon our model for the EGFP-mRFP1 fusion protein (i.e., 60% EGFP-mRFP1† and 40% EGFP-mRFP1 with a FRET efficiency of E = 0.52), we expect an apparent brightness of EGFP in the mixture of 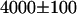 cpsm (Eq. 6). A measurement of the apparent brightness in cells expressing the fusion protein resulted in a brightness of
cpsm (Eq. 6). A measurement of the apparent brightness in cells expressing the fusion protein resulted in a brightness of 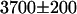 cpsm, which is in good agreement with the value predicted by our model.
cpsm, which is in good agreement with the value predicted by our model.
We then investigated the acceptor mRFP1 using a 605–645 nm bandpass filter to completely block the EGFP emission. As a control, we measured the brightness of cells expressing only mRFP1 and obtained a brightness of 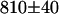 cpsm. Next we measured the mRFP1's brightness in cells expressing the fusion protein and obtained a brightness value of
cpsm. Next we measured the mRFP1's brightness in cells expressing the fusion protein and obtained a brightness value of 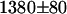 cpsm. This brightness value corresponds to a FRET efficiency of
cpsm. This brightness value corresponds to a FRET efficiency of 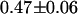 for the EGFP-mRFP1 fusion protein (Eq. 1), which is in good agreement with our dual-color PCH and lifetime measurements. This result also implies that all of the fluorescing mRFP1 are associated with an EGFP. If this were not the case, then the brightness returned would have been reduced relative to the expected value for
for the EGFP-mRFP1 fusion protein (Eq. 1), which is in good agreement with our dual-color PCH and lifetime measurements. This result also implies that all of the fluorescing mRFP1 are associated with an EGFP. If this were not the case, then the brightness returned would have been reduced relative to the expected value for 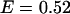 because it contains contributions from mRFP1 without FRET and mRFP1 with FRET. A mixture of these two species would produce an apparent brightness intermediate of the monomer mRFP1 brightness and that of mRFP1 in the EGFP-mRFP1 fusion protein and this was not observed.
because it contains contributions from mRFP1 without FRET and mRFP1 with FRET. A mixture of these two species would produce an apparent brightness intermediate of the monomer mRFP1 brightness and that of mRFP1 in the EGFP-mRFP1 fusion protein and this was not observed.
We also analyzed the apparent brightness spectrum obtained from cells expressing the fusion protein. To reproduce the apparent brightness spectrum shown in Fig. 4, we needed to use a composition of 65% EGFP-mRFP1† together with 35% EGFP-mRFP1 with 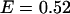 . The brightness spectrum of EGFP-mRFP1† is identical to the brightness spectrum of EGFP, and the brightness spectrum of EGFP-mRFP1 in the presence of FRET is calculated from a superposition of the brightness spectra of EGFP and mRFP1. A single-species model of EGFP-mRFP1 with 17% FRET efficiency described the EGFP contribution to the brightness spectrum but failed to describe the mRFP1 portion (Fig. 4). In fact, the apparent brightness spectrum could not be reproduced without including excess EGFP for any
. The brightness spectrum of EGFP-mRFP1† is identical to the brightness spectrum of EGFP, and the brightness spectrum of EGFP-mRFP1 in the presence of FRET is calculated from a superposition of the brightness spectra of EGFP and mRFP1. A single-species model of EGFP-mRFP1 with 17% FRET efficiency described the EGFP contribution to the brightness spectrum but failed to describe the mRFP1 portion (Fig. 4). In fact, the apparent brightness spectrum could not be reproduced without including excess EGFP for any 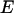 -value. Thus, the analysis of the brightness spectra is consistent with the dual-color PCH and fluorescence lifetime analysis (Table 1).
-value. Thus, the analysis of the brightness spectra is consistent with the dual-color PCH and fluorescence lifetime analysis (Table 1).
FIGURE 4.

Brightness spectrum 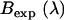 of EGFP-mRFP1 (solid black) expressed in COS cells. Also shown is the expected apparent brightness spectrum
of EGFP-mRFP1 (solid black) expressed in COS cells. Also shown is the expected apparent brightness spectrum 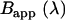 for a sample composed of 65% EGFP and 35% of fluorescent EGFP-mRFP1 with a 0.52 FRET efficiency (gray solid). A single-species model (
for a sample composed of 65% EGFP and 35% of fluorescent EGFP-mRFP1 with a 0.52 FRET efficiency (gray solid). A single-species model (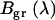 ) with
) with 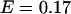 (dashed black) fails to describe the experimental spectrum in its entirety.
(dashed black) fails to describe the experimental spectrum in its entirety.
Dual-color PCH, fluorescence spectra, and lifetime analysis consistently indicate that our sample is best described by a mixture of functional EGFP-mRFP1 and of a second component with a nonfluorescent mRFP1, denoted as EGFP-mRFP1†. One of the possible explanations for the lower fraction of fluorescent mRFP1 in the fusion protein is photobleaching that occurs while selecting and centering the cell on the microscope stage in preparation for the two-photon experiments. It is known that mRFP1 is considerable less photostable than EGFP (29). So we selected and centered transfected cells in epifluorescence mode using an EGFP excitation filter to avoid the direct excitation of mRFP1. This process takes <10 s. However, photobleaching of mRFP1 might still occur because of energy transfer from EGFP. To rule out photobleaching via FRET, we monitored the two-photon fluorescence intensity ratio before and after exposing cells expressing the fusion protein to epifluorescence excitation for 10 s. Bleaching of mRFP1 would decrease the signal in the red channel, which alters the intensity ratio. We observed no change in the intensity ratio, which clearly demonstrates that photobleaching is not responsible for the observed presence of EGFP-mRFP1† in cells.
To check whether the problem we encountered with the fusion protein is due to dark states of the mRFP1 or due to improper folding of the mRFP1 in the fusion protein, we constructed an mRFP1-EGFP fusion protein and an mRFP1-mRFP1 fusion protein. The two explanations mentioned above lead to different predictions (discussed later) for the apparent brightness of the mRFP1-EGFP and the mRFP1-mRFP1 fusion proteins. The single-channel brightness of mRFP1-EGFP (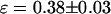 ) was identical within errors to EGFP-mRFP1's single-channel brightness (
) was identical within errors to EGFP-mRFP1's single-channel brightness (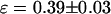 ). The mRFP1 homodimer had an apparent brightness
). The mRFP1 homodimer had an apparent brightness 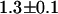 times larger than monomeric mRFP1.
times larger than monomeric mRFP1.
Resolving the fusion protein by dual-color PCH in the presence of photodepletion
In the analysis discussed above, we used global dual-color PCH analysis of several histograms to resolve EGFP-mRFP1† from EGFP-mRFP1. Each histogram was obtained from a different cell using ∼30 s data acquisition times. We wanted to test whether the two-species mixture can be resolved from a single measurement. So we measured cells expressing either EGFP, mRFP1, or the fusion protein for ∼5.5 min. To further improve the signal statistics, we increased the brightness by using a higher excitation power. The higher excitation power caused photobleaching as evidenced by a systematic decrease in the fluorescence intensity as a function of measurement time. The gradual decrease in fluorescence intensity is due to cumulative depletion of the finite amount of fluorophore present in the cell. mRFP1, for example, exhibited a ∼20% decrease in the fluorescence intensity over a 5 min period. When the entire data set was used to generate a histogram and this histogram was fit, we observed a bias in the dual-channel brightness values and in the concentration compared to the values obtained using a histogram based upon the first 0.5 min of data. The brightness increased and the number of molecules decreased compared to the result obtained for the short data acquisition time. This example illustrates the challenge of interpreting data in the presence of photodepletion. For cells expressing EGFP, we only observed a drop of ∼4% in the fluorescence intensity over a 5 min time period. This result is consistent with reports that state that mRFP1 is significantly more sensitive to photobleaching than EGFP (29). In fact, the amplitude of the intensity drop observed for EGFP is approaching the level of the intensity drifts observed when measuring cells over a few minutes. A fit of the histogram of the entire data set returned, within error, the same brightness and number of molecules as a histogram based on the first 0.5 min of the measurement. In other words, the effect of photobleaching on EGFP is negligible for the experimental conditions employed.
This leaves us with the question of how to analyze the mRFP1 measurements. Because of the decreasing fluorescence intensity, the process observed for mRFP1 in cells is not stationary anymore, which leads to biased parameters. However, if instead of treating the whole data set we only analyze a short data segment, then the drop in the fluorescence intensity is small enough that the data can be regarded as stationary with good approximation. We previously employed, successfully, a similar method when analyzing brightness data in cells (30). We divide the complete data set into 10 consecutive segments, each segment corresponding to 0.5 min of data acquisition. The histogram of each segment is fit individually to obtain the brightness and number of molecules for that segment. The results for mRFP1 are shown in Fig. 5. We see that the brightness of mRFP1 in each channel is constant as a function of acquisition time (Fig. 5 A), whereas the number of molecules decreases by 17% (Fig. 5 B). This result is consistent with our model, where depletion reduces the number of molecules but leaves the brightness of the remaining fluorophores unchanged. We also fit all 10 histograms globally to a single-species model where the brightness was linked between all 10 histograms (reduced 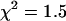 ). To ensure that photobleaching was not already introducing a bias over 0.5 min, we also determined the brightness values from 0.25 min of data and found they agreed within error with the values obtained over 0.5 min.
). To ensure that photobleaching was not already introducing a bias over 0.5 min, we also determined the brightness values from 0.25 min of data and found they agreed within error with the values obtained over 0.5 min.
FIGURE 5.

Data were acquired from a cell expressing mRFP1 for a period of 5.5 min. The data were divided into 10 consecutive segments, each 0.5 min long, and histogrammed. Each of the 10 histograms was fit to a single-species model to obtain the brightness in each channel (A) and the number of molecules (B) as a function of the data segment number (or data acquisition time). The brightness of mRFP1 is larger than that shown in Fig. 3 B due to the use of a higher excitation power. At this excitation power, some photobleaching of mRFP1 occurs. Thus we see a reduction in the number of molecules in consecutive histograms. However, the brightness is unaffected by the photobleaching and remains constant across the data acquisition time.
We now analyze the data taken from cells expressing the fusion protein in the same manner as the mRFP1 data. The data set was divided into 10 segments, and the resulting histograms were globally fit to a single species, where the brightness is linked across different histograms. The single-species model failed to describe the data (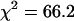 ), whereas a two-species model not only described the data (
), whereas a two-species model not only described the data (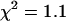 ) but returned brightnesses consistent with a EGFP-mRFP1 fusion protein exhibiting a 54% ± 5% FRET efficiency (Table 1) and with EGFP-mRFP1†, which has the same brightness as monomeric EGFP (Fig. 6 A). The outcome of this analysis agrees nicely with our earlier results, which were based on global PCH analysis of several cell measurements. The concentration of EGFP-mRFP1† increases as a function of acquisition time, whereas the EGFP-mRFP1 concentration decreases (Fig. 6 B). The initial EGFP-mRFP1 fraction of the cell is 42% (Table 1), but by the end of the data acquisition (5.5 min) the fraction is reduced to only 32% (Fig. 6 C). The EGFP-mRFP1† fraction on the other hand grows, because photobleaching of mRFP1 increases the EGFP-mRFP1† population. However, we gain less EGFP-mRFP1† population than we would expect based upon the drop in the EGFP-mRFP1 population, which indicates also some photobleaching of EGFP (∼10%). This result demonstrates that it is possible to directly resolve a mixture of EGFP-mRFP1† and EGFP-mRFP1 in cells from a single dual-color PCH measurement, although photobleaching complicates the analysis. The method described here provides a simple way to account for the depletion of fluorophores due to photobleaching.
) but returned brightnesses consistent with a EGFP-mRFP1 fusion protein exhibiting a 54% ± 5% FRET efficiency (Table 1) and with EGFP-mRFP1†, which has the same brightness as monomeric EGFP (Fig. 6 A). The outcome of this analysis agrees nicely with our earlier results, which were based on global PCH analysis of several cell measurements. The concentration of EGFP-mRFP1† increases as a function of acquisition time, whereas the EGFP-mRFP1 concentration decreases (Fig. 6 B). The initial EGFP-mRFP1 fraction of the cell is 42% (Table 1), but by the end of the data acquisition (5.5 min) the fraction is reduced to only 32% (Fig. 6 C). The EGFP-mRFP1† fraction on the other hand grows, because photobleaching of mRFP1 increases the EGFP-mRFP1† population. However, we gain less EGFP-mRFP1† population than we would expect based upon the drop in the EGFP-mRFP1 population, which indicates also some photobleaching of EGFP (∼10%). This result demonstrates that it is possible to directly resolve a mixture of EGFP-mRFP1† and EGFP-mRFP1 in cells from a single dual-color PCH measurement, although photobleaching complicates the analysis. The method described here provides a simple way to account for the depletion of fluorophores due to photobleaching.
FIGURE 6.

Cell expressing EGFP-mRFP1 was measured for a period of 5.5 min. The data were subsequently divided into 10 segments and 10 histograms were formed. The histograms were globally fit to a two-species model, and the brightness values returned by the fit (⋄) are shown in A and indicate that one is EGFP-mRFP1† (□) and the other is EGFP-mRFP1 (▵) with 54% FRET efficiency. The number of molecules of EGFP-mRFP1† and EGFP-mRFP1 as a function time are shown in B. The lines indicate the error in the concentration. The EGFP-mRFP1† concentration increases as a function of time, whereas the EGFP-mRFP1 concentration decreases because the mRFP1 in the EGFP-mRFP1 fusion protein is being photobleached. (C) The fraction of fully fluorescent EGFP-mRFP1 as a function of time. The initial fraction of EGFP-mRFP1 was 42%, in good agreement with our other measurements.
More importantly, we see that acquiring data from a single cell over longer periods in the presence of photobleaching actually offers an advantage over measuring multiple cells over shorter periods. Recall that we obtained a reduced 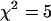 using multiple short histograms from different cells, whereas we obtained a reduced
using multiple short histograms from different cells, whereas we obtained a reduced 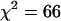 using histograms obtained from the same cell at different times. Adding more short histograms from different cells did not appreciably increase the reduced
using histograms obtained from the same cell at different times. Adding more short histograms from different cells did not appreciably increase the reduced 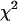 . This difference arises because photobleaching systematically varies the composition of the sample. Different fluorophores typically have different photobleaching behavior, so if more than one type of fluorophore is present, a predominant decrease in one of the species is observed, whereas the other is much less affected. The brightness of the surviving population is unaffected. Thus, global analysis of the data as described above provides a sensitive tool for identifying populations by the time-dependent changes in their population.
. This difference arises because photobleaching systematically varies the composition of the sample. Different fluorophores typically have different photobleaching behavior, so if more than one type of fluorophore is present, a predominant decrease in one of the species is observed, whereas the other is much less affected. The brightness of the surviving population is unaffected. Thus, global analysis of the data as described above provides a sensitive tool for identifying populations by the time-dependent changes in their population.
DISCUSSION
It is our goal to develop dual-color PCH into a quantitative tool for studying protein interactions inside cells. We previously showed that spectral cross talk between fluorophores complicates the resolution of species by dual-color PCH. Thus, we chose EGFP and mRFP1 as the fluorescence pair because of their large spectral separation. Dual-color PCH successfully resolved mixtures of these two proteins inside cells from single measurements. To characterize FRET effects on the dual-color PCH, we cloned a EGFP-mRFP1 fusion protein. Dual-color PCH analysis unexpectedly showed that cells expressing the fusion protein are best described as a mixture where one component represents the fusion protein EGFP-mRFP1 and the other has the same brightness signature as EGFP, which we interpreted as a fusion protein without a fluorescing mRFP1 fluorophore, EGFP-mRFP1†. Note that mRFP1† simply denotes nonfluorescent mRFP1, irrespective of the underlying mechanism. The results from fluorescence lifetime and fluorescence spectra analysis agree with the dual-color PCH analysis.
The ability to resolve a mixture is important if we want to quantify protein-interactions in cells. Here we investigated the potential of the EGFP/mRFP1 pair for this purpose. We resolved mixtures of EGFP and mRFP1 coexpressed in cells quantitatively from a single PCH measurement. We also demonstrated that it is possible to resolve a mixture of EGFP-mRFP1† (which has the same fluorescent properties as EGFP) and EGFP-mRFP1 in cells with a single measurement. This mixture was obtained inadvertently due to the properties of mRFP1 in the fusion protein. We did not experimentally investigate mixtures of mRFP1 and EGFP-mRFP1. Instead, we modeled mRFP1/EGFP-mRFP1 mixtures by calculating dual-color PCHs based upon the measured brightness values and concentrations observed in our cell measurements. By fitting these calculated histograms to a single-species model, we found that the simulated mRFP1/EGFP-mRFP1 mixtures are easier to resolve than EGFP/EGFP-mRFP1 mixtures. Thus, the brightness properties of EGFP and mRFP1 are suitable for the characterization of protein interactions in cells. The widely used CFP/YFP pair is inferior to EGFP/mRFP1 when resolving species experimentally by PCH analysis (unpublished observations). The advantage of the EGFP/mRFP1 pair stems from its considerably smaller spectral overlap compared to the CFP/YFP pair. Unfortunately, the presence of a nonfluorescent population of mRFP1 considerably complicates its practical use. Recently, improved versions of mRFP1 have been introduced that are more robust in fusion proteins than the original mRFP1 (29). We plan to characterize the potential of mCherry and other red fluorescent proteins to serve as a suitable marker for dual-color PCH analysis. In addition, the original mRFP1 is very dim, which limits the ability of PCH to distinguish brightness species. mCherry and other red fluorescent proteins are brighter than the original mRFP1. This increase in brightness will boost the sensitivity of PCH analysis to resolve components inside cells. Our results show that a well-behaved red fluorescent protein together with EGFP would provide a promising pair for dual-color PCH analysis of protein-protein interaction in cells.
It has been observed that some fluorescent proteins switch between bright and dark conformations (31,32). The kinetics of dark states falls into three categories: i), blinking occurs on a timescale faster than the sampling time, ii), blinking occurs during the diffusion time of the molecule through the observation volume, and iii), blinking occurs on timescales larger than the diffusion time. We ignore case i because we would observe an averaged brightness with no consequences for our data analysis. We exclude case ii because blinking was not observed in the autocorrelation function of EGFP or mRFP1 (data not shown). Case iii requires further discussion. Because the blinking rate is very infrequent with respect to the diffusion time, we can treat the sample as a mixture of two static species, one bright and the other dark. The presence of a dark species is not detectable for monomeric mRFP1, because no signal is produced as the protein passes through the observation volume. If mRFP1 is linked to another fluorescent protein, such as EGFP, the dark fraction of mRFP1 reveals itself as a fusion protein where only EGFP fluoresces. Thus the presence of a dark species leads to experimentally testable hypothesis. For example, a homodimer of a protein with dark states would lead to four populations (dark-dark, bright-bright, dark-bright, and bright-dark). It is straightforward to calculate that the apparent single-channel brightness 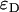 of a homodimer with dark states is given by
of a homodimer with dark states is given by 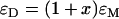 , where
, where 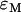 is the brightness of the monomer and
is the brightness of the monomer and 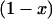 is the dark population fraction of the protein. We previously showed that the brightness of dimeric EGFP doubles, thus the presence of dark states in EGFP is negligible under our experimental conditions (11).
is the dark population fraction of the protein. We previously showed that the brightness of dimeric EGFP doubles, thus the presence of dark states in EGFP is negligible under our experimental conditions (11).
A long-lived dark state of mRFP1 could explain the observed nonfluorescent fraction of the mRFP1-EGFP fusion protein. On the other hand, it has been reported that the fluorescence signal of mRFP1 in fusion proteins can be reduced, particularly when the partner protein is linked to mRFP1's N-terminus (29). Studying the brightness of mRFP1-EGFP in addition to EGFP-mRFP1 distinguishes between the two possibilities. In this case, changing the terminus of the mRFP1 that is fused to EGFP should change the population of nonfluorescent mRFP1. This shift in populations would be evidenced by a difference in the apparent brightnesses of EGFP-mRFP1 and mRFP1-EGFP. However, we observed the same brightness (within error) for both constructs, which is consistent with the dark state model. In addition, if the nonfluorescent mRFP1 is due to a dark state, then we would also expect that the apparent brightness of dimeric mRFP1 would be ∼1.4 times the brightness of monomeric mRFP1, to account for the 60% mRFP1 molecules in the dark configuration. The measured brightness of mRFP1-mRFP1 is 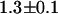 times the brightness of monomeric mRFP1. Thus, the data are consistent with a dark state population of 60%.
times the brightness of monomeric mRFP1. Thus, the data are consistent with a dark state population of 60%.
The formation of protein complexes may bring fluorophores in close enough proximity that FRET occurs. We formulated a theory of FRET's effect on the dual-channel brightnesses and incorporated this into our dual-color PCH model. This model requires the FRET efficiency and the ratio of the donor's cross section to the acceptor's cross section as input. We measured changes in the donor lifetime to determine the FRET efficiency and calculated the cross-sectional ratio from the emission spectra and the optical properties of the microscope. The FRET theory was verified by observing the changes in the brightness values of the donor and acceptor in the presence of FRET. In addition, lifetime, spectral, and dual-color PCH analysis return within error the same FRET efficiency, which provides another consistency check of the model.
Dual-color PCH, lifetime, and spectral analyses all pointed to ∼40% functional EGFP-mRFP1 fusion protein, whereas the rest lacks mRFP1 fluorescence; this result is best explained by a spectroscopic dark state of mRFP1. Thus, dual-color PCH provides an alternative approach to check the integrity of fluorescent protein constructs and identify the presence of dark states. A single species was not sufficient to describe the dual-color histograms of cells expressing the fusion protein, indicating the presence of a mixture of species. Analysis of dual-color PCH in the presence of depletion due to photobleaching even allowed us to directly resolve the mixture from a single measurement. This analysis also provided superior resolution capabilities than using histograms obtained from several cells over shorter acquisition times, as judged by the 10-fold increase in the reduced 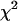 . Our result also highlights the importance of characterizing the behavior of fluorescent proteins in fusion constructs and for combining different fluorescence measurement modalities to detect problems with fluorescent proteins. Failure to do so may result in a biased interpretation of data. For example, calculating the FRET efficiency of our mRFP1-EGFP sample solely based on fluorescence intensity or fluorescence lifetime measurements would yield different and biased values. The problem with the sample only reveals itself after observing the mismatch of the FRET efficiency determined by both methods. Yet the majority of quantitative cellular FRET experiments that are reported in the literature only employ either donor lifetime or fluorescence intensity measurements but not both, and thus would fail to identify the heterogeneity of the sample.
. Our result also highlights the importance of characterizing the behavior of fluorescent proteins in fusion constructs and for combining different fluorescence measurement modalities to detect problems with fluorescent proteins. Failure to do so may result in a biased interpretation of data. For example, calculating the FRET efficiency of our mRFP1-EGFP sample solely based on fluorescence intensity or fluorescence lifetime measurements would yield different and biased values. The problem with the sample only reveals itself after observing the mismatch of the FRET efficiency determined by both methods. Yet the majority of quantitative cellular FRET experiments that are reported in the literature only employ either donor lifetime or fluorescence intensity measurements but not both, and thus would fail to identify the heterogeneity of the sample.
CONCLUSION
We successfully applied dual-color PCH analysis to resolve mixtures of species in living cells from a single measurement. All data are corrected for nonideal detector effects and we included the autofluorescence background in the analysis of cells with low fluorescence intensities. We chose EGFP and mRFP1 as our model system to determine the feasibility of the dual-color PCH technique in cells. We succeeded in resolving mixtures of EGFP and mRFP1 with a single histogram. In addition, we studied an EGFP-mRFP1 fusion protein to investigate the influence of FRET on dual-color brightness values. The behavior of the fusion protein was more complex than anticipated. We concluded from our dual-color PCH, fluorescence lifetime, and spectral measurements that some of the mRFP1 was nonfluorescent in the EGFP-mRFP1 fusion protein. The results of all methods are consistent with this interpretation. We determined the fractional population of the fully fluorescent fusion protein and its FRET efficiency, and resolved the two fusion protein populations from a single dual-color PCH measurement. We conclude that dual-color PCH analysis offers a sensitive method to characterize fusion proteins by testing whether a single species is sufficient to describe the data. Our results show that EGFP and mRFP1 represent a suitable pair for resolving species by dual-color PCH in cells. However, the nonfluorescent population of mRFP1, which we interpret as a dark state of the protein, complicates the analysis considerably. Improved versions of mRFP1 are available and might provide better spectroscopic properties for brightness analysis (29). In summary, our results show that dual-color PCH analysis of a green/red fluorescent protein pair has the potential to quantitatively resolve interacting protein species in cells.
Acknowledgments
This work was supported by grants from the National Institutes of Health (GM64589) and National Science Foundation (MCB-0110831) and by a Doctoral Dissertation Fellowship from the University of Minnesota.
References
- 1.Bacia, K., I. V. Majoul, and P. Schwille. 2002. Probing the endocytic pathway in live cells using dual-color fluorescence cross-correlation analysis. Biophys. J. 83:1184–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Camacho, A., K. Korn, M. Damond, J. F. Cajot, E. Litborn, B. Liao, P. Thyberg, H. Winter, A. Honegger, P. Gardellin, and R. Rigler. 2004. Direct quantification of mRNA expression levels using single molecule detection. J. Biotechnol. 107:107–114. [DOI] [PubMed] [Google Scholar]
- 3.Kim, S. A., K. G. Heinze, K. Bacia, M. N. Waxham, and P. Schwille. 2005. Two-photon cross-correlation analysis of intracellular reactions with variable stoichiometry. Biophys. J. 88:4319–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kim, S. A., K. G. Heinze, M. N. Waxham, and P. Schwille. 2004. Intracellular calmodulin availability accessed with two-photon cross-correlation. Proc. Natl. Acad. Sci. USA. 101:105–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Saito, K., I. Wada, M. Tamura, and M. Kinjo. 2004. Direct detection of caspase-3 activation in single live cells by cross-correlation analysis. Biochem. Biophys. Res. Commun. 324:849–854. [DOI] [PubMed] [Google Scholar]
- 6.Thews, E., M. Gerken, R. Eckert, J. Zapfel, C. Tietz, and J. Wrachtrup. 2005. Cross talk free fluorescence cross correlation spectroscopy in live cells. Biophys. J. 89:2069–2076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Heinze, K. G., A. Koltermann, and P. Schwille. 2000. Simultaneous two-photon excitation of distinct labels for dual-color fluorescence crosscorrelation analysis. Proc. Natl. Acad. Sci. USA. 97:10377–10382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen, Y., M. Tekmen, L. Hillesheim, J. Skinner, B. Wu, and J. D. Müller. 2005. Dual-color photon-counting histogram. Biophys. J. 88:2177–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kask, P., K. Palo, N. Fay, L. Brand, U. Mets, D. Ullmann, J. Jungmann, J. Pschorr, and K. Gall. 2000. Two-dimensional fluorescence intensity distribution analysis: theory and applications. Biophys. J. 78:1703–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen, Y., L. N. Wei, and J. D. Müller. 2005. Unraveling protein-protein interactions in living cells with fluorescence fluctuation brightness analysis. Biophys. J. 88:4366–4377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen, Y., L. N. Wei, and J. D. Müller. 2003. Probing protein oligomerization in living cells with fluorescence fluctuation spectroscopy. Proc. Natl. Acad. Sci. USA. 100:15492–15497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hillesheim, L. N., and J. D. Müller. 2005. The dual-color photon counting histogram with non-ideal photodetectors. Biophys. J. 89:3491–3507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clegg, R. M. 1996. Fluorescence resonance energy transfer. In Fluorescence Imaging Spectroscopy and Microscopy. X. F. Wang and B. Herman, editors. John Wiley & Sons, New York. 179–252.
- 14.Lakowicz, J. R. 1999. Principles of Fluorescence Spectroscopy. Plenum Press, New York.
- 15.van Thor, J. J., and K. J. Hellingwerf. 2002. Fluorescence resonance energy transfer applications using green fluorescent protein. Methods Mol. Biol. 183:101–119. [DOI] [PubMed] [Google Scholar]
- 16.Chen, Y., J. D. Müller, S. Y. Tetin, J. D. Tyner, and E. Gratton. 2000. Probing ligand protein binding equilibria with fluorescence fluctuation spectroscopy. Biophys. J. 79:1074–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Müller, J. D. 2004. Cumulant analysis in fluorescence fluctuation spectroscopy. Biophys. J. 86:3981–3992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu, B., and J. D. Muller. 2005. Time-integrated fluorescence cumulant analysis in fluorescence fluctuation spectroscopy. Biophys. J. 89:2721–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hillesheim, L. N., and J. D. Müller. 2003. The photon counting histogram in fluorescence fluctuation spectroscopy with non-ideal photodetectors. Biophys. J. 85:1948–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hillesheim, L. N. 2005. Dual-color photon counting histogram analysis: theory and application in living cells. PhD thesis. University of Minnesota, Minneapolis, Minnesota.
- 21.Blab, G. A., P. H. M. Lommerse, L. Cognet, G. S. Harms, and T. Schmidt. 2001. Two-photon excitation action cross-sections of the autofluorescent proteins. Chem. Phys. Lett. 350:71–77. [Google Scholar]
- 22.O'Connor, D. V., and D. Phillips. 1984. Time-Correlated Single Photon Counting. Academic Press, New York.
- 23.Lifetime data of selected fluorophores. 2005. http://www.iss.com/Resources/fluorophores.html
- 24.Denk, W., J. H. Strickler, and W. W. Webb. 1990. Two-photon laser scanning fluorescence microscopy. Science. 248:73–76. [DOI] [PubMed] [Google Scholar]
- 25.Patterson, G., R. N. Day, and D. Piston. 2001. Fluorescent protein spectra. J. Cell Sci. 114:837–838. [DOI] [PubMed] [Google Scholar]
- 26.Campbell, R. E., O. Tour, A. E. Palmer, P. A. Steinbach, G. S. Baird, D. A. Zacharias, and R. Y. Tsien. 2002. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA. 99:7877–7882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zipfel, W. R., R. M. Williams, and W. W. Webb. 2003. Nonlinear magic: multiphoton microscopy in the biosciences. Nat. Biotechnol. 21:1369–1377. [DOI] [PubMed] [Google Scholar]
- 28.Wu, B., Y. Chen and J. D. Mueller. 2006. Dual-color time-integrated fluorescence cumulant analysis. Biophys. J. 2687–2698. [DOI] [PMC free article] [PubMed]
- 29.Shaner, N. C., R. E. Campbell, P. A. Steinbach, B. N. G. Giepmans, A. E. Palmer, and R. Y. Tsien. 2004. Improved monomeric red, orange and yellow fluorescent proteins derived from discosoma sp red fluorescent protein. Nat. Biotechnol. 22:1567–1572. [DOI] [PubMed] [Google Scholar]
- 30.Chen, Y., J. D. Müller, Q. Ruan, and E. Gratton. 2002. Molecular brightness characterization of EGFP in vivo by fluorescence fluctuation spectroscopy. Biophys. J. 82:133–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heikal, A. A., S. T. Hess, G. S. Baird, R. Y. Tsien, and W. W. Webb. 2000. Molecular spectroscopy and dynamics of intrinsically fluorescent proteins: coral red (dsRed) and yellow (citrine). Proc. Natl. Acad. Sci. USA. 97:11996–12001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schwille, P., S. Kummer, A. A. Heikal, W. E. Moerner, and W. W. Webb. 2000. Fluorescence correlation spectroscopy reveals fast optical excitation-driven intramolecular dynamics of yellow fluorescent proteins. Proc. Natl. Acad. Sci. USA. 97:151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]