

Abstract
Unlike other restriction enzymes, BfiI functions without metal ions. It recognizes an asymmetric DNA sequence, 5′-ACTGGG-3′, and cuts top and bottom strands at fixed positions downstream of this sequence. Many restriction enzymes are dimers of identical subunits, with one active site for each DNA strand. Others, like FokI, dimerize transiently during catalysis. BfiI is also a dimer but it has only one active site, at the dimer interface. We show here that BfiI remains a dimer as it makes double-strand breaks in DNA and that its single active site acts sequentially, first on the bottom and then the top strand. Hence, after cutting the bottom strand, a rearrangement of either the protein and/or the DNA in the BfiI–DNA complex must switch the active site to the top strand. Low pH values selectively block top-strand cleavage, converting BfiI into a nicking enzyme that cleaves only the bottom strand. The switch to the top strand may depend on the ionization of the cleaved 5′ phosphate in the bottom strand. BfiI thus uses a mechanism for making double-strand breaks that is novel among restriction enzymes.
Sequence-specific cleavage of double-stranded DNA underpins many genetic events, including recombination, transposition, viral DNA integration, and the restriction of DNA (1–4). The enzymes that make double-strand breaks use many different strategies to achieve this end. The simplest may be the orthodox type II restriction enzymes such as EcoRI or EcoRV, dimers of identical subunits that recognize palindromic DNA sequences. In the presence of Mg2+, they cleave both strands at fixed positions in their recognition sites (3). They bind their target sites symmetrically, so that one active site from the dimer is positioned against one DNA strand and likewise the second active site on the other strand. Independent reactions in each active site then generate the double-strand break. Some homing endonucleases, such as I-CreI, also act in this way (4), as do the enzymes that resolve Holliday junctions (2).
This strategy cannot apply to enzymes that recognize nonpalindromic sites and cut both strands away from the recognition site (3). For example, FokI, a type IIS restriction enzyme, recognizes the sequence 5′-GGATG-3′ and cuts top and bottom strands 9 and 13 nt downstream of this site, respectively (5). FokI is a monomer with separate domains for DNA recognition and catalysis. The catalytic domain has a single active site, which is like that in each subunit of an orthodox enzyme, so the FokI monomer cannot cleave both DNA strands (6). To make double-strand breaks, the monomer of FokI bound to its recognition site associates transiently with a second monomer to form a dimer with two active sites (7–9). Many type IIS enzymes operate in this way (10, 11).
An alternative to using a homodimeric enzyme to make double-strand breaks is an enzyme with different subunits. For example, the transposition of Tn7 requires two proteins, TnsA and TnsB, that each cleave one strand of the DNA (12). Interestingly, the structure of TnsA is similar to a single subunit of a type II restriction enzyme (13).
Although double-strand breaks in DNA are commonly made by oligomeric enzymes with one active site in each subunit, this is by no means universal. For instance, the PI-SceI homing endonuclease has two active sites within a single polypeptide chain, one for each strand (14, 15). Conversely, many transposases use a mechanism originally found for V(D)J recombination (16), where a single active site acts on both strands. In this active site, first one strand is cut to leave a 3′ hydroxyl, which then attacks the other strand to generate a hairpin intermediate, which is subsequently resolved before strand transfer to the target DNA (17–19). The hairpin intermediate thus enables one active site to catalyze both double-strand cleavage and further DNA integration. Even so, hairpins are probably not essential to achieve a double-strand break by a monomeric enzyme with one active site. The intron-encoded endonuclease I-TevI is a monomer with one active site and it has been suggested that, after it has cleaved the bottom strand, it distorts the DNA to access the top strand (20). However, this scheme has yet to be confirmed: given the crystal structure of the catalytic domain of I-TevI, both transient dimerization and hairpin schemes are also possible (21).
All of the enzymes noted above use divalent metal ions as cofactors in their DNA cleavage reactions. In contrast, the BfiI restriction endonuclease does not require metal ions: it can cleave DNA in the presence of EDTA (22). BfiI must therefore hydrolyze phosphodiester bonds by a radically different mechanism from all restriction enzymes characterized to date (22). Like FokI (5), BfiI is a type IIS endonuclease: it recognizes an asymmetric sequence, ACTGGG and cleaves top and bottom strands 5 and 4 nt downstream of this site (23). Sequence alignments and site-directed mutagenesis (22, 24) indicate that the N-terminal half of BfiI is similar to an EDTA-resistant DNase that lacks sequence specificity, Nuc of Salmonella typhimurium (25). Nuc, a member of the phospholipase D superfamily, is a homodimer in solution but its crystal structure shows only one active site, at the subunit interface (26). Like Nuc, BfiI is a dimer in solution and also contains just one active site (24). This study reveals how the single active site in the BfiI dimer cuts both DNA strands.
Experimental Procedures
Proteins. WT BfiI and the K107A mutant were purified as described (24). Concentrations were determined from A280 readings using an extinction coefficient of 95,420 M–1·cm–1 for the BfiI dimer.
Oligonucleotides. Some duplexes were made by annealing two oligodeoxyribonucleotides with complementary sequences, both 30 nt long (Table 1). For other duplexes, a 30-nt oligonucleotide was annealed to one of 18 nt and one of 12 nt, or to just one of 18 nt (Table 1). The 18-nt oligonucleotide that forms part of the bottom strand in the 30/(18P_12) duplex was phosphorylated at its 5′ end by T4 polynucleotide kinase (PNK). For 30/(18P_dd12), terminal polynucleotidyl transferase (TdT) and ddATP were used to place a dideoxynucleotide at the 3′ end of the 12-nt oligonucleotide. Oligonucleotides were 5′ labeled with [γ-33P]ATP and PNK or 3′ labeled with [α-32P]ddATP and TdT. The labeled strand is noted by an asterisk: for example, *30/30 and 30/30* carry the label in top and bottom strands, respectively. DNA modifying enzymes were from Fermentas (Vilnius, Lithuania), [γ-33P]ATP and [α-32P]ddATP were from Amersham Pharmacia, and oligonucleotides were from Metabion (Martinsried, Germany).
Table 1. Oligonucleotide substrates.
| Duplex | Sequence | Specification |
|---|---|---|
| 30/30 | 5′-AGCGTAGCACTGGGCTGCTGAACTGTGCTG-3′ | 30-bp cognate substrate for Bfil (recognition sequence underlined); Bio-30/30 has the same sequence, as shown, but with a biotin tag at the 5′ end of its top strand |
| Bio-30/30 | 3′-TCGCATCGTGACCCGACGACTTGACACGAC-5′ | |
| 30/30(S) | 5′-AGCGTAGCACTGGGCTGC-TGAACTGTGCTG-3′ | As 30/30 except for a phosphorothioate linkage at the position indicated by s, the site of bottom-strand cleavage by Bfil |
| 3′-TCGCATCGTGACCCGACGsACTTGACACGAC-5′ | ||
| 30(S)/30 | 5′-AGCGTAGCACTGGGCTGCTsGsAsACTGTGCTG-3′ | As 30/30 except for phosphorothioate linkages at the three positions indicated by s, the sites of top-strand cleavage by Bfil |
| 3′-TCGCATCGTGACCCGACGA-C-T-TGACACGAC-5′ | ||
| 30/(18P_12) | 5′-AGCGTAGCACTGGGCTGC-TGAACTGTGCTG-3′ | Duplex made from three oligos to give a nicked DNA akin to the Bfil product on the bottom strand of 30/30; the nucleotide 5′ to the nick is phosphorylated, to mimic the terminus left by Bfil |
| 3′-TCGCATCGTGACCCGACGp ACTTGACACGAC-5′ | ||
| 30/18P | 5′-AGCGTAGCACTGGGCTGCTGAACTGTGCTG-3′ | Analogous to 30/(18P_12) except made from two oligos to give a 3′-tailed duplex |
| 3′-TCGCATCGTGACCCGACGp-5′ | ||
| 30/(18P_dd12) | 5′-AGCGTAGCACTGGGCTGC-TGAACTGTGCTG-3′ | As 30/(18P_12) except that the nucleotide 3′ to the nick is replaced by a dideoxynucleotide (indicated by dd) |
| 3′-TCGCATCGTGACCCGACGpddACTTGACACGAC-5′ | ||
| 30/(18Δ_12) | 5′-AGCGTAGCACTGGGCTGC-TGAACTGTGCTG-3′ | As 30/(18P_12), except that the nucleotide 5′ to the nick is not phosphorylated |
| 3′-TCGCATCGTGACCCGACGOH ACTTGACACGAC-5′ |
Reactions. Reactions were typically carried out by adding BfiI (0.01–50 nM dimer) to 3.0 nM labeled duplex in 30 mM buffer, 110 mM potassium acetate, and 10 μg/ml BSA at 25°C. The buffer was Mes/KOH (for reactions at pH 6.5–6.8), Tris-acetate (for reactions at pH 7–8.5), or glycine/KOH (for reactions at pH 9–9.5). Aliquots were removed at timed intervals and quenched with phenol/chloroform. The aqueous phase was mixed with loading dye (95% vol/vol formamide, 0.01% bromophenol blue) before denaturing gel electrophoresis through 20% polyacrylamide (in Tris-borate) at 30 V/cm. Radiolabeled DNA was detected and quantified by using a Cyclone Storage Phosphor System and OPTIQUANT 3.0 software (Perkin–Elmer).
Immobilized Oligonucleotides. Streptavidin-coated magnetic beads (Promega, binding capacity ≈1 nmol/mg) were added to 80 μl of either bio-30*/30 or bio-30/30* (4 nM) in 0.5× SCC buffer (75 mM NaCl/7.5 mM sodium citrate, pH 7.2) to a final concentration of 0.12 mg/ml, to give ≈10 pmol biotin-binding sites on the beads for ≈0.32 pmol biotinylated oligonucleotide. After 5 min at 25°C, the beads were washed twice with 0.5× SCC buffer. The amount of labeled DNA in the supernatant was used to estimate the amount on the beads: typically, >70% of the labeled duplex was captured. The beads were resuspended in pH 8.0 reaction buffer before adding BfiI to a final concentration in the range from 5 to 50 nM: cleavage of the immobilized DNA was then monitored as above. In other experiments, magnetic beads carrying a bio-30*/30 duplex were resuspended in 60 μl of pH 6.5 reaction buffer containing 50 nM BfiI. After 5 min at 25°C, unbound BfiI was removed by washing the beads twice with the pH 6.5 buffer (in control experiments, BfiI did not bind to the beads by itself). Finally, the beads were resuspended in 60 μl of pH 8.0 reaction buffer before monitoring top DNA strand cleavage. Alternatively, the beads were suspended in pH 8.0 buffer containing the K107A mutant of BfiI at 100 nM.
Data Analysis. Rate constants for single turnovers were determined by fitting to single exponentials. Steady-state rates were determined by linear regression. Data analysis used KYPLOT 2.0 software (27).
Results
Sequential Nicking. The BfiI endonuclease cleaves supercoiled plasmids that have one or two copies of its recognition site to give first the open-circle form of the DNA, by cleaving the recognition site(s) in one strand: only later does it generate the linear DNA products from cutting both strands at the same site (24). However, it was not determined whether one particular strand was cleaved before the other.
To see whether BfiI acts first on a particular strand, single-turnover reactions were performed with a 30-bp duplex that has the recognition site for BfiI, 30/30 (Table 1). The duplex was 5′ end labeled with 33P in either top or bottom strand to give, respectively, *30/30 and 30/30*: the former reveals the cleavage of the top strand and the latter the bottom strand. The bottom strand was cleaved much more rapidly than the top strand (Fig. 1A). At pH 8.0, the half-time (t1/2) for cutting the bottom strand, 0.8 min, was ≈10 times faster than that for cutting the top strand, 7 min. The difference between the rates on bottom and top strands was even more pronounced at pH 6.5 (Fig. 1 A). At pH 6.5, the rate for cutting the bottom strand (t1/2 ≈ 0.3 min) was about twice as fast as that at pH 8.0 but top-strand cleavage became extremely slow (t1/2 > 100 min). Reduced pH thus selectively blocks top-strand cleavage and effectively transforms BfiI into a site-specific nicking enzyme that acts only on the bottom strand.
Fig. 1.

Cleavage of top and bottom strands. The cartoons above A and B illustrate the DNA substrates used: arrows mark BfiI cleavage sites. (A) The reactions at 25°C contained 3.0 nM duplex, either *30/30 or 30/30*, and 50 nM BfiI dimer, in pH 8.0 or pH 6.5 buffer. At pH 8.0: •, top-strand cleavage (from *30/30); ○, bottom-strand cleavage (from 30/30*). At pH 6.5: ▪, top-strand cleavage; □, bottom-strand cleavage. The solid lines are exponential fits to the data. (B) Reactions at pH 8.0 were as in A except that 30/30 duplex was replaced by 30/30(S) or 30(S)/30, which contain a phosphorothioate at the BfiI cleavage site of the bottom or the top strand, respectively. The 30/30(S) duplex was 5′ end labeled either in the top or the bottom strand: ♦, cleavage of top strand of 30/30(S); ⋄, cleavage of bottom strand of 30/30(S). □, cleavage of the 30(S)/30 duplex 5′ end labeled in the bottom strand. Also shown again here are the pH 8.0 reactions on top (•) and bottom (○) strands of the native substrate, 30/30. The arrows indicate changes in DNA cleavage profiles caused by the bottom-strand modification.
In addition to the previously reported site of action of BfiI on the top strand, 5 nt downstream of the recognition sequence (23), additional cleavages of the top strand were also detected 6 and 7 nt away from the site: they constituted 30% and 10% of the reaction products, respectively (data not shown). The same pattern of top-strand cleavage was observed with two other 30-bp duplexes that have the recognition sequence for BfiI flanked by different sequences, but they were all cleaved in the bottom strand only at the cognate position 4 nt away. A 30-bp duplex that lacks the BfiI site was not cleaved in either strand (data not shown). Another type IIS enzyme, HphI, cuts DNA at varied distances away from its recognition site but, unlike BfiI, HphI displays the same variations in top and bottom stands: it cuts the two strands either 8 and 7 nt away or 9 and 8 nt away (28). Thus, whereas HphI maintains register on top and bottom strands, BfiI action on the top strand seems to be detached from that on the bottom.
The relatively slow cleavage of the top strand (Fig. 1 A) could be caused by BfiI just being more active toward the bottom strand, or it could be caused by an obligatory pathway where BfiI has to cut the bottom strand before the top. These possibilities were distinguished by using a duplex, 30/30(S) (Table 1), with the same sequence as the 30/30 DNA used above but with a phosphorothioate at the cleavage site in the bottom strand. In gel-shift experiments, phosphorothioate substitutions at either this position, or at all three of the sites of top-strand cleavage (the duplex 30(S)/30, Table 1), had no effect on DNA binding by BfiI. In addition, the phosphorothioate substitutions in the top strand of the 30(S)/30 duplex had no effect on the rate of cleavage of the bottom strand (Fig. 1B). However, the phosphorothioate at the target position in the bottom strand hindered severely not only the cleavage of that strand (t1/2 > 4 h, compare 0.8 min for the oxyester duplex), but also the cleavage of the top strand: the amount of product cleaved in the top strand never exceeded the amount cleaved in the bottom strand (Fig. 1B). Thus, BfiI cuts DNA adjacent to its recognition site in a strictly sequential manner: it attacks the top strand only after cutting the bottom strand.
Oligomerization of BfiI. BfiI is a dimer in solution and, like the EDTA-resistant nuclease Nuc (26), it contains a single catalytic center at the subunit interface (24). To make a double-strand break, the BfiI dimer could form an oligomer with two or more active sites (Fig. 2A). This would be like FokI, which binds DNA as a monomer with one active site but then captures a second monomer to form a dimer with two active sites (8, 9, 10). However, a tetramer is the minimal unit of BfiI with two active sites. Alternatively, after cutting the bottom strand, the BfiI–DNA complex could rearrange itself to switch the active site to the top strand (Fig. 2 A).
Fig. 2.

Subunit assembly for DNA cleavage by BfiI. (A) Cartoon representations of possible reaction schemes for double-strand DNA cleavage by BfiI. (B) Reactions at 25°C, in reaction buffer at pH 9.0, contained various concentrations of BfiI dimer (25–100 pM) and 3 nM 30/30 duplex, radiolabeled in either bottom or top strand. Samples were taken from the reactions at timed intervals and analyzed as in Experimental Procedures to determine the extent of cleavage of each strand. The steady-state rates for bottom-strand (○) and top-strand (•) cleavages were determined by linear regression; kcat for the bottom strand = 0.20 ± 0.02 min–1.
Previous gel-filtration studies of BfiI seem to discount the oligomerization model. The BfiI protein is a dimer in solution in the absence of DNA and when bound to its recognition sequence (24). Even so, two dimers could still associate transiently during the cleavage reaction to form a tetramer that is too unstable to detect by gel filtration. However, if this were the case, one would expect a nonlinear relationship between the steady-state rate of DNA cleavage by BfiI and the enzyme concentration: a 2-fold increase in enzyme concentration should increase the cleavage rate by >2-fold, as is the case with FokI (8).
The reactions of BfiI at its recognition site on the 30/30 duplex were examined under steady-state conditions over a range of enzyme concentrations (Fig. 2B). The rates of cleavage of bottom and top strands both increased linearly with the concentration of BfiI. Moreover, even with a 100-fold excess of cognate DNA over enzyme, conditions that largely preclude the binding of two molecules of protein to one molecule of DNA, BfiI still cleaved both strands of the DNA. It is therefore most unlikely that top-strand cleavage requires the BfiI dimer at the recognition site to recruit a second dimer.
Cleavage of Immobilized DNA. Intermolecular interactions between BfiI–DNA complexes should not occur when the BfiI protein is bound to DNA immobilized on a solid surface. The duplex bio-30/30 has the recognition sequence for BfiI and a biotin tag at the 5′ end of the top strand (Table 1). Samples of this duplex, with radiolabels in either the top or the bottom strands, were immobilized on streptavidin-coated magnetic beads. The beads carrying the duplex were added to a solution of BfiI at pH 8.0, and cleavage of the top and bottom strands was monitored as before. At pH 8.0, BfiI cleaved both bottom and top strands of the immobilized DNA, at similar rates to those with the same duplex in solution. The biotinylated DNA is present at a low density on the beads: it occupies <3% of the available biotin-binding sites (see Experimental Procedures). Hence, BfiI reactions do not need intermolecular interactions between BfiI–DNA complexes.
In the above experiments, the amount of BfiI added to the beads exceeded the amount of biotinylated duplex on the beads, so the enzyme bound to the DNA might have captured a second dimer from free solution to cleave the top strand. This hypothesis was tested by the following experiment (Fig. 3A). First, the beads carrying the bio-30/30 duplex, radiolabeled in the top strand, were preincubated with an excess of BfiI for 5 min at pH 6.5. Control experiments using bio-30/30 radiolabeled in the bottom strand showed that, as in free solution (Fig. 1 A), all of the bottom strand was cleaved within this time, but >90% of the top strand remained intact (data not shown). Next, the beads were withdrawn from the enzyme solution and washed with pH 6.5 buffer, to remove any protein adsorbed nonspecifically on the beads or weakly bound to the BfiI–DNA complexes. Finally, the beads carrying the immobilized DNA, which may (or may not) have pulled down some BfiI protein, were resuspended in buffer at pH 8.0 and the cleavage of the top DNA strand was monitored as before. After the shift to pH 8.0, almost all of the top strand was cleaved within 30 min (Fig. 3B), at a similar rate as the duplex in free solution (Fig. 1 A). BfiI protein must therefore have been pulled down with the beads, and bottom- and top-strand cleavages of an individual duplex are most likely caused by the same molecule of BfiI protein.
Fig. 3.

BfiI cleavage of immobilized DNA. (A) The experimental strategy. (B) A solution of 4 nM bio-30*/30 duplex (Table 1), whose top strand carries a biotin at its 5′ end and a 32P label at its 3′ end, was incubated with streptavidincoated magnetic beads in reaction buffer at pH 6.5 containing 50 nM BfiI for 5 min. The beads were washed twice with pH 6.5 buffer and then resuspended in pH 8.0 buffer. At timed intervals after the resuspension, the extent of cleavage of the top strand was monitored (○). In a parallel experiment, top-strand cleavage was measured under the same conditions except that the beads were resuspended in pH 8.0 buffer containing the K107A mutant of BfiI at 100 nM (•). The solid line is the optimal exponential fit to the latter data (rate constant 0.18 ± 0.02 min–1, amplitude 52%).
The washing step at pH 6.5 should eliminate protein adsorbed nonspecifically on the beads and disrupt weak protein aggregates on the DNA, such as a transient tetramer. However, the BfiI molecules pulled down with the beads at pH 6.5 could have dissociated from the DNA at pH 8.0 and then reassociated with each other to give higher-order assemblies (e.g., tetramers) that execute top-strand cleavage. To examine this possibility, bio-30/30 was immobilized on beads and incubated with BfiI at pH 6.5 to cleave the bottom strand. After washing to remove excess protein, the beads were resuspended in pH 8.0 buffer and top-strand cleavage was monitored as before, except that on this occasion the resuspension buffer also contained a large excess of the K107A mutant of BfiI, which has no catalytic activity but binds DNA with similar affinity to the WT enzyme (24). If the WT enzyme bound to the nicked duplex at pH 6.5 dissociates from the DNA at pH 8.0, the inactive mutant will replace it. Yet, even in the presence of excess K107A, a large fraction of the top strand of the immobilized duplex was still cleaved upon resuspension of the beads at pH 8.0 (Fig. 3B). The extent of top-strand cleavage is, however, reduced from almost 100% in the absence of K107A to ≈50% in its presence. This finding indicates that at least ≈50% of the immobilized duplexes retain the WT BfiI dimer, which, after cutting the bottom strand at pH 6.5, proceeds to cut the top strand at pH 8.0.
The above data thus indicate that BfiI operates on both DNA strands by an isomerization rather than an oligomerization mechanism (Fig. 2 A).
A Hairpin Intermediate? The strictly successive order of double-strand DNA breaks by certain transposases is caused by a hairpin mechanism. In this scheme (16–19), a 3′ OH end, created by hydrolytic nicking of the bottom strand, mounts an intramolecular attack on the scissile phosphodiester of the top strand. This joins the 3′ OH from the bottom strand to a 5′ phosphate from the top strand. The resultant hairpin is subsequently resolved into the final product(s) by a further hydrolytic attack.
To examine the possibility of a hairpin intermediate for BfiI, the hydrolysis of the top DNA strand was studied with two duplexes that deviate from the DNA produced by BfiI action on the bottom strand. One, *30/18P (Table 1), lacks any DNA 3′ to the site of bottom-strand cleavage, whereas the other, *30/(18P_dd12), carries a dideoxynucleotide in place of the deoxynucleotide 3′ to the cleavage site. Both lack the 3′ OH group that is essential for hairpin formation. Yet BfiI still acted on the top DNA strand of both of these substrates, in the same manner as the nicked substrate with a 3′ OH group, *30/(18P_12) (data not shown). Hence, double-strand cleavage by BfiI cannot involve a hairpin intermediate.
pH Dependence of Top-Strand Cleavage. The BfiI endonuclease is thus a dimer with a single active site, which cleaves two DNA strands in consecutive reactions. After cutting the bottom strand, a rearrangement of the protein and/or the DNA may switch the active site to the top strand (Fig. 2A). The switch was characterized by analyzing the selective effect of low pH on blocking top-strand cleavage without inhibiting bottom-strand cleavage (Fig. 1A).
Top-strand cleavage was examined by itself, without interference from the reaction on the bottom strand, by using as the substrate the nicked duplex *30/(18P_12). This corresponds to the product from a BfiI reaction on the bottom strand (Table 1). Single turnovers of BfiI on *30/(18P_12) were carried out at saturating enzyme concentrations, at different pH values in the range of 6.5–9.5: exponential rate constants (ktop) for the hydrolysis of the top DNA strand were evaluated as in Experimental Procedures and plotted on a logarithmic scale against the pH (Fig. 4). The rate constants for top-strand cleavage increase with pH across this range, to an asymptote at pH values >9. The simplest scheme to account for this behavior is a single deprotonation event between two forms of the enzyme–substrate complex, ESH and ES, that each carry out the reaction at a particular rate, kESH and kES, respectively. The pH dependency for such a scheme is given by Eq. 1:
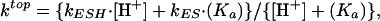 |
[1] |
where Ka is the equilibrium constant for the deprotonation. The optimal fit of the data in Fig. 4 to Eq. 1 yielded the following: pKa = 8.5 ± 0.1, kESH = 0.004 ± 0.001 min–1, and kES = 0.23 ± 0.03 min–1.
Fig. 4.

pH dependence of top-strand cleavage. The cartoons above the graph illustrate DNA substrates used: arrows indicate BfiI cleavage sites. The reactions at 25°C contained 3 nM duplex and 50 nM BfiI in reaction buffers at the indicated pH. The duplexes were: *30/(18P_12), ○ or *30/(18Δ_12), •. For each reaction, a value for ktop was determined by fitting the extent of cleavage with time to a single exponential. The optimal fit of Eq. 1 to the ktop values from *30/(18P_12), shown as a solid line, yielded the following: pKa = 8.5 ± 0.1, kESH = 0.004 ± 0.001 min–1, and kES = 0.23 ± 0.03 min–1.
A single ionizing group in the BfiI–DNA complex, with a pKa of 8.5, thus governs the rate constant for top-strand cleavage. However, this group has only little influence on the rate constant for bottom-strand cleavage: the rates for the bottom strand, measured on 30/30*, were similar at pH 8.0 and pH 6.5 (Fig. 1A). Hence, instead of an amino acid at the active site of BfiI, the ionizing group may be the 5′ phosphate at the site of bottom-strand cleavage. (A pKa of 8.5 is higher than the typical values of ≈6.8 for the second pKa of a phosphomonoester, but ionizing groups at enzyme active sites often have perturbed pKa values.)
To test whether the terminal phosphate at the nick in the bottom strand affects top-strand cleavage, single-turnover reactions were carried on the duplex *30/(18Δ_12), which lacks the 5′ phosphate at the bottom-strand nick (Table 1). The removal of the phosphate from the nick in the bottom strand had a major effect on the pH dependency of top-strand cleavage (Fig. 4). The rate constants on *30/(18Δ_12) remained low throughout the pH range tested, at values similar to those observed on *30/(18P_12) at pH 7. The ≈40-fold increase seen with *30/(18P_12) as the pH was raised to 9.5 was not emulated with *30/(18Δ_12). Instead, *30/(18Δ_12) gave rate constants that were almost independent of pH, varying only 3-fold between pH 6.5 and 9.5. Hence, deprotonation of the 5′ phosphate in the bottom strand controls the rearrangement of enzyme and/or substrate needed for top-strand cleavage by BfiI.
DNA Synapsis by BfiI. As is common for type IIS restriction enzymes (10, 11), BfiI cleaves plasmids with two copies of its recognition sequence more rapidly than plasmids with one site (24). The BfiI dimer also forms synaptic complexes with two cognate DNA molecules (24). BfiI is thus most active on binding two copies of its target sequence although it still has detectable activity when bound to a single site. The majority of the experiments described above were conducted with the BfiI enzyme in large excess over the DNA, so that the cleavage kinetics could be analyzed as first-order reactions. But under these conditions, the BfiI protein will be in a suboptimal state, bound to only one DNA molecule, rather than in its optimal state bound to two copies of its recognition site.
All of the single-turnover experiments in this study that had used enzyme in excess of the DNA were therefore repeated with the BfiI dimer at half the concentration of the duplex, the optimal ratio for forming the synaptic complex with two duplexes bound to one dimer. In all cases, the reactions with the 1:2 ratio of protein/DNA gave similar data to those with BfiI in excess of the DNA. BfiI cleaved bottom and top strands in obligatory succession and had the same pH dependency for top-strand cleavage, regardless of whether it was bound to one or two duplexes.
Discussion
Site-specific deoxyribonucleases generally make double-strand breaks in DNA by one of the following means: by acting as an oligomeric protein, made from either identical or different subunits, with separate active sites in each subunit (2–4, 12); by possessing two distinct active sites in a single polypeptide chain (14, 15); or by using a single active site to cut both strands via a hairpin intermediate (16–19).
BfiI is a type IIS restriction endonuclease that, unlike other restriction enzymes characterized to date, cleaves DNA in the absence of Mg2+ (22). The catalytic domain of BfiI is similar to Nuc (22, 24), a nonspecific nuclease from S. typhimurium that can function in EDTA (25). The crystal structure of Nuc reveals a dimer with a single active site at the subunit interface, containing residues from both subunits (26). BfiI is also a homodimer with a single active site (24), presumably arranged like that in Nuc. BfiI was shown here to cut the two strands of the DNA adjacent to its recognition site in obligatory succession: first the bottom strand and then the top (Fig. 1). So how does its single active site cleave both DNA strands?
One possibility is that BfiI makes double-strand DNA breaks by the same scheme as the Mg2+-dependent type IIS enzyme FokI. FokI is a monomer with one active site (6). It binds DNA as a monomer but, to cleave both strands, it associates with another monomer to form a dimer (7–10). However, the linear relationship between BfiI concentration and reaction rate (Fig. 2B), and the reactions on magnetic beads switched from nonpermissive to permissive pH values (Fig. 3), show that BfiI does not change its oligomerization state to make double-strand breaks in DNA.
A further possibility is that the single active site in BfiI cuts both strands via a hairpin intermediate. To form a hairpin, the 3′ OH group from phosphodiester hydrolysis in one strand acts as the nucleophile to attack the target phosphodiester in the other strand. However, the duplexes 30/18P and 30/(18P_dd12) lack the 3′ OH group at the site of bottom-strand cleavage by BfiI (Table 1), yet BfiI still cleaved the top strand of these substrates. BfiI therefore cannot act via the same sort of hairpin intermediate as in V(D)J recombination or Tn10/Tn5 transposition (16–19).
It seems instead that BfiI makes double-strand breaks in DNA by means of two separate hydrolytic reactions, that cleave sequentially first the bottom and then the top strand, with both reactions occurring in the same active site, the only site in this dimeric enzyme. The use of one catalytic center to cut both strands, rather than one center for each strand, distinguishes BfiI from all other restriction enzymes characterized to date. However, a corollary to the use of one center for both strands is that, after cleaving the bottom strand, the BfiI–DNA complex must undergo a rearrangement that switches the catalytic site to the scissile phosphate in the top strand. The target phosphodiester bonds for BfiI in the two strands are separated by ≈17 Å in B-form DNA. Hence, the switch to the top strand demands major conformational changes of the enzyme or the DNA or both (Fig. 2A). The switch seems to be driven by the 5′ phosphate created by bottom-strand cleavage carrying a negative charge of two: it does not occur if that phosphate is either removed or carries a negative charge of minus one caused by protonation (Fig. 4). In the latter case, BfiI is transformed into a site-specific nicking enzyme. However, its ability to cleave both strands can be restored at high pH values. This makes BfiI different from the other nicking endonucleases characterized to date (29, 30).
A further consideration in using the same catalytic center to carry out separate reactions on both strands is that the switch to the top strand might seem to require a 180° rotation of the center, to match the antiparallel polarity of the second strand. However, the rotation may be unnecessary for BfiI. In the Nuc enzyme, and thus presumably also BfiI (22, 24), the single active site is located on the symmetry axis between the protein subunits so that every active-site residue from one subunit has its counterpart from the other subunit (26). For Nuc, His-94 from one subunit may act as the attacking nucleophile and His-94 from the other subunit as a proton donor, but the nucleophile could be from either the A or the B subunit and the proton donor from the converse (26). Hence, in BfiI, the equivalent residue (His-105) from one subunit might attack the target phosphodiester in the bottom strand in line with its 3′ leaving group: subsequently, His-105 from the other subunit attacks the top strand, again in line with the leaving group. It is not known yet how Nuc cleaves double-stranded DNA. The mechanism proposed here for BfiI may be the first indication of how an endonuclease from the phospholipase D family cleaves both DNA strands.
Cleavage of the bottom DNA strand by the I-TevI homing endonuclease was shown previously to induce DNA bending, and it was suggested that this distortion places the top strand into the single active site of this monomeric enzyme (20). However, alternative schemes for making double-strand breaks in DNA have yet to be eliminated for I-TevI (21). The alternatives could perhaps be tested by examining I-TevI reactions on duplexes attached to magnetic beads, on transferring the beads from nonpermissive to permissive conditions (namely Fig. 3). Whereas the BfiI dimer has a symmetrical active site, the catalytic domain of I-TevI (21) has an asymmetric center that can handle only one polarity of the DNA chain. Hence, if the two DNA strands at the target site for I-TevI are to enter successively into its active site, the active site must be rotated through 180° between the reactions on the bottom and the top strands.
Acknowledgments
We thank V. Stonyte and L. Manakova for help with the BfiI purification and A. Lagunavicius and M. Zaremba for discussions. This work was funded by a Collaborative Research Initiative Grant from the Wellcome Trust. V.S. is a Howard Hughes Medical Institute International Research Scholar. V.S. also acknowledges support from the Lithuania Science Foundation.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Mizuuchi, K. (1997) Genes Cells 2, 1–12. [DOI] [PubMed] [Google Scholar]
- 2.Lilley, D. M. & White, M. F. (2001) Nat. Rev. Mol. Cell Biol. 2, 433–443. [DOI] [PubMed] [Google Scholar]
- 3.Pingoud, A. & Jeltsch, A. (2001) Nucleic Acids Res. 29, 3705–3727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Galburt, E. A. & Stoddard, B. L. (2002) Biochemistry 41, 13851–13860. [DOI] [PubMed] [Google Scholar]
- 5.Sugisaki, H. & Kanazawa, S. (1981) Gene 16, 73–78. [DOI] [PubMed] [Google Scholar]
- 6.Wah, D. A., Hirsch, J. A., Dorner, L. F., Schildkraut, I. & Aggarwal, A. K. (1997) Nature 388, 97–100. [DOI] [PubMed] [Google Scholar]
- 7.Wah, D. A., Bitinaite, J., Schildkraut, I. & Aggarwal, A. K. (1998) Proc. Natl. Acad. Sci. USA 95, 10564–10569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bitinaite, J., Wah, D. A., Aggarwal, A. K. & Schildkraut, I. (1998) Proc. Natl. Acad. Sci. USA 95, 10570–10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vanamee, E. S., Santagata, S. & Aggarwal, A. K. (2001) J. Mol. Biol. 309, 69–78. [DOI] [PubMed] [Google Scholar]
- 10.Bath, A. J., Milsom, S. E., Gormley, N. A. & Halford, S. E. (2002) J. Biol. Chem. 277, 4024–4033. [DOI] [PubMed] [Google Scholar]
- 11.Soundararajan, M., Chang, Z., Morgan, R. D., Heslop, P. & Connolly, B. A. (2002) J. Biol. Chem. 277, 887–895. [DOI] [PubMed] [Google Scholar]
- 12.Biery, M. C., Lopata, M. & Craig, N. L. (2000) J. Mol. Biol. 297, 25–37. [DOI] [PubMed] [Google Scholar]
- 13.Hickman, A. B., Li, Y., Mathew, S. V., May, E. W., Craig, N. L. & Dyda, F. (2000) Mol. Cell 5, 1025–1034. [DOI] [PubMed] [Google Scholar]
- 14.Christ, F., Schoettler, S., Wende, W., Steuer, S., Pingoud, A. & Pingoud, V. (1999) EMBO J. 18, 6908–6916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Moure, C. M., Gimble, F. S. & Quiocho, F. A. (2002) Nat. Struct. Biol 9, 764–770. [DOI] [PubMed] [Google Scholar]
- 16.McBlane, J. F., van Gent, D. C., Ramsden, D. A., Romeo, C., Cuomo, C. A., Gellert, M. & Oettinger, M. A. (1995) Cell 83, 387–395. [DOI] [PubMed] [Google Scholar]
- 17.Kennedy, A. K., Haniford, D. B. & Mizuuchi, K. (2000) Cell 101, 295–305. [DOI] [PubMed] [Google Scholar]
- 18.Kennedy, A. K., Guhathakurta, A., Kleckner, N. & Haniford, D. B. (1998) Cell 95, 125–134. [DOI] [PubMed] [Google Scholar]
- 19.Davies, D. R., Goryshin, I. Y., Reznikoff, W. S. & Rayment, I. (2000) Science 289, 77–85. [DOI] [PubMed] [Google Scholar]
- 20.Mueller, J. E., Smith, D., Bryk, M. & Belfort, M. (1995) EMBO J. 14, 5724–5735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Van Roey, P., Meehan, L., Kowalski, J. C., Belfort, M. & Derbyshire, V. (2002) Nat. Struct. Biol. 9, 806–811. [DOI] [PubMed] [Google Scholar]
- 22.Sapranauskas, R., Sasnauskas, G., Lagunavicius, A., Vilkaitis, G., Lubys, A. & Siksnys, V. (2000) J. Biol. Chem. 275, 30878–30885. [DOI] [PubMed] [Google Scholar]
- 23.Vitkute, J., Maneliene, Z., Petrusyte, M. & Janulaitis, A. (1998) Nucleic Acids Res. 26, 3348–3349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lagunavicius, A., Sasnauskas, G., Halford, S. E. & Siksnys, V. (2003) J. Mol. Biol. 326, 1051–1064. [DOI] [PubMed] [Google Scholar]
- 25.Ponting, C. P. & Kerr, I. D. (1996) Protein Sci. 5, 914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stuckey, J. A. & Dixon, J. E. (1999) Nat. Struct. Biol. 6, 278–284. [DOI] [PubMed] [Google Scholar]
- 27.Yoshioka, K. (2002) Comp. Stat. 17, 425–437. [Google Scholar]
- 28.Kleid, D., Humayun, Z., Jeffrey, A. & Ptashne, M. (1976) Proc. Natl. Acad. Sci. USA 73, 293–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Morgan, R. D., Calvet, C., Demeter, M., Agra, R. & Kong, H. M. (2000) Biol. Chem. 381, 1123–1125. [DOI] [PubMed] [Google Scholar]
- 30.Higgins, L. S., Besnier, C. & Kong, H. (2001) Nucleic Acids Res. 29, 2492–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]