

Abstract
Oncogenic Kit mutations are found in somatic gastrointestinal (GI) stromal tumors (GISTs) and mastocytosis. A mouse model for the study of constitutive activation of Kit in oncogenesis has been produced by a knock-in strategy introducing a Kit exon 11-activating mutation into the mouse genome based on a mutation found in a case of human familial GIST syndrome. Heterozygous mutant KitV558Δ/+ mice develop symptoms of disease and eventually die from pathology in the GI tract. Patchy hyperplasia of Kit-positive cells is evident within the myenteric plexus of the entire GI tract. Neoplastic lesions indistinguishable from human GISTs were observed in the cecum of the mutant mice with high penetrance. In addition, mast cell numbers in the dorsal skin were increased. Therefore KitV558Δ/+ mice reproduce human familial GISTs, and they may be used as a model for the study of the role and mechanisms of Kit in neoplasia. Importantly, these results demonstrate that constitutive Kit signaling is critical and sufficient for induction of GIST and hyperplasia of interstitial cells of Cajal.
Kit encodes a growth factor receptor with ligand-dependent tyrosine kinase activity (1–3). Kit ligand (KitL) is the only known ligand of the Kit receptor (4). KitL binding to the receptor mediates receptor dimerization, activation of kinase activity, and autophosphorylation. Subsequently, Kit activates several signaling cascades, leading to cell proliferation, cell survival, and other cellular responses. Kit and KitL are encoded at the White spotting (W) and Steel (Sl) loci in the mouse, respectively (4–6). Mutations at the murine W and Sl loci generate deficiencies in several major cell systems during embryogenesis and in the postnatal animal: in hematopoiesis, melanogenesis, gametogenesis, and intestinal pacemaker cells. In hematopoiesis Kit receptor signaling is critical in the stem cell hierarchy, erythropoiesis, mast cell development and function, megakaryopoiesis, and lymphopoiesis (7–10). Interstitial cells of Cajal (ICC) function as pacemaker cells in the gastrointestinal (GI) tract and they mediate inputs from the enteric nervous system to smooth muscle cells. ICC express the Kit receptor tyrosine kinase and inhibition of Kit function interferes with the autonomous movement of the GI tract (11–13).
GI stromal tumor (GIST) is the most common mesenchymal neoplasm of the human intestinal tract. GISTs are a heterogeneous group of tumors, which historically had been classified either as leiomyoma, leiomyosarcoma, or GI autonomic nerve tumors (14). GISTs express Kit and they are thought to derive from a Kit+ or Kitlow ICC progenitor or ICC through somatic mutation, based on immunophenotypic and ultrastructural similarities (15).
Originally, we had identified Kit as the oncogene of an acute transforming feline retrovirus, the HZ4-FeSV (1). However, a role for Kit in human neoplasia has been emerging only more recently. First, human and murine mast cell lines were found to carry Kit-activating mutations in the activation loop of the kinase and subsequently the same mutation was found in human mastocytosis/mast cell leukemia (16, 17) and germ cell tumors (18). Finally, analysis of GISTs revealed that these tumors express Kit and that a substantial number of them contain Kit-activating mutations (15, 19). Whereas most of the Kit-activating mutations in GIST are associated with exon 11, others are associated with exons 9, 13, and 17 (20). Importantly, in vitro characterization of several mutations found in mastocytosis and GISTs by using expression of mutant Kit cDNA in culture confirmed the constitutive activity of the mutant receptors in model cell systems (15, 21). A transgenic mouse model for the study of a role of Kit in oncogenesis in which a mutant Kit receptor was expressed by a heterologous promoter has been reported (22). But only acute lymphocytic leukemia and lymphomas were observed and no malignancies known to be associated with Kit activation were found.
Cases of human familial GIST syndrome with associated ICC hyperplasia (HP), hyperpigmentation, and/or urticaria pigmentosa, with germ-line Kit mutations, have been reported (23, 24). The finding that Kit-activating mutations may be inherited implied that it might be possible to develop a mouse carrying an inherited Kit gain-of-function mutation as a model for the study of a role for Kit in oncogenesis.
Materials and Methods
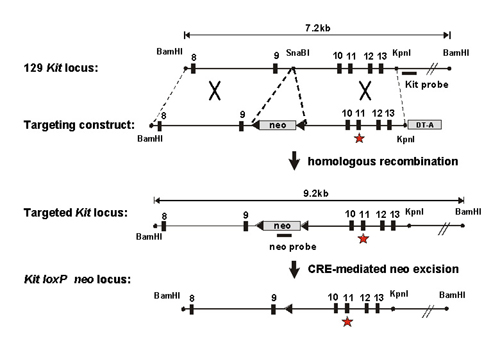
Generation of Heterozygous Mutant KitV558Δ/+ Mice. Site-directed mutagenesis was performed on a 2.4-kb SpeI–KpnI genomic Kit fragment including exons 10–13, deleting Val-558 in exon 11. The mutant Kit genomic fragment was inserted into a fragment including Kit exons 8–13. A neomycin resistance gene expression cassette flanked by loxP sites was inserted into a SnaBl site in intron 9. For negative selection a diphtheria toxin A gene cassette provided by Frank Costantini (Columbia University, New York) was placed at the 3′ end of the targeting construct. 129/SvJ embryonic stem (ES) cells (GSI-1 P/O, Genome Systems, St. Louis) were electroporated with linearized targeting construct following standard protocols. Neomycin-resistant ES cell clones were isolated and analyzed for homologous recombination. First, ES cell clones were screened by a PCR strategy (primer-set A/B) in which a 5-kb fragment including the neo cassette and exon 14 outside the targeting region was amplified. Correctly targeted ES cell clones then were verified by Southern blot analysis and the presence of the KitV558Δ was confirmed by sequence analysis. Correctly targeted ES cell clones were microinjected into C57BL/6J blastocysts and male mice displaying 85–100% chimerism were backcrossed to C57BL/6J females for germ-line transmission.
The floxed neo cassette was excised in vivo by mating heterozygous mutant males with EIIa-cre transgenic females (25). EIIa-cre transgenic mice on a C57BL/6J background (N10) were kindly provided by Monica Bessler (Washington University Medical School, St. Louis). Excision of the neo cassette was monitored by using DNA digested with BamHI and Southern blot analysis using Kit- and Neo-specific probes. In addition, the residual lox site and adjacent multiple cloning sites were identified by PCR (lox-PCR: primer-set C/D). Primers were: A, 5′-AAGAACTCGTCAAGAAGGCGATAGAAGGCG-3′; B, 5′-CTCCGTTGAGTGCAGAAGGTTC-3′; C, 5′-ACGATGTGGGCAAGAGTT-3′; and D, 5′-GATACTGTTAACATTTCGATACAGATGTTTAGC-3′.
Mast Cell Cultures. Bone marrow-derived mast cells (BMMCs) from KitV558Δ/+ and control mice were derived and cultured as published (26). Kit cell surface expression was monitored by using anti-mouse Kit antibody (StemCell Technologies, Vancouver) and fluorescence-activated cell sorter analysis. Before stimulation, BMMCs were washed free of growth factor and cultured in serum-free medium (Stemspan SF Expansion Medium, Stemcell Technologies) for 6 h.
Cell Proliferation and Apoptosis. Proliferation assays were performed essentially as described (27). Briefly, 105 starved BMMCs were seeded at 0.2 ml per well in triplicate in 96-well plates, followed by stimulation with KitL (PeProtech, Boston) as indicated or with IL-3 (20 ng/ml) (Sigma) for 24 h. After 20 h, 0.5 μCi of [3H]thymidine was added for 4 h. Cells were harvested on glass fiber filters and β-emission was determined. For apoptosis assays, cells were stimulated with IL-3 (20 ng ml) for 12 h. They then were plated at 106 cells per 2 ml per well in six-well plates, starved for 1 h, and stimulated with KitL (100 ng/ml), IL-3 (20 ng/ml), or deprived for 50 h. Cells were harvested and analyzed by flow cytometry using the Annexin V-FITC detection Kit I (PharMingen).
Immunoprecipitation and Western Blotting. Starved BMMCs were stimulated with KitL (100 ng/ml) or not for 5 min at 37°C. Cells were lysed in Nonidet P-40 lysis buffer containing 50 mM Tris·HCl (pH 8.0), 150 mM NaCl, 5 mM EDTA, 1% Nonidet P-40, 2 mM Na3VO4, 1 mM PMSF, 10 mM NaF, and 20 μl/ml proteinase inhibitor mixture (Sigma). Cleared lysates were precipitated overnight with a goat anti-Kit antibody (Santa Cruz Biotechnology) and fractionated by SDS/PAGE. For Western blotting, rabbit anti-Kit (Oncogene Science), rabbit anti-Tyr(P), and mouse anti-Tyr(P) 20 and 99 (Santa Cruz Biotechnology) were used. For preparation of tumor lysate, 1 g of snap-frozen tumor specimen was ground to powder in liquid nitrogen, resuspended in 5 ml of Nonidet P-40 lysis buffer, Dounce-homogenized 20 times, and incubated on ice for 30 min. Cleared lysate was subjected to immunoprecipitation, SDS/PAGE, and Western blotting as described for mast cells.
Histological Analysis and Immunohistochemistry. Paraffin sections (8 μm) from 4% paraformaldehyde-fixed tissues were stained or used for immunohistochemistry. For immunohistochemical detection the MOM kit (Vector Laboratories) was used. Anti-Ki-67 antibody (NCL-Ki-67-MM1) from Vector Laboratories was used at 0.4 μg/ml. For BrdUrd labeling, mice were injected i.p. with BrdUrd (Sigma, 50 μg/g of body weight) and after 3 h the injection was repeated and mice were killed 12 h later. Anti-BrdUrd antibody (BMC9318) from Roche Diagnostics was used at 6 μg/ml. Polyclonal rabbit anti-Kit antibody (DAKO) was used at 1:500. Synaptophysin staining was done according to the manufacturer's instructions (BioGenex, San Ramon, CA).
Determination of Mast Cell Numbers and Peripheral Blood Parameters. Skin mast cell numbers were determined as described (28). Mast cells per 0.5 mm2 skin between the epidermis and the panniculus were counted in several independent sections and averaged. Peripheral blood parameters were analyzed as described (28).
Electron Microscopy. For ultrastructural studies tissue was fixed in 2% glutaraldehyde, postfixed in 1% osmium tetroxide, and embedded in epoxy resin by using standard procedures. Sections were examined with a Philips EM410 electron microscope.
Results
A Single Point Mutation in the Juxtamembrane Domain of the Kit Receptor Gene, KitV558Δ, Obtained by Gene Targeting. To gain insight into the role of Kit-activating mutations in oncogenesis in vivo in mice we introduced an amino acid deletion mutation, KitV558Δ, into the murine Kit gene by using knock-in gene targeting technology. The V558 deletion mutation (human V559) in the juxtamembrane domain of the Kit receptor had been identified in a human familial GIST case (23) and in vitro studies had shown that this mutation results in constitutive activation of the Kit receptor tyrosine kinase. Furthermore, Val-559 substitution mutations (mouse V558) constitute a major subset (11%) of somatic mutations found in GIST (C.R.A., unpublished work). The juxtamembrane domain of human and mouse Kit are conserved completely and it seemed reasonable to assume that the V558 deletion mutation would produce the same functional consequences in mice and humans. To introduce the V558 deletion mutation into the Kit gene in ES cells a targeting construct was made that contained the valine deletion mutation in Kit exon 11 and a neomycine resistance (neo) cassette flanked by loxP sites for subsequent removal in vivo of the neo cassette inserted into intron 9 (Fig. 6, which is published as supporting information on the PNAS web site, www.pnas.org). Homologous replacement in ES cells produced three correctly targeted ES cell clones identified by PCR, Southern blot, and sequencing analysis. These ES cell clones were microinjected into C57BL/6J blastocysts, and chimeras were produced that gave rise to germ-line transmission.
We have previously noticed that inclusion of a neo cassette in intronic Kit sequences interferes with the expression of the Kit gene (29). Therefore we removed the neo cassette by cremediated excision in vivo as described (29). The presence of the KitV558Δ mutation was confirmed by DNA sequencing. Both heterozygous KitV558Δ/+ male and female mice were fertile, but with increasing age, fertility was impaired.
Myenteric Plexus (Myp) HP and GISTs in the GI Tract of Heterozygous KitV558Δ/+ Mice. Starting at 4 weeks of age heterozygous mutant KitV558Δ/+ mice developed symptoms of disease and eventually died from pathology in the GI tract. A gross examination of the entire GI tract of mutant and WT mice showed variable distension of the distal ileum (mega-ileum) ending at the level of the cecum in 12/14 mutants (Fig. 1A). The remaining small intestine, stomach, colon, and anus had normal appearance. In 9/14 mutants the cecum was enlarged and distended and contained clear fluid or pus-like contents. In all mutants a firm white nodular mass varying in size from 1 mm to 2 cm was found in the cecum. None of the control mice showed GI tract pathology. Furthermore, in 7/14 mutants a well-defined, black-pigmented area was identified at the gastroesophageal junction. Survival of mutant KitV558Δ/+ mice was ≈50% at 9 months (Fig. 1B). Moreover, all animals we have killed independent of fatal disease had GI pathology. Therefore, the penetrance of GI disease in KitV558Δ/+ mice approaches 100%.
Fig. 1.

(A) Photograph showing the intestinal tract of KitV558Δ/+ and WT mice. Nodular tumor mass in cecum of mutant mouse is indicated by arrow. Arrows indicate mega-ileum and pigmented distal esophagus at the gastro-esophageal junction. (B) Survival plot of KitV558Δ/+ mice.
Tissues from six mutant and three WT mice processed for microscopic examination representative sections from the esophagus, small intestine, cecum, and large intestine were examined histologically (Table 1, which is published as supporting information on the PNAS web site). In five of six mutants patchy thickening/HP of the esophageal MyP was identified. All six mutants showed similar MyP HP of the stomach (Fig. 2 C and D) and in three cases the proximal duodenum was involved as well. No MyP HP was detected in the normal looking distal duodenum, jejunum, and distended ileum (mega-ileum) (Fig. 2 A). In the cecum invariably patchy MyP HP was observed. Examination of the dilated cecum showed atrophy of the normal colonic folds, lumen distension, and in one case abscess formation associated with acute serositis (Fig. 2B). Patchy MyP HP within the large intestine was evident in all samples (Fig. 2 E–G). The hyperplastic MyP showed strong and diffuse staining with Kit antibody, suggesting that the main cellular component of the hyperplastic plexus is represented by ICC (Figs. 2G and 3C). Sections from the dark portion of the distal esophagus, observed in five of six mutants (Table 1), showed the presence of pigmented cells specifically within the MyP (Fig. 7 A and B, which is published as supporting information on the PNAS web site). Fontana–Masson stain confirmed the presence of melanin pigment.
Fig. 2.

Histological analysis of GI tract of KitV558Δ/+ mutant mice. Paraffin sections obtained from mutant (A–C, E, and G) and WT (D and F) GI tract were stained with hematoxylin/eosin. (A) Uninvolved small intestine, normal-appearing MyP is indicated by an arrow. (B) Focal abscess in cecum is shown, indicated by an asterisk. (C) MyP HP in stomach is shown, hyperplastic lesion is indicated by an arrow. (D) Normal MyP in stomach is indicated by an arrow. (E and F) Hyperplastic and normal MyP in mutant and WT colon are shown. (G) Anti-Kit staining of MyP HP in colon. (Magnifications: ×100, A–G; ×200, Insets.)
Fig. 3.

Histological and immunohistochemical analysis of GISTs in KitV558Δ/+ mutant mice and human familial GISTs. Paraffin sections were stained with hematoxylin/eosin (A and B), anti-Kit antibody (C and D), or antisynaptophysin antibody (E). (F) Hematoxylin/eosin staining of paraffin sections including GIST lesion and adjacent MyP HP. (G) Hematoxylin/eosin staining of MyP HP. (H) Hematoxylin/eosin staining of GIST. (I) Anti-Kit staining. Black arrow indicates GIST lesion and white arrows indicate MyP HP. (Magnifications: ×40, A and F; ×20, C; ×100, B, E, and I; ×200, D, G, and H.)
The neoplastic lesions detected in the cecum were characterized by uniform spindle cell morphology and involved the muscularis propria and submucosa of the intestinal wall (Table 1, Fig. 3 A–D). Kit immunohistochemistry showed strong and diffuse reactivity of the spindle cells (Fig. 3 C and D), and synaptophysin immunohistochemistry outlined perikarya and neurites of the enteric neurons, extending between tumor cells (Fig. 3E). The morphologic appearance and the strong Kit immunoreactivity of the lesions support the diagnosis of GIST. The proliferative characteristics of the neoplastic cells were studied by using Ki67 staining and BrdUrd labeling. The proliferation index like in human tumors is heterogenous, varying from 5% to 10% in most areas and up to 20% in focal areas (not shown).
For comparison, histological sections of neoplastic and hyperplastic lesions of a human patient with familial GIST, resulting from a missense mutation, Kit-W557R (G.S., unpublished work), are shown (Fig. 3 F–I). Morphologically, the appearance of GIST in a background of patchy MyP HP and the Kit staining pattern in the human familial GIST patient are indistinguishable from that of the disease observed in KitV558Δ/+ mice.
Tissue from one mutant mouse was processed for electron microscopy. Ultrastructurally, the MyP HP was found to be composed of numerous, small, undifferentiated spindle cells with short, branching filopodia-like cell processes (Fig. 4A), consistent with ICC HP. The ICC showed scant cytoplasm, with small clusters of dense mitochondria and subplasmalemmal vacuoles. Rare ganglion cells were identified and intermixed, and the number of axonal processes was significantly reduced, because of replacement by ICC. Ultrastructurally, the GIST was composed of oval to slightly spindle-shaped cells, with a moderate amount of cytoplasm (Fig. 4B). The cytoplasm was rich in dense mitochondria and prominent Golgi apparati and lacked organelles characteristic of lineage differentiation. Furthermore, the cells displayed short microvilli-like cell processes and no obvious basal lamina was noted. However, rare rudimentary cell junctions were present.
Fig. 4.

Electron micrographic analysis of MyP HP and GIST. (A) Thickened MyP in cecum, showing large ganglion cell (GLC) and numerous small undifferentiated spindle cells with narrow branching cell processes (ICC) are indicated by arrow; cells have scant cytoplasm with dense mitochondria and subplasmalemmal vacuoles. SMC, smooth muscle cell. (B) GIST. Discohesive plump spindle cells with round- to oval-shaped uniform nuclei and moderate amount of cytoplasm separated by a loose stroma; cells have short “microvilli-like” cell processes, numerous dense mitochondria, and prominent Golgi apparati. (Magnification: ×4,960.)
The pigmented area of the esophageal MyP included large, ovoid cells with abundant cytoplasm filled with numerous stage IV melanosomes (not shown) and they were surrounded by a discontinuous linear basal lamina (Fig. 7 D and E). The pigmented cells were seen intermixed with other cell components of the MyP, such as Schwann cells, axonal processes, and ICC.
Increased Numbers of Tissue Mast Cells and Peritoneal Mast Cells in Heterozygous KitV558Δ/+ Mutant Mice. KitL and Kit have multiple roles in hematopoiesis including the stem cell compartment, erythropoiesis, and mast cells (9, 10). Interestingly, hematocrit values and red blood cell, white blood cell, granulocyte, and platelet numbers in heterozygous mutant mice did not deviate from normal (not shown). This finding is in agreement with the observation that patients with familial GISTs appear to have normal values for steady-state hematopoiesis.
In contrast to the unchanged hematopoietic parameters, numbers of tissue mast cells in the dorsal skin and the peritoneum in KitV558Δ/+ mice appeared to be increased 4-fold (P = 0.012) (Fig. 5A). In addition, examination of sections of dorsal skin showed extracellular granules in the vicinity of mast cells, implying that mast cell degranulation occurs quite readily (Fig. 5B).
Fig. 5.

Characteristics of mast cells in the skin of KitV558Δ/+ mice and proliferation and cell survival characteristics of BMMCs isolated from KitV558Δ/+ mice. (A) Paraffin-embedded sections of dorsal skin were stained with toluidine blue and mast cells were counted (mast cells per mm2). (B) Histological section showing increased numbers of skin mast cells. Arrows show granules released into the interstitial space. (C) Deprivation-induced apoptosis. KitV558Δ/+ and WT BMMCs were incubated in serum-free medium (SFM) and in SFM containing KitL (100 ng/ml) or IL-3 (20 ng/ml) for 50 h. Percentage of apoptosis was determined by Annexin V staining and fluorescence-activated cell sorter analysis. Experiments were done in triplicate. Similar results were obtained in three independent experiments. (D) Phosphorylation of Kit receptors in KitV558Δ/+ and WT BMMCs in the absence and presence of KitL. Starved cells were treated with and without KitL (100 ng/ml) for 5 min at 37°C. Cell lysates were immunoprecipitated with anti-Kit antibody, blotted with antiphosphotyrosine antibody (Upper), and stripped and reprobed with anti-Kit antibody (Lower). (E) Proliferation of KitV558Δ/+ and WT BMMCs. Cells were stimulated with KitL (as indicated), IL-3, or medium alone for 20 h, [3H]thymidine (2.5 μCi/ml) was then added for 4 h. Data are expressed as means ± standard error of triplicate samples. (F) Phosphorylation of Kit receptor in tumor lysate. Kit proteins in tumor lysates (from different animals: GIST nos. 1, 2, and 3) were immunoprecipitated and fractionated by SDS/PAGE. Membranes were blotted with antiphosphotyrosine antibody (Upper) and anti-Kit antibody (Lower). Samples from WT BMMCs stimulated with KitL and unstimulated are shown for comparison.
Survival and Proliferation Characteristics of Mutant KitV558Δ/+ BMMCs. KitL induces survival and proliferation of BMMCs (27). It seemed reasonable that BMMCs obtained from KitV558Δ/+ mice exhibit different requirements for KitL for cell survival and cell proliferation. Surprisingly, young mutant BMMCs had a reduced requirement for the survival response of KitL, but in contrast they had an unchanged requirement for KitL to promote cell cycle progression (Fig. 5 C and E). Furthermore, analysis of the phosphorylation status of the Kit receptor in the unstimulated mutant BMMCs was indistinguishable from that in WT BMMCs (Fig. 5D). Interestingly, upon extended culture mutant mast cells (3 months and older), but not WT controls, became factor independent for both the proliferative and survival response and the Kit receptor was autophosphorylated (I.E., unpublished observation).
We next analyzed the phosphorylation status of the Kit protein in GIST lysates. In agreement with the expectation that in KitV558Δ/+ mice the Kit receptor is constitutively active, we found that the Kit receptor is autophosphorylated in all tumor samples (Fig. 5F).
Discussion
Activating mutations in the Kit receptor tyrosine kinase are associated with neoplasms and HPs in humans and dogs (15, 17, 30). Whereas GISTs and canine mastocytoma most often harbor mutations in the juxtamembrane domain of the Kit receptor, the mutations in human mastocytosis predominantly involve the activation loop of the Kit kinase (17, 31). In GIST there is a high association with Kit-activating mutations, suggesting that the activated Kit receptor has a critical role in tumor development (20). In agreement with this notion familial cases of GIST have been reported with Kit-activating mutations in the germ line (23). We have introduced a mutation, KitV558Δ/+, found in a case of human familial GIST syndrome into the mouse germ line to produce a mouse model for GIST and Kit mutation-associated hematopoietic diseases. Significantly, KitV558Δ/+ mice reproduce the human conditions associated with this mutation consisting of MyP HP, GIST, and mastocytosis. No metastasis was noticed, neither to the liver nor the abdominal cavity, as seen in advanced somatic human GIST. The penetrance of the GI phenotype in KitV558Δ/+ mice is remarkable. These observations strongly suggest that constitutive Kit signaling is critical and sufficient for induction of HP and neoplasia in these mice.
GIST is thought to derive from ICC. ICC localize to the MyP (ICC-MY) and to within the muscle layers (ICC-IM) throughout the intestinal tract, from the distal esophagus to the rectum (32). However, HP in the mutant animals was patchy and apparent only within the MyP, and neoplastic lesions of GISTs were found predominantly in the MyP of the cecum. ICC-MY have a dual role in promoting slow wave activity and creating peristalsis and in neurotransmission. In contrast, ICC-IM and deep muscular plexus-ICC mostly have a role in neurotransmission (32). Therefore, the site and differing functional properties may contribute to the oncogenic potential of the different ICC subpopulations. The anatomy of the cecum in humans and the mouse differ considerably in that the mouse cecum is longer. Thus the involvement of the mouse cecum as a primary site of pathology as opposed to the stomach or small intestine in human sporadic GISTs might result from different anatomical features of the cecum in mice and humans. The patchy appearance of hyperplastic and neoplastic lesions may suggest either that mutant Kit receptor signaling is limiting to initiate oncogenesis or that additional events are required for initiation of HP and oncogenesis. Recent cytogenetic characterization of human sporadic GISTs suggests a series of changes that may be characteristic for GIST progression (33). Cytogenetic profiling of the mouse tumors may help identify critical steps in GIST development. The histological and ultrastructural characteristics of the ICC HP and the GIST lesions mirror those of the human disease. Comparable with human GISTs, Kit expression within the tumor and hyperplastic changes was diffuse and strong. Ultrastructurally, the tumor cells lacked obvious differentiation toward any specific lineage, such as smooth muscle or neuronal. The undifferentiated appearance of the cells with branching cell processes and dense mitochondria were reminiscent electron microscopically of ICC.
During embryonic development a mesenchymal progenitor gives rise to both longitudinal smooth muscle cells and ICC-MY (34, 35). The Kit receptor is expressed in the progenitors and subsequently in ICC (35, 36, 37). It has been proposed that the decision of whether an ICC or a smooth muscle cell fate is followed depends on Kit receptor signaling. If this were true then one might predict that smooth muscle cell differentiation be diminished in mutant mice because of constitutive Kit receptor signaling. However, the longitudinal smooth muscle layer in the mutant mice did not differ from normal controls. This, on one hand, may mean that this hypothesis is not valid; on the other hand, it may be that the signal from the KitV558Δ receptor is too weak to affect the cell fate decision.
The distended ileum and cecum in the mutant mice in part could be caused by partial blockage of the GI tract; alternatively, the mutant Kit receptor may affect the formation of functional ICC networks, and consequently this may affect slow wave activity and/or the neurotransmission function and give rise to mega-ileum. Therefore, it will be of interest to characterize the ICC networks in more detail, the functional characteristics of ICC, and slow wave activity and neurotransmission in the mutant mice.
Neural crest cells after onset of migration have increasingly restricted potential for cell proliferation and differentiation (38). However, there is evidence for plasticity of differentiated cell fates of neural crest derived cell populations. Whereas a bi-potential progenitor gives rise to both melanocytes and Schwann cells, in peripheral nerve cell cultures in vitro-differentiated Schwann cells may transdifferentiate into melanocytes (39). We presume that the melanogenic cells in the thickened MyP of the distal esophagus in mutant mice are derived from Schwann cells. It is possible that a combination of environmental cues and an activated Kit receptor in Schwann cells promote this transdifferentiation.
The Kit receptor is critically involved in the development and the proliferation and survival of mast cells (4, 40). Furthermore, Kit promotes mediator release and enhances IgE-dependent mast cell activation. Thus, repeated s.c. administration of KitL to normal mice increases mast cell numbers at various anatomical sites and induces mast cell activation and associated inflammatory responses (41, 42). Human mastocytosis including urticaria pigmentosa are known to be associated with Kit receptor-activating mutations (17, 31). Increased mast cell numbers at various sites including the skin, bone marrow, and spleen typically characterizes this disease. Although activating mutations in human mastocytosis are predominantly confined to the activation loop of the kinase, juxtamembrane domain mutations have been noticed as well (17, 43). In agreement with this, familial GIST cases with a juxtamembrane domain mutation have been reported to have associated mastocytosis and/or urticaria pigmentosa. The finding of increased numbers of skin mast cells in the mutant mice is therefore significant, again reflecting the human disease. Interestingly, histological analysis of dorsal skin revealed that tissue mast cells appear to have released granules into the dermis, presumably reflecting mast cell activation.
In vitro characterization of the KitV558Δ mutation in BaF/3 cells indicated that the KitV558Δ mutation promoted cell proliferation and thus abolished the growth factor dependence of the BaF/3 cells (23). Our results from the in vitro analysis of mutant KitV558Δ/+ BMMCs appeared to be more complex. Young BMMC cultures were refractory to growth factor deprivation-induced apoptosis. However, these cultures did not promote cell cycle progression. It was only after prolonged culture (3 months) that the mutant BMMCs acquired complete independence for KitL-induced survival and cell cycle progression. Alternative splicing produces two variant Kit proteins, differing by 4 aa in the extracellular domain of the receptor (44). Evidence is accumulating to indicate that the short Kit protein product is more active than the long Kit protein. Therefore, it may be possible that the Kit-long variant is expressed predominantly in the young mutant BMMC and that the older cultures express the Kit-short variant, which may explain the progression from partial to complete growth factor independence of these cultures.
Kit has a critical role in hematopoietic stem cells and the progenitor compartments of all hematopoietic cell lineages. Therefore, one might have expected that the KitV558Δ mutation would affect several aspects of hematopoiesis and possibly lymphopoiesis (10, 45). However, lineage negative hematopoietic stem cell subsets as well as T-cell progenitor subsets were normal and steady-state hematopoietic parameters were not affected by the mutation. In agreement with this observation, lymphoid and myeloid cells in a patient with systemic mastocytosis were found to carry the mutant Kit gene, suggesting that the mutation was contained in a self-renewing stem cell subset with lymphoid, myeloid, and mast cell potential and indicating that the Kit mutation may not directly affect lymphoid and myeloid maturation (46).
Targeted drug therapy in the treatment of malignancies has recently had its first clinical breakthrough successes. The tyrosine kinase inhibitor STI571 (Gleevec, Novartis), an inhibitor of the ABL, PDGFR, and Kit kinases, is successfully used to treat patients with chronic myelogenous leukemia (47). In addition, STI571 is being used to treat patients with GISTs, a disease with no prior treatment options (48). These results highlight the critical role for both bcr-abl and a constitutively active Kit receptor in the etiology of the two diseases. The KitV558Δ/+ mouse model should provide a useful tool to investigate the consequences of drug treatment on tumor development.
Supplementary Material
Acknowledgments
We thank Drs. Elizabeth Lacy and Willie Mark for advice with the gene targeting experiments; Dr. Alexandra Joyner for the pKSloxPNT plasmid; Drs. Heiner Westphal and Monica Bessler for the EIIa-cre mice; Harry Satterwhite for expert assistance with hematological determinations; Sandra Gonzales, Melissa Besada, and Scott Kerns of the Molecular Cytology Facility at the Sloan-Kettering Institute for help with histological analysis; and Ann Baron for ultrastructural analysis. We also thank Drs. Murray Brennan, Mike Gershon, Robert Maki, Ronald DeMatteo, and James Woodruff for insightful discussion and encouragement. This work was supported by National Institutes of Health Grants HL/DK55748 and DH38908 (to P.B.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: KitL, Kit ligand; ICC, interstitial cells of Cajal; GI, gastrointestinal; GIST, GI stromal tumor; ES, embryonic stem; BMMC, bone marrow-derived mast cell; HP, hyperplasia; MyP, myenteric plexus.
References
- 1.Besmer, P., Murphy, J. E., George, P. C., Qiu, F. H., Bergold, P. J., Lederman, L., Snyder, H. W., Jr., Brodeur, D., Zuckerman, E. E. & Hardy, W. D. (1986) Nature 320, 415–421. [DOI] [PubMed] [Google Scholar]
- 2.Qiu, F. H., Ray, P., Brown, K., Barker, P. E., Jhanwar, S., Ruddle, F. H. & Besmer, P. (1988) EMBO J. 7, 1003–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yarden, Y., Kuang, W. J., Yang-Feng, T., Coussens, L., Munemitsu, S., Dull, T. J., Chen, E., Schlessinger, J., Francke, U. & Ullrich, A. (1987) EMBO J. 6, 3341–3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Besmer, P. (1991) Curr. Opin. Cell Biol. 3, 939–946. [DOI] [PubMed] [Google Scholar]
- 5.Chabot, B., Stephenson, D. A., Chapman, V. M., Besmer, P. & Bernstein, A. (1988) Nature 335, 88–89. [DOI] [PubMed] [Google Scholar]
- 6.Geissler, E. N., Cheng, S. V., Gusella, J. F. & Housman, D. E. (1988) Proc. Natl. Acad. Sci. USA 85, 9635–9639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Russell, E. S. (1979) Adv. Genet. 20, 357–459. [PubMed] [Google Scholar]
- 8.Besmer, P., Manova, K., Duttlinger, R., Huang, E. J., Packer, A., Gyssler, C. & Bachvarova, R. F. (1993) Development (Cambridge, U.K.) Suppl., 125–137. [PubMed]
- 9.Galli, S. J., Zsebo, K. M. & Geissler, E. N. (1994) Adv. Immunol. 55, 1–96. [DOI] [PubMed] [Google Scholar]
- 10.Besmer, P. (1997) Kit-Ligand-Stem Cell Factor (Dekker, New York).
- 11.Maeda, H., Yamagata, A., Nishikawa, S., Yoshinaga, K., Kobayashi, S. & Nishi, K. (1992) Development (Cambridge, U.K.) 116, 369–375. [DOI] [PubMed] [Google Scholar]
- 12.Ward, S. M., Burns, A. J., Torihashi, S. & Sanders, K. M. (1994) J. Physiol. (London) 480, 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huizinga, J. D., Thuneberg, L., Kluppel, M., Malysz, J., Mikkelsen, H. B. & Bernstein, A. (1995) Nature 373, 347–349. [DOI] [PubMed] [Google Scholar]
- 14.Miettinen, M. & Lasota, J. (2001) Virchows Arch. 438, 1–12. [DOI] [PubMed] [Google Scholar]
- 15.Hirota, S., Isozaki, K., Moriyama, Y., Hashimoto, K., Nishida, T., Ishiguro, S., Kawano, K., Hanada, M., Kurata, A., Takeda, M., et al. (1998) Science 279, 577–580. [DOI] [PubMed] [Google Scholar]
- 16.Furitsu, T., Tsujimura, T., Tono, T., Ikeda, H., Kitayama, H., Koshimizu, U., Sugahara, H., Butterfield, J. H., Ashman, L. K., Kanayama, Y., et al. (1993) J. Clin. Invest. 92, 1736–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nagata, H., Worobec, A. S., Oh, C. K., Chowdhury, B. A., Tannenbaum, S., Suzuki, Y. & Metcalfe, D. D. (1995) Proc. Natl. Acad. Sci. USA 92, 10560–10564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tian, Q., Frierson, H. F., Krystal, G. W. & Moskaluk, C. A. (1999) Am. J. Pathol. 154, 1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Taniguchi, M., Nishida, T., Hirota, S., Isozaki, K., Ito, T., Nomura, T., Matsuda, H. & Kitamura, Y. (1999) Cancer Res. 59, 4297–4300. [PubMed] [Google Scholar]
- 20.Rubin, B. P., Singer, S., Tsao, C., Duensing, A., Lux, M. L., Ruiz, R., Hibbard, M. K., Chen, C. J., Xiao, S., Tuveson, D. A., et al. (2001) Cancer Res. 61, 8118–8121. [PubMed] [Google Scholar]
- 21.Tuveson, D. A., Willis, N. A., Jacks, T., Griffin, J. D., Singer, S., Fletcher, C. D., Fletcher, J. A. & Demetri, G. D. (2001) Oncogene 20, 5054–5058. [DOI] [PubMed] [Google Scholar]
- 22.Kitayama, H., Tsujimura, T., Matsumura, I., Oritani, K., Ikeda, H., Ishikawa, J., Okabe, M., Suzuki, M., Yamamura, K., Matsuzawa, Y., et al. (1996) Blood 88, 995–1004. [PubMed] [Google Scholar]
- 23.Nishida, T., Hirota, S., Taniguchi, M., Hashimoto, K., Isozaki, K., Nakamura, H., Kanakura, Y., Tanaka, T., Takabayashi, A., Matsuda, H. & Kitamura, Y. (1998) Nat. Genet. 19, 323–324. [DOI] [PubMed] [Google Scholar]
- 24.Beghini, A., Tibiletti, M. G., Roversi, G., Chiaravalli, A. M., Serio, G., Capella, C. & Larizza, L. (2001) Cancer 92, 657–662. [DOI] [PubMed] [Google Scholar]
- 25.Lakso, M., Pichel, J. G., Gorman, J. R., Sauer, B., Okamoto, Y., Lee, E., Alt, F. W. & Westphal, H. (1996) Proc. Natl. Acad. Sci. USA 93, 5860–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Timokhina, I., Kissel, H., Stella, G. & Besmer, P. (1998) EMBO J. 17, 6250–6262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yee, N. S., Paek, I. & Besmer, P. (1994) J. Exp. Med. 179, 1777–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tajima, Y., Moore, M. A. S., Soares, V., Ono, M., Kissel, H. & Besmer, P. (1998) Proc. Natl. Acad. Sci. USA 95, 11903–11908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kissel, H., Timokhina, I., Hardy, M. P., Rothschild, G., Tajima, Y., Soares, V., Angeles, M., Whitlow, S. R., Manova, K. & Besmer, P. (2000) EMBO J. 19, 1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.London, C. A., Galli, S. J., Yuuki, T., Hu, Z. Q., Helfand, S. C. & Geissler, E. N. (1999) Exp. Hematol. 27, 689–697. [DOI] [PubMed] [Google Scholar]
- 31.Brockow, K. & Metcalfe, D. D. (2001) Curr. Opin. Allergy Clin. Immunol. 1, 449–454. [DOI] [PubMed] [Google Scholar]
- 32.Sanders, K. M., Ordog, T. & Ward, S. M. (2002) Am. J. Physiol. 282, G747–G756. [DOI] [PubMed] [Google Scholar]
- 33.El-Rifai, W., Sarlomo-Rikala, M., Andersson, L. C., Knuutila, S. & Miettinen, M. (2000) Cancer Res. 60, 3899–3903. [PubMed] [Google Scholar]
- 34.Lecoin, L., Gabella, G. & Le Douarin, N. (1996) Development (Cambridge, U.K.) 122, 725–733. [DOI] [PubMed] [Google Scholar]
- 35.Kluppel, M., Huizinga, J. D., Malysz, J. & Bernstein, A. (1998) Dev. Dyn. 211, 60–71. [DOI] [PubMed] [Google Scholar]
- 36.Torihashi, S., Ward, S. M. & Sanders, K. M. (1997) Gastroenterology 112, 144–155. [DOI] [PubMed] [Google Scholar]
- 37.Wu, J. J., Rothman, T. P. & Gershon, M. D. (2000) J. Neurosci. Res. 59, 384–401. [DOI] [PubMed] [Google Scholar]
- 38.Baroffio, A., Dupin, E. & Le Douarin, N. M. (1988) Proc. Natl. Acad. Sci. USA 85, 5325–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nataf, V. & Le Douarin, N. M. (2000) Pigment Cell Res. 13, 172–178. [DOI] [PubMed] [Google Scholar]
- 40.Kitamura, Y., Go, S. & Hatanaka, K. (1978) Blood 52, 447–452. [PubMed] [Google Scholar]
- 41.Tsai, M., Takeishi, T., Thompson, H., Langley, K. E., Zsebo, K. M., Metcalfe, D. D., Geissler, E. N. & Galli, S. J. (1991) Proc. Natl. Acad. Sci. USA 88, 6382–6386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wershil, B. K., Tsai, M., Geissler, E. N., Zsebo, K. M. & Galli, S. J. (1992) J. Exp. Med. 175, 245–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Buttner, C., Henz, B. M., Welker, P., Sepp, N. T. & Grabbe, J. (1998) J. Invest. Dermatol. 111, 1227–1231. [DOI] [PubMed] [Google Scholar]
- 44.Reith, A. D., Ellis, C., Lyman, S. D., Anderson, D. M., Williams, D. E., Bernstein, A. & Pawson, T. (1991) EMBO J. 10, 2451–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rodewald, H. R., Ogawa, M., Haller, C., Waskow, C. & DiSanto, J. P. (1997) Immunity 6, 265–272. [DOI] [PubMed] [Google Scholar]
- 46.Yavuz, A. S., Lipsky, P. E., Yavuz, S., Metcalfe, D. D. & Akin, C. (2002) Blood 100, 661–665. [DOI] [PubMed] [Google Scholar]
- 47.Druker, B. J., Sawyers, C. L., Kantarjian, H., Resta, D. J., Reese, S. F., Ford, J. M., Capdeville, R. & Talpaz, M. (2001) N. Engl. J. Med. 344, 1038–1042. [DOI] [PubMed] [Google Scholar]
- 48.Demetri, G. D., von Mehren, M., Blanke, C. D., Van den Abbeele, A. D., Eisenberg, B., Roberts, P. J., Heinrich, M. C., Tuveson, D. A., Singer, S., Janicek, M., et al. (2002) N. Engl. J. Med. 347, 472–480. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}