

Abstract
DNA methylation-mediated epigenetic regulation plays critical roles in regulating mammalian gene expression, but its role in normal brain function is not clear. Methyl-CpG binding protein 1 (MBD1), a member of the methylated DNA-binding protein family, has been shown to bind methylated gene promoters and facilitate transcriptional repression in vitro. Here we report the generation and analysis of MBD1-/- mice. MBD1-/- mice had no detectable developmental defects and appeared healthy throughout life. However, we found that MBD1-/- neural stem cells exhibited reduced neuronal differentiation and increased genomic instability. Furthermore, adult MBD1-/- mice had decreased neurogenesis, impaired spatial learning, and a significant reduction in long-term potentiation in the dentate gyrus of the hippocampus. Our findings indicate that DNA methylation is important in maintaining cellular genomic stability and is crucial for normal neural stem cell and brain functions.
DNA methylation at CpG dinucleotides is crucial for silencing inactive X-chromosomes, imprinted genes, and parasitic DNA (1). DNA methylation regulates gene expression through two mechanisms: (i) methylation at CpG sites blocks the binding of transcription factors and leads to transcriptional inactivation; and (ii) methyl-CpGs are bound by a family of methyl-CpG binding proteins (MBDs), including MBD1, MBD2, MBD3, MBD4, and MeCP2. Binding of MBDs and further recruitment of histone deacetylase (HDAC) repressor complexes result in histone deacetylation and inactive chromatin structures that are repressive for transcription (1). The most extensively studied member of this family is MeCP2, whose mutation causes neurological deficits in both humans (Rett Syndrome; ref. 2) and rodents (3, 4, 5). Mice lacking MBD2 show mild maternal behavior deficits (6). MBD3-/- mice die at an early embryonic stage (6). Mice deficient in MBD4, a mismatch repair enzyme, show deficits in DNA repair and increased tumor formation (7).
MBD1 shares homology with other MBDs only in the methyl-CpG binding domain. Extensive in vitro studies have shown that MBD1 binds specifically to methylated gene promoters through its MBD domain and carries out transcriptional repression that requires its transrepression domain (8, 9). This process requires an unknown HDAC that is different from the HDAC1 mediating the MeCP2 functions (10). MBD1 has functionally unclear zinc finger motifs (CXXC1, CXXC2, and CXXC3) that share homology with DNA methyltransferase-1 (Dnmt1) (11), human trithorax protein (12, 13), and human-CpG binding protein (14). The splice variants of MBD1 differ in the number of zinc fingers and in their N-terminal and C-terminal sequences (15). The functional relevance of these splice variants is unknown. In the mammalian genome, MBD1 is concentrated at the heterochromatin sites in centromeric regions, where DNA is highly methylated (10). A single MBD1 gene is located on chromosome 18 of both the human and mouse genome (16). MBD1 is expressed in many tissues, including brain (9, 10). Despite the intense studies performed by using in vitro assays, the in vivo function of MBD1 is still unclear. Because MBD1 has no sequence specificity, except for methylated CpG, its in vivo target genes are not known.
Maintaining normal DNA methylation levels is critical during mammalian development, as demonstrated by the embryonic lethality of Dnmt1-/- mice (17). Mutation of the de novo DNA methyltransferase 1-36 (Dnmt3b) causes immunodeficiency, centromeric instability, and facial anomalies (ICF) syndrome in humans, with characteristics of genomic instability (18). Both Dnmt1 and MeCP2 are highly expressed in the CNS, including postmitotic neurons (19). The function of DNA methylation in the adult CNS is not clear. DNA methylation may maintain a subtle balance of global gene expression pattern, which is crucial for neuronal function (20, 21). Previous work has demonstrated that DNA methylation can maintain genomic stability of cells (22). Cells lacking the Dnmt1 gene have 50–100 times higher levels of intracisternal A particle (IAP), a type of endogenous virus whose expression levels are frequently elevated in cancer cells with genomic instability (22). However, further studies on other DNA methylation-related proteins are needed to link DNA methylation, genomic stability and CNS functions.
To understand the function of DNA methylation and MBD proteins in the mammalian CNS, we generated MBD1-deficient (MBD1-/-) mice. The animals developed normally and appeared healthy as adults. We found that MBD1 was expressed in neurons throughout the brain, with the highest concentration in the hippocampus, one of the two regions that have persistent structural plasticity throughout life in mammals (23). MBD1 was expressed at a moderate level in dentate gyrus (DG) neurons and at higher levels in certain immature cells in the subgranular layer (SGL) of the DG. This finding led us to investigate the neurogenesis of adult MBD1-/- mice. We found that cultured MBD1-/- adult neural stem cells (ANCs) had reduced neurogenesis and increased genomic instability, and MBD1-/- mice had reduced neurogenesis, impaired spatial learning ability, and marked reduction in DG-specific long-term potentiation (LTP). Our work demonstrates that an MBD protein is important for maintaining genomic stability. The neurological defects in MBD1-/- mice are clear examples of the importance of epigenetic regulation in CNS function.
Methods
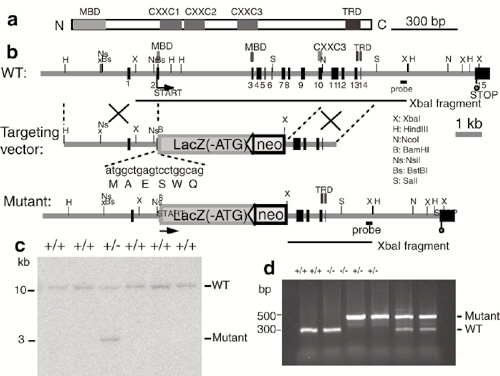
Gene Targeting Strategy. See Supporting Materials and Methods, which is published as supporting information on the PNAS web site, www.pnas.org.
Animals and Tissues. All mice used were under the 129S4 genetic background, between 2 and 5 months old, and age- and sex-matched between genotypes in each study. All animal procedures were performed according to protocols approved by The Salk Institute for Biological Studies Animal Care and Use Committee. For histology, mice were anesthetized and brains were processed as described (24). For RNA, PCR, and microarray analysis, fresh brains or hippocampi were dissected and frozen immediately on dry ice.
In Situ Hybridization, Histology, and Immunohistochemistry. PCR product of MBD1 (411 bp, containing CXXC3) was cloned and used as a template for 35S-labeled riboprobe. In situ hybridization was carried out as described (25). X-Gal (5-bromo-4-chloro-3-indolyl β-D-galactoside) staining was performed for 2 h at 37°C. Floating brain slices (40 μM) were used for X-Gal staining and Nissl staining, and images were collected as described (26). Double labeling was analyzed by using a Bio-Rad radiance confocal imaging system.
Cell Culture and in Vitro Differentiation Analysis. See Supporting Materials and Methods, which is published as supporting information on the PNAS web site.
Expression Profiling and Real-Time Quantitative PCR. See Supporting Materials and Methods.
Western Blotting. The rabbit anti-p73 IAP (gag protein) antibody (1:3,000) used in Western blot was kindly provided by Kira K. Lueders (Laboratory of Biochemistry, National Cancer Institute, Bethesda). The rabbit anti-MBD1 antibody (M254; Santa Cruz Biotechnology) was used at a ratio of 1:1,000. Quantification of IAP Western blotting was done by using NIH IMAGE software with normalization to β-actin level.
Bisulfite Sequencing and Genomic Southern Blot Analysis. See Supporting Materials and Methods.
Karyotyping and Spectral Karyotyping (SKY) Analysis. Chromosome spreads were made as described (27). Chromosome images were captured by using a Nikon E800 microscope and Spot RT charge-coupled device camera (Diagnostic Instruments, Sterling Heights, MI). Chromosome counting was carried out blind by three different individuals and notes were compared before decoding. SKY analysis was performed essentially as described (28, 29).
In Vivo Proliferation and Neurogenesis Analysis. Female mice, between 2 and 4 months of age (age-matched between genotypes), were injected with 50 mg/kg BrdUrd daily for 10 days. For proliferation analysis, 10 WT, 6 MBD1+/-, and 7 MBD1-/- mice were killed on the 11th day. For cell survival analysis, six WT, eight MBD1+/-, and seven MBD1-/- mice were killed 4 weeks postinjection. Cell proliferation, neurogenesis, and hippocampal volume analyses were performed as described (24).
Behavior and LTP Analysis. A battery of neurological tests, including rotarod and field activity analyses, was performed as described (30, 31). Morris water maze learning tests were performed according to the published paradigm (24). Finally, 29 WT and 20 MBD1-/- mice were used for data analysis. Eleven MBD1-/- and 12 WT female mice were used for LTP analysis. LTP of DG and CA1 was recorded and analyzed as described (24).
Results
Generation of MBD1-/- Mice. To investigate the in vivo function of MBD1, we generated MBD1-/- mice by deleting exons 2–10 of the MBD1 gene (see Fig. 6, which is published as supporting information on the PNAS web site). The remaining coding region is not functional as a transcriptional repressor in in vitro assays (data not shown). In MBD1+/- and MBD1-/- mice, β-gal expression is under the control of endogenous MBD1 promoter. Both Northern blots (data not shown) and in situ hybridization (Fig. 1a) showed no MBD1 transcript in MBD1-/- brain tissue and reduced MBD1 transcript in MBD1+/- mouse brains. Histological analysis indicated that MBD1-/- mice had no detectable developmental defects. Therefore, we focused our analysis on adult mice. Adult MBD1-/- mice appear healthy and fertile, with a normal life span. We performed a battery of neurological tests and found no significant defects in adult MBD1-/- mice. Because the CNS has been shown to be more sensitive than other organs to the mutation of another MBD, MeCP2 (4, 5), we focused our analysis on the brain of adult MBD1-/- mice.
Fig. 1.

Histological analysis of the brains of MBD1-/- mice. (a) In situ hybridization was used to detect the expression of MBD1 and β-gal mRNA. Both probes show strong hybridization in hippocampus. (b and c) Nissl staining of the brains of WT (b) and MBD1-/- (c) mice. (Scale bar, 1.0 mm.) (d) X-Gal staining showing MBD1 expression pattern in the brains of MBD1+/- (+/-) and MBD1-/- (-/-) mice. X-Gal staining is negative in the brains of WT mice (+/+). (e) High magnification of Nissl staining (Upper) and X-Gal staining (Lower) of the DG of hippocampus. (Scale bar, 0.1 mm.)
MBD1-/- Is Localized in Both Neurons and Immature Cells, but Not in Astrocytes. The brain of MBD1-/- mice has structure and cellular arrangements that are indistinguishable from those of WT mice, as shown by Nissl staining (Fig. 1 b and c). Using X-Gal staining, we found that MBD1 protein was expressed throughout the brain, with the highest concentration in the hippocampus (Fig. 1d). No X-Gal staining was found in any white matter tracts, suggesting an absence of staining in oligodendrocytes. The highest density of X-Gal staining was found in the CA1 and DG neuronal layers. Antibody staining confirmed that MBD1 was colocalized with many but not all neurons (NeuN+; Fig. 2a, arrowhead), but not astrocytes (glial fibrillary acidic protein; GFAP+). Interestingly, a small number of large, NeuN-negative (NeuN-) cells in the hilar region (Fig. 2a, asterisk) and SGL of DG, where adult neurogenesis persists (23), expressed higher levels of MBD1 than did NeuN+ granule cells (Fig. 2a, arrow). Some of these large NeuN- cells expressed both nestin and MBD1 (Fig. 2b, arrowhead). Therefore, MBD1 is expressed in neurons and a subpopulation of NeuN-, nestin+ immature cells in the hippocampus.
Fig. 2.

MBD1 is expressed in neurons and in some immature cells. (a) MBD1 protein, detected by β-gal expression, is localized in NeuN+ granule cells of the DG (arrowhead), but not in GFAP+ astrocytes. Some NeuN- cells in the hilar region (*) and in the SGL (arrow) express MBD1 at higher levels than NeuN+ cells in the granule cell layers. (b) MBD1 is expressed in some of the nestin+ cells in the SGL of the DG (arrowhead). (Scale bars, 20 μm.)
MBD1-/- Neural Stem Cells Have Decreased Neuronal Differentiation. Because MBD1 expression is highly concentrated in the hippocampus and in a subpopulation of immature cells, we speculate that MBD1 may play an important role in ANC functions and hippocampal neuroplasticity. We isolated ANCs from the brains of MBD1-/- mice and WT controls. At both mRNA and protein levels, a full-length and a truncated (lacking CXXC3) form of MBD1 was expressed in proliferating WT ANCs but were undetectable in MBD1-/- ANCs (see Fig. 7, which is published as supporting information on the PNAS web site). MBD1-/- ANCs grow mainly as a monolayer in medium containing N2 supplement, but form some spheres at higher density. Under proliferating conditions, MBD1-/- ANCs more readily form spheres than do WT cells. On induction for differentiation, both MBD1-/- and WT cells can differentiate into all three major CNS cell types: neurons, astrocytes, and oligodendrocytes. However, MBD1-/- cells differentiated into 41% fewer type III β-tubulin-positive (TuJI+) neurons than did WT cells (see Fig. 7; WT, 7.45 ± 1.61%; MBD1-/-,4.39 ± 1.56%; P = 0.014, paired t test). The data were generated from three lines of independently isolated primary ANCs, and each ANC cell line was analyzed at both passage 4 and passage 11 (see Table 1, which is published as supporting information on the PNAS web site). The percentage of differentiated astrocytes (GFAP+) was not significantly different between MBD1-/- and WT cells (WT, 42.89 ± 5.78%, MBD1-/-, 45.81 ± 3.61%, P = 0.72, paired t test). Therefore, MBD1-/- ANCs have a small but significant decrease in capacity for neuronal differentiation.
MBD1-/- ANCs Have Increased Endogenous Viral Expression. Previous work (10) has shown that MBD1 does not have sequence specificity, except for in methylated-CpGs. To understand the genetic basis of decreased neuronal differentiation of MBD1-/- ANCs, we used high-density oligonucleotide microarrays (U74Av2 arrays; Affymetrix, Santa Clara, CA) to identify genes whose expression levels in mouse hippocampus were affected by the absence of MBD1. Using the pairwise comparison method with a cutoff at 1.5-fold, we found that only eight probe sets of 7,931 probe sets with detectable levels showed differential expression (see Fig. 8, which is published as supporting information on the PNAS web site). All eight probe sets passed rigorous statistical analyses. As expected, MBD1 expression was detected in WT samples, but was undetected in MBD1-/- samples. Interestingly, all six probe sets showing small but significant increases (≈2-fold) in expression in MBD1-/- samples represented the endogenous virus, IAP (Fig. 8). Differential expression patterns of IAPs in hippocampus were confirmed independently by using both real-time PCR and cDNA microarray methods (data not shown). The expression level of IAP mRNA in MBD1-/- ANCs was 2.8-fold higher than in the WT samples (Fig. 3a; WT, 1.36 ± 0.72%; MBD1-/-,3.73 ± 0.29%; P < 0.05, t test), and no difference in IAP expression was found in MBD1-/- primary hippocampal astrocytes. No difference was found in the expression levels of MBD2, MBD3, MBD4, and MeCP2 between WT and MBD1-/- mice. Increased expression of IAP GAG protein was also detected by using Western blotting (Fig. 3b). In the absence of Trichostatin A (TSA), an inhibitor of HDAC, the IAP protein level was 5.4-fold higher in MBD1-/- cells than in WT cells. Interestingly, TSA treatment caused a much larger increase in expression of IAP in MBD1-/- cells (a 3.02-fold increase) than in WT cells (a 0.37-fold increase), indicating increased sensitivity to disruption of the HDAC-mediated repression of IAP in MBD1-/- cells (Fig. 3d). The LTR regions of IAP provirus in the mouse genome are heavily methylated in somatic cells (32), and global demethylation in Dnmt1-/- mice causes a large increase in IAP transcription (22). We analyzed the methylation pattern of the LTR of IAP by using both genomic Southern blot and bisulfite sequencing and found there was no significant difference in methylation status of IAP-LTR between WT and MBD1-/- cells. Therefore, unlike in the case of Dnmt1-/- animals, the increased IAP expression in MBD1-/- mice was not a result of demethylation per se. We also performed a Southern blot with a minor satellite probe to detect global methylation status and found that the global DNA methylation level was not different in MBD1-/- ANCs (data not shown).
Fig. 3.

MBD1-/- ANCs have higher expression of IAP and increased aneuploidy. (a) Real-time PCR shows significantly increased expression of IAP in MBD1-/- ANCs (P = 0.038). (b) Western blot showing increased IAP GAG protein expression in MBD1-/- ANCs. The levels of IAP GAG increased after treatment by TSA. (c and d) Distribution of WT (c) and MBD1-/- (d) ANCs with different chromosome numbers. Arrows point to the number of cells with normal chromosome count (40 chromosomes per cell in this case). Note that most WT aneuploid cells lost chromosomes (e), whereas most MBD1-/- cells gained chromosomes (d). (e) Increased percentage of aneuploid cells in MBD1-/- ANCs (P = 0.02). (f) Example of a MBD1-/- cell analyzed by SKY. (43, XY, +2, +10, and +18). White arrowhead indicates a fragment from chromosome 2. *; t test, P < 0.05.
Increased Genomic Instability in MBD1-/- ANCs. Enhanced IAP expression has been found in cancer cells with chromosomal aberrations (33, 34) and Dnmt1-/- cells with increased mutation rates (35). We speculated that increased IAP expression in MBD1-/- ANCs could be either an indication or cause of genomic instability. Genomic instability includes both chromosomal instability (e.g., chromosomal breaks or translocation) and aneuploidy (33). To determine whether MBD1-/- ANCs had genomic instability, we analyzed karyotypes of three pairs of WT and MBD1-/- ANCs. A total of 80 MBD1-/- and 81 WT prometaphase/metaphase spreads were analyzed. It has been shown that neuroblasts have a relatively high percentage (33.2% for fresh isolated neuroblasts and 14% for neuroblasts cultured in FGF-2) of aneuploid cells compared with other cell types (28). We found that prometaphase/metaphase ANCs from WT mice had 21.3% aneuploidy (28). However, MBD1-/- cells had much higher aneuploidy (Fig. 3e; MBD1-/-, 46.0 ± 4.6%; WT, 21.3 ± 4.1%; P < 0.05, t test, n = 3). Furthermore, chromosomal distribution showed that, among MBD1-/- aneuploid cells, 67.6% gained chromosomes, whereas only 27.8% of WT aneuploid cells gained chromosomes (Fig. 3 c and d). The aneuploidy observed in WT neuroblasts was mainly from chromosome loss (28). To determine the type of genomic instability in ANCs, we analyzed an additional 29 MBD1-/- and 30 WT prometaphase/metaphase cells by SKY (Fig. 3f). SKY confirmed the significantly higher aneuploidy of the MBD1-/- cells. Furthermore, 9 of 16 aneuploid MBD1-/- cells analyzed by SKY gained chromosome 10, where MBD3 gene is located (Fig. 3f), whereas none of the 11 aneuploid WT cells gained chromosome 10. The MBD1-/- cell shown in Fig. 3f also gained chromosome 18, which hosts MBD1 and MBD2 genes in WT cells. Even though no chromosomal translocation was found among the cells analyzed, we did observe that three MBD1-/- cells and no WT cells analyzed by SKY gained a fragment of chromosome 2 (Fig. 3f, white arrowhead), suggesting the possibility of chromosomal break. The above data indicate that MBD1 is important for maintaining genomic stability in ANCs.
Adult MBD1-/- Mice Have Reduced Hippocampal Neurogenesis. The reduced neuronal differentiation and increased genomic instability of cultured MBD1-/- ANCs suggest that the MBD1-/- stem cells may have similar deficits in vivo. We found that the forebrains of MBD1-/- mice weighed 15.2% less than those of WT mice (Fig. 4a; WT, 276.3 ± 7.0 mg, n = 20; MBD1-/-, 234.2 ± 6.0 mg, n = 19; P < 0.0001, t test), whereas there was no significant difference in body weight between adult WT and MBD1-/- mice. The decreased brain weight could be either a result of reduced brain size or reduced cell density. Because MBD1 is expressed at a high level in adult hippocampus, we analyzed the cell density of the granule cell layer of the DG. Using stereology, we found that the cell density in the DG of MBD1-/- mice was 8.1% less than in that of WT mice (Fig. 4b; WT, 2.00 ± 0.06 × 10-3 cells per μm3, n = 7; MBD1-/-, 1.84 ± 0.02 × 10-3 cells per μm3, n = 7; P = 0.03, t test). There was no significant difference in the hippocampal volume between WT and MBD1-/- mice (P = 0.5). To determine whether there was any change in cell proliferation that could be responsible for the decreased cell density in the DG of MBD1-/- mice, BrdUrd was injected into adult mice to label proliferating cells in the brain. We found that MBD1-/- mice had nearly normal cell proliferation (BrdUrd+ cells) in the DG, analyzed at 1 day postinjection of BrdUrd (P = 0.37). However, the number of BrdUrd+ cells analyzed at 4 weeks postinjection was 54.9% lower in the DG of MBD1-/- mice compared with that of WT mice (WT, 546.0 ± 51.7 cells per DG, n = 6; MBD1+/-, 456.8 ± 46.9, n = 8; MBD1-/-, 241.7 ± 28.2, n = 8, P < 0.00; Fig. 4c), indicating a significantly decreased survival for newborn cells.
Fig. 4.

Adult MBD1-/- mice have reduced hippocampal neurogenesis. (a) Forebrains of MBD1-/- mice (-/-; n = 20) weigh 15.2% less than those of WT mice (+/+; n = 19; P < 0.0001). There was no significant difference in forebrain weight between WT and MBD1+/- mice (±; n = 14; P > 0.05). (b) Cell density in the DG of MBD1-/- mice is 8.1% lower than in WT mice (P = 0.014). (c) Survival of newborn cells in the DG is reduced in MBD1-/- mice (P < 0.001). (d–h) Phenotypes of BrdUrd+ cells in the hippocampus of WT (d) and MBD1-/- (e) mice. BrdUrd is red, neuron (NeuN+) is green, and astrocyte (GFAP+) is blue. (f and g) Examples of a BrdUrd+ and NeuN+ cell (f) and a BrdUrd+ and GFAP+ cell (g), which were counted to generate data in h.(h) Phenotype summary of BrdUrd+ cells. There is a significant reduction in the percentage of new neurons (P = 0.001) and an increase in percentage of new glia (P < 0.05) and unknown cell types (P < 0.01) in MBD1-/- mice. Filled bars, WT; etched bars, MBD1+/-; and open bars, MBD1-/-. *, t test; P < 0.05; **, P < 0.01; and ***, P < 0.001.
To determine whether reduced newborn cell survival in DG translated into reduced neurogenesis in the MBD1-/- mice, we analyzed the phenotypes of the surviving cells. In addition to reduced cell survival, we found that the percentage of new neurons (NeuN+ and BrdUrd+) in MBD1-/- mice was 42.9% lower compared with WT mice (Fig. 4h; WT, 45.4 ± 2.8%, n = 6; MBD1-/-, 25.9 ± 3.2%, n = 8, P = 0.001). Taken together, the 54.9% decreased BrdUrd+ cells and the 42.9% decreased percentage of new neurons indicate that there is a 74.3% decrease in the number of new neurons in the DG of adult MBD1-/- mice than in their WT littermates. In contrast, the percentages of new astrocytes (GFAP+ and BrdUrd+) and cells with unknown phenotype (BrdUrd+, NeuN-, and GFAP-) were higher in MBD1-/- mice (astrocytes; WT, 3.3 ± 1.5%; MBD1-/-, 7.2 ± 1.4%, P < 0.05; unknown; WT, 51.3 ± 2.3%; MBD1-/-, 66.9 ± 3.3%, P < 0.01), indicating there was little change in the absolute number of new astrocytes, when adding in the reduced cell survival in the DG of MBD1-/- mice (Fig. 4h). Therefore, adult MBD1-/- mice had significantly reduced hippocampal neurogenesis, whereas there was little change in astrocytogenesis.
MBD1-/- Mice Have Deficits in Spatial Learning and DG-Specific LTP. The hippocampus is involved in learning and memory in mammals (36), and changes in learning have been associated with changes in the levels of neurogenesis in adult DG (24, 31). We used the Morris water maze test to probe the learning ability of MBD1-/- mice and found that it took a significantly longer time for them to find the hidden platform, even after 9 days of training (P < 0.001; ANOVA post hoc test; Fig. 5a). There was no difference in total swimming distance (P > 0.05) and visual ability between WT and MBD1-/- mice. For the 4-h (Fig. 5b) and 24-h (data not shown) posttraining probe tests, the platform was removed and mice were placed in the water tank at the opposite quadrant (Fig. 5b, white quadrant) for 60 sec. WT mice spent more time in the target quadrant searching for the platform, whereas MBD1-/- mice spent more time in the start (opposite) quadrant searching for the platform, indicating impaired learning in MBD1-/- mice (P < 0.01, t test). The number of entries to the target (white rectangle inside the black quadrant, Fig. 5b) and mean distance to the target were also significantly different between WT and MBD1-/- mice (data not shown). To make sure that the defects of MBD1-/- mice were learning specific, we tested motor coordination and locomotor activity of MBD1-/- mice by using the rotarod test and open field test, respectively (30). We found that the MBD1-/- mice had no significant deficits in either motor coordination (P = 0.12) or locomotor activity (P = 0.07). Therefore, the MBD1-/- mice appeared to have specific spatial learning deficits.
Fig. 5.

MBD1-/- mice have learning deficits and impaired LTP in the DG. (a) Morris water maze trial tests for 9 consecutive days with four trials per day. •, WT; ○, MBD1-/-. It took MBD1-/- mice a significantly longer time (latency) to find the hidden platform (P < 0.001, ANOVA post hoc test). (b) Morris water maze probe test performed on day 9, 4 h after the last trial test. (Inset) The arrangement of the water maze. Target is the white square inside the dark quadrant. The platform was removed. Each mouse was released in the white quadrant for a 60-sec test. MBD1-/- mice spent significantly less time in the target quadrant (P < 0.001) compared with WT mice. (c and d) LTP in the CA1 region (c) and the DG (d)of WT and MBD1-/- mice. (c) Neither the induction nor the time course for LTP was significantly different in the CA1 region of slices obtained from WT and MBD1-/- mice (P > 0.05). (d) LTP in the DG was severely attenuated in slices obtained from MBD1-/- animals, whereas normal, and significantly greater, LTP was obtained in slices from WT animals (P < 0.05).
We have previously demonstrated that increased neurogenesis and learning are associated with an increase in DG-specific LTP (24). To determine whether the MBD1-/- had altered synaptic plasticity in the DG, we examined the LTP in both the DG and CA1 regions of the hippocampus (Fig. 5 c and d). MBD1-/- mice exhibited a normal LTP in the CA1 region (Fig. 5; WT, 18.8 ± 12%, n = 7; MBD1-/-, 14.6 ± 6%, n = 4, P > 0.05) but a severely attenuated LTP in the DG region (WT, 33.6 ± 4.6%, n = 9; MBD1-/-,0.6 ± 18%, n = 11, P < 0.05). Therefore, in addition to decreased hippocampal neurogenesis, adult MBD1-/- mice had defective spatial learning and impaired DG-specific LTP. Even though a direct link between adult hippocampal neurogenesis and learning ability has not been established, our results are consistent with the previous observations (24) that enhanced neurogenesis in the adult DG correlates with better learning and increased LTP in the DG.
Discussion
We have generated and analyzed mice lacking a functional MBD1 gene to understand the role of MBD1 in neural function. Using both in vitro and in vivo methods, we discovered that MBD1-/- ANCs have a small but significant decrease in neuronal differentiation and significantly increased genomic instability. Adult MBD1-/- mice have decreased hippocampal neurogenesis and spatial learning ability. Our findings indicate that DNA methylation-mediated epigenetic regulation is important in maintaining genomic stability in neural cells and is crucial for normal CNS function.
Much effort has been put into understanding the genetic regulations of adult neurogenesis, but little work has been done to elucidate the epigenetic regulations in this process. The high concentration of MBD1 mRNA and protein in the adult hippocampus suggests they may play important roles in hippocampal functions, including neuroplasticity. The fact that MBD1 is expressed in neurons and immature cells, but not in astrocytes and oligodendrocytes, suggests that MBD1 may be more involved in neurogenesis than in glial genesis. This suggestion is consistent with the finding that hippocampal neurogenesis, but not astrocytogenesis, is decreased in MBD1-/- mice. To date, the only significant deficits that we have detected in MBD1-/- mice are in the nervous system. The deficits in the MeCP2-deficient mice are also mainly in CNS (4, 5). However, unlike the MeCP2-deficient mice, which display Rett Syndrome-like motor function impairment without learning defects (3), MBD1-/- mice have no significant movement deficits but display a severe learning impairment. This difference may be because of the differential localization and functions of these two MBDs.
We also observed a 15.2% decrease in the forebrain weight of adult MBD1-/- mice, which could be a result of reduced brain volume and/or reduced cell density. Stereological analysis of the hippocampus indicates that reduced cell density, rather than hippocampal size, is contributing to the change in brain weight. However, we have observed that a significant fraction of MBD1-/- mice have smaller forebrains and we do not exclude the possibility that the reduced size of other brain structures is contributing to the reduced total brain weight. Even though we have not observed developmental deficits in MBD1-/- mice, it is possible that increased aneuploidy of neural stem cells during development and subsequent increased cell death lead to smaller forebrains in many adult MBD1-/- mice. However, MBD1 appears to have only a mild effect during neural development, as evidenced by the MBD1-/- mice having no obvious structural defects in the brain and hippocampus.
In MBD1-/- mice, we do see a small but significant increase of IAP expression at both mRNA and protein levels. Under normal conditions, IAP proviral DNA is heavily methylated and repressed in the DNA of somatic cells (32). In Dnmt1-/- animals, there is global demethylation and a large increase in IAP transcription (22). MeCP2 has been shown to repress proviral expression in cultured cells (37). The increased IAP expression in MBD1-/- suggests that MBD1 is involved, to some degree, in IAP repression. Increased IAP expression was not observed in liver, spleen, or fibroblast cells from mice lacking MBD2 (6), a finding that could be due either to different functions between MBD1 and MBD2 or to the different sensitivity of tissues and cells to DNA methylation changes. It is not clear whether increased IAP will lead to a higher mutation rate (1). However, increased IAP expression has been found in cancer cells with chromosomal aberrations (33, 34) and in Dnmt1-/- cells with increased mutation rates (35).
Genomic instability, including aneuploidy (33), has been extensively studied in cancer etiology, because most cancer cells are aneuploid. The mechanisms underlying cell aneuploidy, and whether and how it may cause cancer, are still unclear. Maintaining genomic stability is one of the proposed functions for extensive genomic methylation in mammalian cells (18, 38). Our data provide more evidence for such a function. How can genomic stability affect CNS function? In WT animals, aneuploidy-induced cell death may be important for the development of the nervous system (28) or tumor prevention in the adult (neuronal tumors are much rarer than tumors from other tissues). In MBD1-/- animals, abnormally high levels of aneuploidy may be the reason for decreased newborn cell survival in the DG, which may explain why the MBD1-/- animals have decreased forebrain weight and cell density in the DG of the adult hippocampus. Recent work (29) has shown that mice lacking ATM, a serine protein kinase implicated in DNA repair, have increased genomic instability and the ANCs isolated from these mice do not differentiate into neurons or oligodendrocytes. Reduced brain weight and neuronal size were seen in MeCP2-/- mice (4). In Dnmt1 conditional knockout mice, hypomethylated neural cells were quickly eliminated from the brain (20). It will be interesting to determine the karyotypes of ANCs from Dnmt1-/- and MeCP2-/- mice. The results may provide further understanding of the mechanism of DNA methylation in maintaining genomic stability in the CNS. Increased aneuploidy in mature neurons may also contribute to the learning and LTP deficits seen in MBD1-/- mice. Thus, the genomic stability of mature neurons should be investigated in the MBD1-/- mice.
Supplementary Material
Acknowledgments
We thank B. Miller and A. Garcia for technical support; M. Baker for DNA sequencing and real-time PCR services; Dr. P. Taupin, Dr. J. Hsieh, and Dr. V. Chu for critical review of the manuscript; and M. L. Gage for editing the manuscript. This work is partially funded by the National Institutes of Health/National Institute on Aging, the Christopher Reeve Paralysis Foundation, the Lookout Fund, and the ALS Foundation. X.Z. was supported by a National Institutes of Health postdoctoral fellowship. B.R.C. was supported by Canadian Natural Sciences and Engineering Research Council and Foundation for Innovation awards.
Abbreviations: Dnmt, DNA methyltransferase; MDB, methyl-CpG binding protein; ANC, adult neural stem cell; HDAC, histone deacetylase; IAP, intracisternal A particle; DG, dentate gyrus; LTP, long-term potentiation; SKY, spectral karyotyping; X-Gal, 5-bromo-4-chloro-3-indolyl β-D-galactoside.
References
- 1.Bird, A. (2002) Genes Dev. 16, 6-21. [DOI] [PubMed] [Google Scholar]
- 2.Amir, R. E., Van den Veyver, I. B., Wan, M., Tran, C. Q., Francke, U. & Zoghbi, H. Y. (1999) Nat. Genet. 23, 185-188. [DOI] [PubMed] [Google Scholar]
- 3.Shahbazian, M., Young, J., Yuva-Paylor, L., Spencer, C., Antalffy, B., Noebels, J., Armstrong, D., Paylor, R. & Zoghbi, H. (2002) Neuron 35, 243-254. [DOI] [PubMed] [Google Scholar]
- 4.Chen, R. Z., Akbarian, S., Tudor, M. & Jaenisch, R. (2001) Nat. Genet. 27, 327-331. [DOI] [PubMed] [Google Scholar]
- 5.Guy, J., Hendrich, B., Holmes, M., Martin, J. E. & Bird, A. (2001) Nat. Genet. 27, 322-326. [DOI] [PubMed] [Google Scholar]
- 6.Hendrich, B., Guy, J., Ramsahoye, B., Wilson, V. A. & Bird, A. (2001) Genes Dev. 15, 710-723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Millar, C. B., Guy, J., Sansom, O. J., Selfridge, J., MacDougall, E., Hendrich, B., Keightley, P. D., Bishop, S. M., Clarke, A. R. & Bird, A. (2002) Science 297, 403-405. [DOI] [PubMed] [Google Scholar]
- 8.Ohki, I., Shimotake, N., Fujita, N., Jee, J., Ikegami, T., Nakao, M. & Shirakawa, M. (2001) Cell 105, 487-497. [DOI] [PubMed] [Google Scholar]
- 9.Fujita, N., Shimotake, N., Ohki, I., Chiba, T., Saya, H., Shirakawa, M. & Nakao, M. (2000) Mol. Cell. Biol. 20, 5107-5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ng, H. H., Jeppesen, P. & Bird, A. (2000) Mol. Cell. Biol. 20, 1394-1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bestor, T. H. & Verdine, G. L. (1994) Curr. Opin. Cell Biol. 6, 380-389. [DOI] [PubMed] [Google Scholar]
- 12.Zeleznik-Le, N. J., Harden, A. M. & Rowley, J. D. (1994) Proc. Natl. Acad. Sci. USA 91, 10610-10614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gu, Y., Nakamura, T., Alder, H., Prasad, R., Canaani, O., Cimino, G., Croce, C. M. & Canaani, E. (1992) Cell 71, 701-708. [DOI] [PubMed] [Google Scholar]
- 14.Voo, K. S., Carlone, D. L., Jacobsen, B. M., Flodin, A. & Skalnik, D. G. (2000) Mol. Cell. Biol. 20, 2108-2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fujita, N., Takebayashi, S., Okumura, K., Kudo, S., Chiba, T., Saya, H. & Nakao, M. (1999) Mol. Cell. Biol. 19, 6415-6426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hendrich, B., Abbott, C., McQueen, H., Chambers, D., Cross, S. & Bird, A. (1999) Mamm. Genome 10, 906-912. [DOI] [PubMed] [Google Scholar]
- 17.Li, E., Bestor, T. H. & Jaenisch, R. (1992) Cell 69, 915-926. [DOI] [PubMed] [Google Scholar]
- 18.Hassan, K. M., Norwood, T., Gimelli, G., Gartler, S. M. & Hansen, R. S. (2001) Hum. Genet. 109, 452-462. [DOI] [PubMed] [Google Scholar]
- 19.Tucker, K. L. (2001) Neuron 30, 649-652. [DOI] [PubMed] [Google Scholar]
- 20.Fan, G., Beard, C., Chen, R. Z., Csankovszki, G., Sun, Y., Siniaia, M., Biniszkiewicz, D., Bates, B., Lee, P. P., Kuhn, R., et al. (2001) J. Neurosci. 21, 788-797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jackson-Grusby, L., Beard, C., Possemato, R., Tudor, M., Fambrough, D., Csankovszki, G., Dausman, J., Lee, P., Wilson, C., Lander, E. & Jaenisch, R. (2001) Nat. Genet. 27, 31-39. [DOI] [PubMed] [Google Scholar]
- 22.Walsh, C. P., Chaillet, J. R. & Bestor, T. H. (1998) Nat. Genet. 20, 116-117. [DOI] [PubMed] [Google Scholar]
- 23.Gage, F. H. (2000) Science 287, 1433-1438. [DOI] [PubMed] [Google Scholar]
- 24.van Praag, H., Christie, B. R., Sejnowski, T. J. & Gage, F. H. (1999) Proc. Natl. Acad. Sci. USA 96, 13427-13431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lein, E. S., Hohn, A. & Shatz, C. J. (2000) J. Comp. Neurol. 420, 1-18. [DOI] [PubMed] [Google Scholar]
- 26.Zhao, X., Lein, E. S., He, A., Smith, S. C., Aston, C. & Gage, F. H. (2001) J. Comp. Neurol. 441, 187-196. [DOI] [PubMed] [Google Scholar]
- 27.Barch, M. J., Knutsen, T. & Spurbeck, J. L. (1997) The AGT Cytogenetics Laboratory Manual (Lippincott, Philadelphia).
- 28.Rehen, S. K., McConnell, M. J., Kaushal, D., Kingsbury, M. A., Yang, A. H. & Chun, J. (2001) Proc. Natl. Acad. Sci. USA 98, 13361-13366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Allen, D. M., van Praag, H., Ray, J., Weaver, Z., Winrow, C. J., Carter, T. A., Braquet, R., Harrington, E., Ried, T., Brown, K. D., et al. (2001) Genes Dev. 15, 554-566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Crawley, J. N. (1999) Brain Res. 835, 18-26. [DOI] [PubMed] [Google Scholar]
- 31.Kempermann, G., Brandon, E. P. & Gage, F. H. (1998) Curr. Biol. 8, 939-942. [DOI] [PubMed] [Google Scholar]
- 32.Yoder, J. A., Walsh, C. P. & Bestor, T. H. (1997) Trends Genet. 13, 335-340. [DOI] [PubMed] [Google Scholar]
- 33.van Gent, D. C., Hoeijmakers, J. H. & Kanaar, R. (2001) Nat. Rev. Genet. 2, 196-206. [DOI] [PubMed] [Google Scholar]
- 34.Laird, P. W. & Jaenisch, R. (1996) Annu. Rev. Genet. 30, 441-464. [DOI] [PubMed] [Google Scholar]
- 35.Chen, R. Z., Pettersson, U., Beard, C., Jackson-Grusby, L. & Jaenisch, R. (1998) Nature 395, 89-93. [DOI] [PubMed] [Google Scholar]
- 36.Kandel, E. R. (2001) Science 294, 1030-1038. [DOI] [PubMed] [Google Scholar]
- 37.Lorincz, M. C., Schubeler, D. & Groudine, M. (2001) Mol. Cell. Biol. 21, 7913-7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Esteller, M. & Herman, J. G. (2002) J. Pathol. 196, 1-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}