

Abstract
It is now accepted that the structural transition from cellular prion protein (PrPC) to proteinase K-resistant prion protein scrapie (PrPSc) is the major event leading to transmissible spongiform encephalopathies. Although the mechanism of this transition remains elusive, glycosylation has been proposed to impede the PrPC to PrPSc conversion. To address the role of glycosylation, we have prepared glycosylated and unglycosylated peptides derived from the 175–195 fragment of the human prion protein. Comparison of the structure, aggregation kinetics, fibril formation capabilities, and redox susceptibility of Cys-179 has shown that the N-linked glycan (at Asn-181) significantly reduces the rate of fibrillization by promoting intermolecular disulfide formation via Cys-179. Further-more, the aggressive fibrillization of a C179S mutant of this fragment highlights the significant role of disulfide stability in retarding the rate of fibril formation. The implications of these studies are discussed in the context of fibril formation in the intact prion protein.
Spongiform encephalopathies are a group of fatal neurodegenerative diseases that can be manifested sporadically, genetically inherited, or in some cases transmitted (1–3). In contrast to many diseases where the nature of the infectious particle is virus or bacterium, currently, the only agent associated with these conditions is the structural isoform of the cellular prion protein (PrPC) known as the scrapie conformation (PrPSc) (1, 4). PrPC is an N-linked glycoprotein that is normally attached to the cell membrane by a glycosylphosphatidylinositol anchor (5). PrPC also contains an intramolecular disulfide bond (Cys-179—Cys-214) that provides structural stability to the C terminus of the protein (6, 7). Secondary structure analyses have shown that PrPC is a predominantly helical protein, whereas PrPSc is mainly β-sheet, suggesting that the conversion into PrPSc involves a major structural change (8). Although the structure of the unglycosylated PrPC monomer has been solved by NMR spectroscopy (7, 9), elucidation of the PrPSc structure has been hampered by its highly aggregated state.
As with many membrane-associated proteins, initiation of the biosynthesis of PrP begins with translocation into the endoplasmic reticulum (ER) where the amino-terminal signal sequence is cleaved (1). The ER houses the machinery for asparagine-linked glycosylation as well as for disulfide formation (10, 11). Proteins in the secretory pathway that are misfold in the ER (12, 13) are subject to retrograde transport into the cytosol, where they are degraded by proteasomes (14, 15). In particular, protein isoforms (or mutants) that are less stable during their maturation are most likely to undergo this retrograde transport (14). Lindquist and coworkers have recently shown that inhibition of the proteasome causes PrP to accumulate in the cytosol, where it can adopt a PrPSc-like conformation (16, 17). Furthermore, expression of an unglycosylated, cytosolic form of PrP has extremely toxic effects on neurobalstoma cells as well as in transgenic mice (17, 18). It has also been shown that inhibition of glycosylation by using tunicamycin or exposure of neuroblastoma cells expressing PrP to DTT can cause the protein to acquire PrPSc-like conformations (18).
Many factors have been associated with the conversion of PrPC to PrPSc. For example, fluctuations in metal ion concentrations (19, 20), glycosylphosphatidylinositol anchor stability (21), extracellular molecules such as glycosaminoglycans (22, 23), pH (24), N-linked glycosylation (18, 25), and the redox environment have all been proposed to be implicated (18, 26, 27). Some of these factors (e.g., variations in pH, posttranslational modifications, or redox environment in the secretory pathway) (13, 17) could also be associated with protein localization or processing within the cell. Therefore, the available data suggest that the structural transition between PrPC and PrPSc might not be induced simply by one specific factor, but rather due to the additive effect of a combination of factors. In some cases, there is a possibility that these factors could be interrelated and affecting each other. The evidence showing that a reducing environment can promote the generation of PrPSc-like species (6, 16–18) suggests that factors affecting the stability of the disulfide bridge could also have an effect in preventing or promoting the PrPC to PrPSc transition. Previous glycopeptide studies reveal that N-linked glycosylation can impact the conformation of protein fragments (28–30). In particular, we have shown that glycosylation can alter the thermodynamics of disulfide bond formation, favoring the oxidized form (31). In this study we prepare a peptide and a glycopeptide corresponding to residues 175–195 of the human PrP to investigate the effects of the core carbohydrate unit (chitobiose) of N-linked glycans on the stability and conformational properties of the fragment. The 175–195 segment is derived from helix 2 of the prion protein and contains one of the two glycosylation sites (Asn-181) found in the protein and a cysteine at position 179 (see Fig. 1) (7). Although a detailed study of the intact helix-turn-helix fragment, including both Cys-179 and Cys-214, would have been desirable, the technical limitations in the chemical synthesis of such large glycosylated peptide fragments currently precludes the preparation of such a target. Our results show that N-linked glycosylation, with the disaccharide chitobiose, has a significant effect on the fibril formation capabilities of the PrP175–195 fragment; glycosylation significantly decelerates the rate of fibrillization. Furthermore, the carbohydrate moiety appears to alter the redox properties of C179, causing the peptide to be stabilized as the intermolecular disulfide homodimer, which is highly resistant to fibril formation. These studies may have important implications when considering the effect of asparagine-linked glycosylation and redox environment on prion proteins. A model for relating the results of studies with the prion protein fragment with the intact prion protein is presented in the discussion section.
Fig. 1.

(A) Ribbon diagram generated from the human PrP 121–230 NMR structure (7). Asn-181 and Asn-197 (glycosylation sites) are colored by atom. Disulfide bridge between Cys-197 and Cys-214 is shown in yellow. Fragment 175–195 is shown in blue. (B) Synthetic unglycosylated (PrPUP) and glycosylated (PrPGP) peptides derived from the PrP 175–195 fragment.
Materials and Methods
Peptide Synthesis. The unglycosylated peptide (PrPUP) was synthesized on PAL-PEG-PS resin by using a MilliGen/Biosearch 9050 automated peptide synthesizer. The peptides were prepared as the C-terminal amide and the N-terminal acetyl derivative to better mimic internal sequences in PrP. Standard 9-fluorenylmethoxycarbonyl (Fmoc) chemistry and HBTU/ HOBT activation was used for all residues except cysteine. In this case, preactivated Fmoc-l-Cys(Trt)-OPfp was used in the absence of base to prevent racemization. For glycopeptide synthesis, incorporation of the GlcNAc–GlcNAc disaccharide was achieved by using a presynthesized Fmoc-l-Asn[β-chitobiose-(TBDMS)5]-OH derivative (32). Peptides were cleaved from the resin by using 92% trifluoroacetic acid, 2% triisopropylsilane, 3% ethanedithiol and 3% H2O, purified by using preparative reverse-phase HPLC and characterized by using electrospray mass spectrometry (ESMS) and quantitative amino acid analysis (see supporting information, which is published on the PNAS web site, www.pnas.org). The C179S mutant was synthesized by using similar procedures.
CD Spectroscopy. CD spectra were recorded at 25°C on an Aviv 202 CD spectrophotometer in Quartz cuvettes with an optical path length of 0.1 cm. Peptide stock solutions were prepared by dissolving the peptides in water to a concentration of 210 μM at pH 4 to prevent rapid fibril formation (both peptides remain soluble indefinitely at this pH; see supporting information). Aliquots of the peptides were then mixed with sodium phosphate buffer pH 7.5 and DTT to final concentrations of 70 μM peptide, 1 mM buffer, and 1 mM DTT. The buffer and DTT contribution were subtracted for all spectra. Any contribution to the signal from the carbohydrate was subtracted by using chitobiose amine as a standard. The signal was normalized to mean residue ellipticity based on the peptide concentration.
Fourier Transform Infrared (FTIR) Spectroscopy. FTIR spectra were recorded on a Nicolet Magna 560 infrared spectrometer using a rectangular cuvette with CaF2 windows and 50-μm spacers. Peptides were dissolved in D2O (pD ≈ 3.9) and lyophilized twice before analysis to remove residual H2O. For analysis, peptides were redissolved in D2O to a concentration of 1.3 mM (pD 3.9) and deuterated phosphate buffer (pD 7.9) and DTT were added to a concentration of 5 mM buffer and DTT. FTIR spectra were recorded after aggregation.
Negative Staining Electron Microscopy (EM). Samples used for EM analysis were obtained from aggregation studies (usually 24 h after incubation). Five microliters aliquots were taken from the incubation samples and placed on carbon-coated 300-mesh nickel grids and adsorbed for 2 min. The grids were then stained with 2% uranyl acetate for 2 min, and after drying, were viewed in a Philips EM 410 operated at 80 KV at magnifications of ×55,000, ×24,000, and ×10,000.
Aggregation Kinetics. Although thioflavin T, Congo red, and visible light scattering assays gave similar results, a visible light scattering assay in 96-well plate format was preferentially used to study the aggregation because of its capability for multiple sample handling. Kinetic analysis was performed at 25°C on a Molecular Devices Spectramax 190 plate reader. Peptide solutions were prepared as described (see CD Spectroscopy), and kinetic studies were initiated by mixing the peptide in water at pH 4 with phosphate buffer at pH 7.5. The 96-well plate was agitated for 1 s every 20 min, and absorbance at 425 nm was recorded immediately after agitation. The analyses were performed in duplicate or triplicate, and the average and standard deviation values were reported.
Evaluation of the Aggregation Kinetics Data. The model from Nielsen (33) was used to fit the data and calculate the lag times for the aggregation kinetics (Eq. 1).
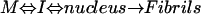 |
[1] |
Here, M is the peptide monomer and I is an intermediate. In this model, fibril formation is described as a sigmoidal curve defined by an initial lag phase (where no increase in the 425-nm absorbance is observed), followed by a growth phase (when major aggregates start to form), and finally an equilibrium phase when the signal reaches a plateau (when aggregation is complete) (33). The absorbance at 425 nm was plotted as a function of time, and the data were fitted to the model described by Eq. 2 using kaleidagraph (33)
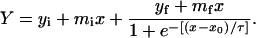 |
[2] |
Here, Y is the absorbance at 425 nm, x is the time, x0 is the time to reach 50% of the maximum absorbance and 1/τ gives the apparent rate constant for fibril growth (kapp). The lag time is then calculated from x0–2τ. The data analysis showed good agreement with this model (R values of 0.99).
Congo Red Binding. Aliquots of 40 μl were taken from the aggregation kinetics samples after 24 h of incubation. Congo red (in 10 mM phosphate buffer at pH 7.5) was added to the aliquots to a final concentration of 10 μM for visual inspection and 5 μM for UV–visible analysis. The solutions were incubated for 10 min and then analyzed in a Beckman DU 7500 spectrophotometer using a 1-cm path length cuvette. A spectrum of the unbound dye was also recorded. Spectra of the peptide without dye were recorded for subtraction from the Congo red–peptide spectra to reduce the high baseline caused by the peptide aggregation.
ESMS. An Applied Biosystems Mariner ESMS was used to record the mass spectra of the samples. After 5 days of incubation, 10-μl aliquots from the soluble peptide samples were directly injected into the ESMS running in a 50% methanol/water (0.05% acetic acid) solvent system at 0.25 ml/min. Aggregated samples were first centrifuged, and 10 μl of the supernatant was used for analysis.
NMR Spectroscopy. Peptides (1 mM) were dissolved in a cold (7°C) solution of 5 mM NaOAc-d3, pH 4.5, 10% D2O in water (0.01% DMSO was used as internal reference). All of the NMR experiments were performed at pH 4.5 and 7°C to prevent fibrillization. Spin systems for both peptides were assigned by using total correlation spectroscopy (TOCSY), NOESY, and COSY experiments (34) with Watergate gradient suppression. The 3JNHα coupling constant values were calculated from DQF-COSY experiments at 7°C (34). NOESY spectra were acquired by using 400-ms mixing times. Spectra were recorded on a 600 MHz Bruker DRX 600 Avance spectrometer. The NMR data were processed by using felix 97 software (MSI).
Results
CD and FTIR of PrPUP and PrPGP. Spectra taken immediately after dissolution displayed random-coil characteristics for both unglycosylated (PrPUP) and glycosylated (PrPGP) peptides (Fig. 2 Left). However, after incubation for 24 h, the CD spectra displayed a characteristic β-sheet signature (Fig. 2 Left). Precipitates were also noted in the samples at this stage. Because CD spectroscopy does not provide structural information on the precipitated material, the total secondary structure in the suspension was examined by FTIR. The maxima at 1,615 cm–1 for both peptide and glycopeptide confirmed the presence of aggregated β-sheets as the major secondary structure present in the suspension (Fig. 2 Right). These results reflect the natural tendency of both PrPUP and PrPGP to undergo a structural transition into aggregated β-sheets.
Fig. 2.

(Left) CD spectra of PrPUP at 0 (dotted line) and 24 (solid line) h and PrPGP at 0 (intermediate dashed line) and 24 (dashed line) h of incubation in 1 mM phosphate buffer (pH 7.5) and 1 mM DTT. (Right) Amide I′ region of the FTIR spectra for PrPUP (solid line) and PrPGP (dashed line) after aggregation at pH 7.5 and 5 mM DTT.
Effect of Glycosylation on the Kinetics of Aggregation and Disulfide Stabilization. Previous studies suggest that glycosylation delays the PrPC to PrPSc transition in cells (35). It has also been demonstrated that disulfide stability in PrPC can play an important role in stabilizing the native fold of the protein (6). Although fragment 175–195 includes only one cysteine residue, Cys-179 has the potential to form intermolecular disulfide bonds that could affect the ability of the PrP fragment to form fibrils. To study the effects of glycosylation and reducing environment on the aggregation kinetics of the PrP fragment, we studied the fibrillization of the peptides at various DTT concentrations. A summary of the aggregation lag times for both peptide and glycopeptide are shown in Table 1. The aggregation kinetics for both peptides showed a well defined dependence on DTT concentrations. For both PrPUP and PrPGP, the aggregation lag time decreased with increasing concentrations of DTT. However, PrPGP showed slower aggregation than PrPUP when incubated at similar DTT concentrations (Fig. 3). PrPGP also showed a higher resistance to aggregation at lower DTT concentrations. Although at higher DTT concentrations the difference between the aggregation lag times for PrPUP and PrPGP was not so dramatic (3.4 h for PrPUP and 6.2 h PrPGP at 10 mM DTT), the difference in the aggregation lag times was dramatically increased at lower DTT concentrations or in the absence of DTT. Whereas PrPUP begins to aggregate at 6.9 h in the absence of DTT, no aggregation was observed for PrPGP by 5 days under comparable conditions. It required 100 times more DTT to accelerate the aggregation kinetics of PrPGP to be similar to PrPUP (0.1 mM for PrPUP and 10 mM DTT for PrPGP). Also, no major decrease in the aggregation lag time was observed for PrPUP after the DTT concentration was increased above 1 mM. On the other hand, the aggregation lag times for PrPGP continued to decrease between 1.0 and 10.0 mM DTT concentrations. These results show that for both species, a reducing environment accelerates fibril formation. However, the glycopeptide appears to be more stable in the soluble form than the unglycosylated peptide in a reducing environment.
Table 1. Summary of aggregation lag times for 70 μM PrPUP and PrPGP in 1 mM sodium phosphate buffer (pH 7.5) and different reducing conditions.
| Reducing agent | PrPUP lag time, h | PrPGP lag time, h |
|---|---|---|
| None | 6.9 ± 0.8 | >120 |
| 0.1 mM DTT | 5.9 ± 0.9 | >24 |
| 1.0 mM DTT | 3.4 ± 0.6 | 8.8 ± 0.2 |
| 10.0 mM DTT | 3.4 ± 0.2 | 6.2 ± 0.5 |
| 0.1 mM GSH | 4.6 ± 0.8 | >120 |
Fig. 3.

Comparison of aggregation kinetics for 70 μM PrPUP (solid line) and 70 μM PrPGP (dashed line) in 1 mM phosphate buffer (pH 7.5) and different DTT concentrations: 10 mM DTT (A), 1 mM DTT (B), 0.1 DTT (C), or no DTT (D).
The effect of reduced and oxidized glutathione on the aggregation kinetics of the peptides was also investigated. In the cell, oxidized and reduced glutathione levels are regulated to maintain control of the redox environment. Similar conditions to those used for studies with DTT were used. Both peptides (PrPUP and PrPGP) were incubated in 1 mM sodium phosphate buffer (pH 7.5) and 0.1 mM reduced or oxidized glutathione, and the aggregations kinetics were monitored. Peptide solutions incubated with oxidized glutathione displayed complete inhibition of aggregation with both PrPUP and PrPGP. In contrast, when the peptides were incubated with reduced glutathione, PrPUP showed significant aggregation (with aggregation lag time of 4.6 ± 0.8 h), whereas PrPGP aggregation was inhibited at least for 5 days (Table 1). These results reinforce the proposal that the glycopeptide has a higher resistance to the redox environment than the unglycosylated peptide.
EM Analysis of PrPUP and PrPGP. EM studies were implemented to provide direct information about the specific supramolecular structures generated with PrPUP and PrPGP. EM analysis for PrPUP and PrPGP solutions incubated with 1 mM DTT for 24 h indicated the presence of fibrils (Fig. 4A Upper and Lower, respectively). Only minor differences are noted in the analyses; in general, PrPUP solutions displayed more extended fibril networks than PrPGP, and PrPGP showed a higher quantity of small-sized filaments near the major fibrils. On the other hand, PrPUP and PrPGP solutions incubated in the absence of DTT showed significant differences (Fig. 4B Upper and Lower, respectively). PrPUP grids showed a high concentration of fibrils. In contrast, no major fibrils, only minor precipitates, were observed for PrPGP under the same conditions. These results were also corroborated by the Congo red assay (supplementary material) (36). Similarly, EM analysis of PrPUP incubated with 0.1 mM reduced glutathione displayed fibrils, whereas only minor preciptates were observed for PrPGP under similar conditions (Fig. 4C Upper and Lower, respectively). No fibrils were observed in the peptide solutions incubated with oxidized glutathione. These results are in good agreement with the aggregation kinetics and suggest that fibril formation could be inhibited by oxidation of the peptide. Additionally, the N-linked chitobiose moiety may alter the properties of the glycopeptide, rendering it more resistant to fibril formation. This effect is more pronounced in a nonreducing environment, where the glycosylated peptide (PrPGP) is inhibited from forming fibrils (in contrast to the unglycosylated peptide).
Fig. 4.

EM pictures for PrPUP (Upper) and PrPGP (Lower). (A) Peptides incubated for 24 h in 1 mM phosphate buffer (pH 7.5) and 1 mM DTT. (Bar = 100 nm.) (B) Peptides incubated for 24 h in 1 mM phosphate buffer (pH 7.5) in the absence of DTT. (Bar = 200 nm.) (C) Peptides incubated for 24 h in 1 mM phosphate buffer (pH 7.5) and 0.1 mM reduced glutathione. (Bar = 200 nm.)
Effect of C179S Mutation. To confirm that the inhibition of fibril formation in the peptide fragment is related to Cys-179 (probably by cystine formation), we synthesized the C179S mutant of the unglycosylated PrP 175–195 peptide fragment. Aggregation kinetics, Congo red binding assays and EM analyses were performed on this mutant and compared with the native unglycosylated peptide. Interestingly, aggregation kinetics showed that the C179S mutant aggregates instantaneously without a significant lag time. Also, the aggregation kinetics for the mutant did not show any significant dependence on DTT concentration (Fig. 5 Left). Similar results were observed when incubation was carried out with oxidized glutathione; although oxidized glutathione inhibits the fibrillization of PrPUP, it did not have any effect on the aggregation kinetics of C179S. Visual inspection of the peptide solutions incubated with the Congo red dye showed a very intense pinkish-red color for the C179S mutant compared with the wild type PrPUP (Fig. 5 Right Inset). The high reactivity of the mutant with Congo red was then corroborated by UV-visible analysis (Fig. 5 Right). The wild type and the C179S mutant caused a significant red shift on the Congo red maxima from 488 nm to 520 nm (32 nm). However, titrating similar amounts of peptides into the Congo red solution caused a noticeably higher absorbance. In addition, EM analysis of the mutant revealed a high amount of entangled fibrils (Fig. 6). These results indicate that the C179S mutant is more ``aggressive'' in the fibrillization process and suggest that Cys-179 plays a major role in controlling the formation of fibrils. These results also reinforced the idea that the fibrillization pathway can be inhibited by intermolecular cystine formation.
Fig. 5.

Effect of C179S mutation on the fibril formation of the PrP175–195 fragment. (Left) Aggregation kinetics for C179S in the presence of 10 mM DTT (red) and absence of DTT (blue), and for wild-type PrPUP incubated with 10 mM DTT (dashed line with filled squares) and absence of DTT (dashed lines with open squares). (Right) UV-visible spectra of Congo red (dotted line) and Congo red titrated with C179S (solid line) and PrPUP (dashed line) after 24 h of incubation in the absence of DTT. (Inset) Visual inspection of Congo red solutions titriated with the mutant (S) and wild-type (WT) solutions after 24 h of incubation in the absence of DTT.
Fig. 6.

EM of C179S (A) compared to wild-type PrPUP (B). Both samples were placed on the EM grid 24 h after incubation in 1 mM phosphate buffer (pH 7.5) in the absence of DTT. (Bars = 200 nm.)
Evidence for a Redox Mechanism by Mass Spectrometry. The previous data suggest that a peptide oxidation could be involved in the inhibition of fibril formation and that glycosylation promotes this oxidation. Therefore, we examined the solutions in which fibrillization was inhibited after 5 days of incubation by ESMS. Although PrPUP formed fibrils in the absence of DTT, the suspension was analyzed to compare the results with PrPGP incubated in the absence of DTT. For this case, it was necessary to first centrifuge the PrPUP suspension to remove major precipitates and only use the supernatant for the analysis. Although most of the unglycosylated sample was aggregated, a minor peak corresponding to the PrPUP monomer ([M + 3H+]/3 795.7 (observed); 795.9 (calculated) was observed in the supernatant (Fig. 7A). On the other hand, ESMS analysis of the PrPGP solution showed masses corresponding to the cystine-linked glycopeptide dimer [[M + 5H+]/5 1,117.7 (observed); 1,117.0 (calculated), [M + 6H+]/6 931.4 (observed); 931.0 (calculated), [M + 7H+]/7 798.3 (observed); 798.2 (calculated)] (Fig. 7B). The presence of the intermolecular cystine dimer in the glycopeptide sample and its absence in the unglycosylated peptide sample, suggest that this is the major species responsible for inhibition of the peptide self-assembly into fibrils. Therefore, dimerization via intermolecular cystine formation could serve as an alternate process that competes with fibril formation. These observations correlate with the previous results showing the dependence of the reductive environment on fibril formation and the enhanced effect of the carbohydrate moiety against the fibrillization pathway, potentially by promoting the oxidation.
Fig. 7.

ESMS spectra of peptide solutions after 5 days of incubation at pH 7.5. (A) Supernatant of PrPUP after incubation in the absence of DTT. (B) PrPGP after incubation in the absence of DTT. (C) PrPUP after incubation with 0.1 mM oxidized glutathione. (D) PrPGP after incubation with 0.1 mM oxidized glutathione.
ESMS analysis of solutions incubated with oxidized glutathione showed similar results for both peptides; the peptide–glutathione adduct linked via disulfide bond was the major species present in the samples (Fig. 7 C and D). For PrPUP we observed: [M + 3H+]/3 897.4 (observed); 897.7 (calculated), [M + 4H+]/4 673.3 (observed); 673.5 (calculated). For PrPGP samples incubated with oxidized or reduced glutathione, the peptide-glutathione adduct linked through the disulfide bond was observed (Fig. 7D). Only minor precipitates were observed in the PrPGP sample incubated with reduced glutathione after the 5 days of incubation. The masses observed for PrPGP incubated with oxidized glutathione were: [M + 3H+]/3 775.3 (observed); 775.1 (calculated), [M + 4H+]/4 1,033.4 (observed); 1,033.1 (calculated). When incubated with reduced glutathione we observed: [M + 3H+]/3 775.1 (observed); 775.1 (calculated), [M + 4H+]/4 1,033.1 (observed); 1,033.1 (calculated). These results reinforce the idea that an alternate mechanism promoting the formation of intermolecular disulfide (either to form a homodimer or a mixed disulfide with glutathione) could play a major role in inhibiting the fibril formation pathway in this prion peptide fragment. These observations also strengthen the proposal that glycosylation could be decelerating the fibrillization mechanism by promoting this alternate intermediate.
Structural Differences Between PrPUP and PrPGP by NMR. Previous studies have shown that N-linked glycosylation can impact the local structure of peptides by promoting adoption of more compact conformations (34). In systems where the peptide local conformations are affected, but not the complete secondary structure, 3JHNα coupling constants have provided an indication of local conformational preferences (34, 37, 38). Although both PrPUP and PrPGP displayed unstructured characteristics, comparison of the 3JHNa coupling constants between the peptides only showed noticeable differences near the glycosylation site (at Asn-181) (supporting information). A significant increase in the 3JHNa coupling constants at Asn-181 from 7.0 Hz in PrPUP to 8.7 Hz in PrPGP was observed. Because 3JHNα coupling constants provide information about the dihedral angles of the peptide backbone, similar elevations on this parameter in analogous systems have been previously attributed to the induction of more compact local structures (34, 37, 38). The observed differences suggest that a minor local conformational change in the peptide near the glycosylation site may be caused by the chitobiosyl moiety. Although this minor effect does not seem to influence the total peptide structure significantly, it could potentially be related to the induction of intermolecular cystine formation with Cys-179, which is only two residues away from the glycosylation site.
Discussion
In this study we have synthesized and studied unglycosylated and glycosylated peptides corresponding to the 175–195 fragment of the human prion protein. The glycosylated peptide was modified with the core chitobiose disaccharide found in all N-linked glycoproteins. Comparison of the structure, aggregation kinetics, fibril formation capabilities and redox susceptibility of Cys-179 has provided insight into the effects of the N-linked carbohydrate on the stability of the PrP fragment. Although both peptides are capable of undergoing a random coil to β-sheet transition to form fibrils at and above physiological pH, glycosylation caused a significant deceleration in the rate of fibril formation. The fibrillization of both peptides also showed a major dependence on reductive environment; however, the glycopeptide showed a higher resistance to aggregation under similar reducing conditions. Mass spectrometry then confirmed that the greater stability displayed by the glycopeptide was caused by a higher tendency of this peptide to exist as the intermolecular homodimer linked via a disulfide bond (Fig. 7). The proposal that intermolecular disulfide bond formation stabilizes the peptide as a soluble species was also supported by the aggressive fibrillization of the C179S mutant.
The fact that the PrP 175–195 fragment is capable of forming fibrils at physiological pH, but the fibrilization is inhibited when the peptide is dimerized through disulfide formation is note-worthy when put in the context of the intact protein. In the native protein, part of this fragment is occluded by helix 3. Masking of the peptide surface is strongly stabilized by the Cys-179—Cys 214 disulfide bond (Fig. 1A). Therefore, if exposure of a surface of the 175–195 fragment can induce fibrilization (as displayed by the peptide), to expose that peptide surface, the disulfide must first be reduced. This is in agreement with our results on the peptide fragment and previous reports on the total protein that show that a reducing environment promotes the fibrilization process or a PrPSc-like conformation (6, 16–18). Together these results suggest that factors stabilizing the Cys-179—Cys-214 disulfide are of extreme importance in preventing the PrPC to PrPSc transition. As observed from the primary sequence and the NMR structure of the prion protein (7), the two glycosylation sites are in close proximity to the disulfide bond. Asn-181 is only two residues away from Cys-179, and the other glycosylation site (Asn-197) is located at the turn between the two helices held together by the disulfide (Fig. 1). Although for the 175–195 fragment, formation of the disulfide bond must be intermolecular, a similar behavior is observed; the formation of the disulfide bond inhibits the fibrillization process. Therefore, these results suggest that glycosylation may have a similar effect in the native protein; stabilizing the PrPC isoform in an indirect manner by promoting the intramolecular disulfide stability. Fig. 8 summarizes the effects of glycosylation on the PrP 175–195 fragments and the possible effects that glycosylation might have on the prion protein. In the peptide fragment, intermolecular disulfide bond formation competes with fibrillization (Fig. 8A). The N-linked chitobiose moiety seems to drive the fragment toward the dimer state by promoting the disulfide bond stability. In the intact protein, exposure to a reducing environment, as for example in the cytoplasm when the protein is retrograde transported (14, 16, 17), the intramolecular disulfide bond can be reduced. The surface of the 175–195 fragment with high propensity for fibrillization is then exposed, which promotes the protein fibrillization. It is also possible that final intermolecular disulfide rearrangements can then take place to form the ultimate, protease-resistant PrPSc (27). Although exposure to a reducing environment can also induce PrPSc formation on the glycosylated PrPC, glycosylation could stabilize the intramolecular disulfide bond in the protein decreasing the fraction of PrPSc formation. In the intact protein, the glycans could have a significant impact on the properties of the disulfide bond. They could shield the cystine, which would stabilize the oxidized form of the protein, or they could perturb the redox potential of the cysteines, thus promoting the oxidation (31).
Fig. 8.

Disulfide stabilization by N-linked glycans in the PrP 175–195 fragment (A) and in the context of the protein (B). In the 175–195 fragment, glycosylation promotes the formation of the intermolecular cystine, which inhibits the fibrilization of the peptide. Glycosylation could have a similar effect on the prion protein by stabilizing the intramolecular disulfide bond.
With the results presented here, several proposals can be put forward to explain how glycosylation might affect the fibrillization mechanism. (i) The carbohydrate moiety could cause a minor destabilization on the peptide structure required for the specific intermolecular interactions leading to fibril formation. (ii) Glycosylation could cause a steric effect that inhibits intermolecular association leading to fibrillization. Although this could be a nonspecific factor, this effect could be very significant considering that the size of the PrP carbohydrates in vivo are large and occupy a significant surface area of the protein. (iii) N-linked glycosylation could also be diverting the fibrillization mechanism by indirectly affecting other aspects of the peptide structure. For example, it could influence the oxidation potential of the cysteine residue thus promoting the formation of the intermolecular cystine dimer that could compete with fibril formation.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant GM-39334. We also acknowledge support of the Massachusetts Institute of Technology NMR Facility by National Institutes of Health Grant 1S10RR133886-01.
Abbreviations: PrPC, cellular prion protein; PrPSc, proteinase K-resistant prion protein scrapie; PrPUP, unglycosylated peptide; PrPGP, glycosylated peptide; ESMS, electrospray MS; FTIR, Fourier transform infrared; EM, electron microscopy.
References
- 1.Prusiner, S. B. (1998) Proc. Natl. Acad. Sci. USA 95, 13363–13383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weissmann, C. (1994) Trends Cell Biol. 4, 10–14. [DOI] [PubMed] [Google Scholar]
- 3.Caughey, B. & Chesebro, B. (1997) Trends Cell Biol. 7, 56–62. [DOI] [PubMed] [Google Scholar]
- 4.Prusiner, S. B. (1982) Science 216, 136–144. [DOI] [PubMed] [Google Scholar]
- 5.Stahl, N., Borchelt, D. R., Hsiao, K. & Prusiner, S. B. (1987) Cell 51, 229–240. [DOI] [PubMed] [Google Scholar]
- 6.Jackson, G. S., Hosszu, L. L. P., Power, A., Hill, A. F., Kenney, J., Saibil, H., Craven, C. J., Waltho, J. P., Clarke, A. R. & Collinge, J. (1999) Science 283, 1935–1937. [DOI] [PubMed] [Google Scholar]
- 7.Zahn, R., Liu, A., Lührs, T., Riek, R., Schroetter, C. V., Gracía, F. L., Billeter, M., Calzolai, L., Wider, G. & Wüthrich, K. (2000) Proc. Natl. Acad. Sci. USA 97, 145–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pan, K.-M., Baldwin, M., Nguyen, J., Gasset, M., Serban, A., Groth, D., Melhorn, I., Huang, Z., Fletterick, R. J., Cohen, F. E., et al. (1993) Proc. Natl. Acad. Sci. USA 90, 10962–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.James, T. L., Liu, H., Ulyanov, N. B., Farr-Jones, S., Zhang, H., Donne, D. G., Kaneko, K., Groth, D., Melhorn, I., Prusiner, S. B., et al. (1997) Proc. Natl. Acad. Sci. USA 94, 10086–10091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dempski, R. E. D. & Imperiali, B. (2002) Curr. Opin. Chem. Biol. 6, 844–850. [DOI] [PubMed] [Google Scholar]
- 11.Sevier, C. S. & Kaiser, C. A. (2002) Nat. Rev. Mol. Cell Biol. 3, 836–847. [DOI] [PubMed] [Google Scholar]
- 12.Kopito, R. R. (1997) Cell 88, 427–430. [DOI] [PubMed] [Google Scholar]
- 13.Ellgaard, L., Molinari, M. & Helenius, A. (1999) Science 286, 1882–1888. [DOI] [PubMed] [Google Scholar]
- 14.Ma, J. Y. & Lindquist, S. (2001) Proc. Natl. Acad. Sci. USA 98, 14955–14960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yedidia, Y., Horonchik, L., Tzaban, S., Yanai, A. & Taraboulos, A. (2001) EMBO J. 20, 5383–5391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ma, J. Y. & Lindquist, S. (2002) Science 298, 1785–1788. [DOI] [PubMed] [Google Scholar]
- 17.Ma, J. Y., Wollmann, R. & Lindquist, S. (2002) Science 298, 1781–1785. [DOI] [PubMed] [Google Scholar]
- 18.Ma, J. Y. & Lindquist, S. (1999) Nat. Cell Biol. 1, 358–361. [DOI] [PubMed] [Google Scholar]
- 19.Wong, B.-S., Vénien-Bryan, C., Williamson, R. A., Burton, D. R., Gambeti, P., Sy, M.-S., Brown, D. R. & Jones, I. M. (2000) Biochem. Biophys. Res. Commun. 276, 1217–1224. [DOI] [PubMed] [Google Scholar]
- 20.Stöckel, J., Safar, J., Wallace, A. C., Cohen, F. E. & Prusiner, S. B. (1998) Biochemistry 37, 7185–7193. [DOI] [PubMed] [Google Scholar]
- 21.Taraboulus, A., Scott, M., Semenov, A., Avraham, D., Laszlo, L. & Prusiner, S. B. (1995) J. Cell Biol. 129, 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Priola, S. A. & Caughey, B. (1994) Mol. Neurobiol. 8, 113–120. [DOI] [PubMed] [Google Scholar]
- 23.Leteux, C., Chai, W., Nagai, K., Herbert, C., Lawson, A. M. & Feizi, T. (2001) J. Biol. Chem. 276, 12539–12545. [DOI] [PubMed] [Google Scholar]
- 24.Hornemann, S. & Glockshuber, R. (1998) Proc. Natl. Acad. Sci. USA 95, 6010–6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rudd, P. M., Wormald, M. R., Wing, D. R., Prusiner, S. B. & Dwek, R. A. (2001) Biochemistry 40, 3759–3766. [DOI] [PubMed] [Google Scholar]
- 26.Capellari, S., Zaidi, S. I. A., Urig, C. B., Perry, G., Smith, M. A. & Petersen, R. B. (1999) J. Biol. Chem. 274, 34846–34850. [DOI] [PubMed] [Google Scholar]
- 27.Welker, E., Wedemeyer, W. J. & Sheraga, H. A. (2001) Proc. Natl. Acad. Sci. USA 98, 4334–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O'Connor, S. E. & Imperiali, B. (1996) Chem. Biol. 3, 803–812. [DOI] [PubMed] [Google Scholar]
- 29.Imperiali, B. & O'Connor, S. E. (1999) Curr. Opin. Chem. Biol. 3, 643–649. [DOI] [PubMed] [Google Scholar]
- 30.Live, D. H., Kumar, R. A., Beebe, X. & Danishefsky, S. J. (1996) Proc. Natl. Acad. Sci. USA 93, 12759–12761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rickert, K. W. & Imperiali, B. (1995) Chem. Biol. 2, 751–759. [DOI] [PubMed] [Google Scholar]
- 32.Bosques, C. J., Tai, V. W.-F. & Imperiali, B. (2001) Tetrahedron Lett. 42, 7207–7210. [Google Scholar]
- 33.Nielsen, L., Khurana, R., Coats, A., Frokjaer, S., Brange, J., Vyas, S., Uversky, V. N. & Fink, A. L. (2001) Biochemistry 2001, 6036–6046. [DOI] [PubMed] [Google Scholar]
- 34.O'Connor, S. E. & Imperiali, B. (1997) J. Am. Chem. Soc. 119, 2295–2296. [Google Scholar]
- 35.Lehmann, S. & Harris, D. A. (1997) J. Biol. Chem. 272, 21479–21487. [DOI] [PubMed] [Google Scholar]
- 36.Westermark, G. T., Johnson, K. H. & Westermark, P. (1999) Methods Enzymol. 309, 3–25. [DOI] [PubMed] [Google Scholar]
- 37.Live, D. H., Wang, Z. G., Iserloh, U. & Danishefsky, S. J. (2001) Org. Lett. 3, 851–854. [DOI] [PubMed] [Google Scholar]
- 38.O'Connor, S. E. & Imperiali, B. (1998) Chem. Biol. 5, 427–437. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.