

Abstract
The key event in prion diseases seems to be the conversion of the prion protein PrP from its normal cellular isoform (PrPC) to an aberrant “scrapie” isoform (PrPSc). Earlier studies have detected no covalent modification in the scrapie isoform and have concluded that the PrPC → PrPSc conversion is a purely conformational transition involving no chemical reactions. However, a reexamination of the available biochemical data suggests that the PrPC → PrPSc conversion also involves a covalent reaction of the (sole) intramolecular disulfide bond of PrPC. Specifically, the data are consistent with the hypothesis that infectious prions are composed of PrPSc polymers linked by intermolecular disulfide bonds. Thus, the PrPC → PrPSc conversion may involve not only a conformational transition but also a thiol/disulfide exchange reaction between the terminal thiolate of such a PrPSc polymer and the disulfide bond of a PrPC monomer. This hypothesis seems to account for several unusual features of prion diseases.
Transmissible spongiform encephalopathies are fatal neurodegenerative diseases characterized by vacuolation of brain tissue and deposition of amyloid fibrils (1–3). Transmissible spongiform encephalopathies are unusual in that they have long incubation times and low infectivities and may be infectious, sporadic, or inherited (1–3). Even more remarkably, transmissible spongiform encephalopathies do not result from typical pathogens such as bacteria and viruses; rather, the key event seems to be the conversion of a protein (the prion protein PrP) from its normal cellular isoform (PrPC) to an abnormal scrapie isoform (PrPSc; refs. 1–3). Although the structures of the cellular isoform for several species have been well characterized by NMR (4, 5), structural data on the scrapie isoform are scarce; however, it is generally believed that the PrPC → PrPSc conversion is purely conformational and involves no chemical reactions (3, 6–9). Ample experimental evidence indicates that the PrPC → PrPSc conversion does involve a significant change in conformation; the scrapie isoform has an increased resistance to proteases and significantly more β-sheet structure than the cellular isoform (10–12). We argue here, however, that the PrPC → PrPSc conversion may also involve a covalent reaction of the (sole) intramolecular disulfide bond of PrPC. Specifically, the available biochemical data are consistent with the hypothesis that infectious prions are composed of PrPSc polymers linked by intermolecular disulfide bonds. Thus, the PrPC → PrPSc conversion may involve not only a conformational transition but also a thiol/disulfide exchange reaction between the terminal thiolate of such a PrPSc polymer and the disulfide bond of a PrPC monomer.
Experimental Data on Intermolecular Disulfide Bonds
The structure of the PrPSc isoform is difficult to study experimentally, primarily because the infectious scrapie isoform PrPSc forms high-molecular-weight aggregates under most solution conditions (1–3). If these aggregates are solubilized by strongly denaturing conditions, their infectivity is lost irreversibly. Incubation of these aggregates in low levels of denaturants does yield some monomeric protein; however, these monomers are not infectious (8, 12–15), and it has been suggested that these denatured monomers are merely PrPC molecules that have associated tightly to the PrPSc aggregates (8).
The addition of chemical denaturants causes PrP monomers with an intact (intramolecular) disulfide bond to dissociate from infectious PrPSc aggregates (16, 17). These dissociation data have been interpreted as evidence that the disulfide bond in infectious PrPSc prions is intramolecular (1, 7, 16, 17), because it was assumed that intermolecular disulfide bonds would prevent the dissociation into monomers and that the reduction of the intermolecular disulfide bonds would result in monomers with a reduced disulfide bond. These data seem to be the only basis for the generally held belief that the intramolecular disulfide bond of PrPC does not undergo covalent modification in the PrPC → PrPSc conversion.
However, this interpretation of the dissociation data neglects the process of disulfide reshuffling, by which the disulfide bonds of a molecule can rearrange by thiol/disulfide exchange reactions with an intramolecular thiol (18). Our oxidative folding studies have shown that denaturation can initiate disulfide reshuffling within a protein with free thiol groups by eliminating the tertiary structure protecting its disulfide bonds from intramolecular thiol/disulfide exchange reactions (19, 20). Thus, the denaturation of an intermolecularly disulfide-bonded PrP polymer would allow it to depolymerize into (denatured) PrP monomers with an intramolecular disulfide bond through the attack of its terminal thiolates on the preceding intermolecular disulfide bond (Fig. 1a). Cyclic PrP dimers and other cyclic oligomers could also be produced by such intramolecular thiol/disulfide exchange reactions, but the monomeric product should be favored over such oligomers by loop entropy and by any residual native (PrPC) enthalpic interactions, as have been detected by hydrogen–deuterium exchange (21).
Figure 1.

(a) Depolymerization reaction in which a terminal thiolate of a disulfide-bonded polymer attacks the preceding intermolecular disulfide bond, producing a shortened polymer and a monomer with an intact intramolecular disulfide bond. (b) Polymerization reaction in which a terminal thiolate of the disulfide-bonded polymer attacks the intramolecular disulfide bond of the cellular form PrPC, lengthening the polymer. Presumably, the PrPC molecule is destabilized by its association to the scrapie-form polymer, and hence, its disulfide bond is not as well protected as in free solution. (c) Reduction of an intermolecular disulfide bond. Experiments have demonstrated that the rates of reshuffling and reduction depend strongly on the stability of the tertiary structure protecting the disulfide bond (18–20).
These arguments demonstrate that the dissociation data do not exclude the possibility that prions are composed of PrPSc molecules linked by intermolecular disulfide bonds. Moreover, several experiments (described below) implicate disulfide-bond reactions in the PrPC → PrPSc conversion.
Mechanism of the PrPC → PrPSc Conversion
We propose here a mechanism of prion growth, in which the PrPC → PrPSc conversion involves disulfide polymerization (Fig. 1b). Specifically, the terminal thiolate group of a PrPSc polymer attacks the intramolecular disulfide bond of a PrPC monomer to form an intermolecular disulfide bond, thus lengthening the PrPSc polymer while maintaining a terminal thiolate group. The initial thiolate attack is facilitated by the experimentally observed association of the PrPC monomer and PrPSc polymer (14, 22), which increases the effective concentrations of the thiol and intramolecular disulfide bond for each other and which may destabilize the tertiary structure protecting the PrPC disulfide bond. The subsequent conformational transition to the β-enriched scrapie isoform (second step of Fig. 1b) evidently protects the intermolecular disulfide bond (17, 23), inhibiting the back-reaction of depolymerization. Thus, in this model, the smallest infectious unit is an oligomer of PrPSc that protects its intermolecular disulfide bond (s) from thiolate attack and has at least one free thiolate at which the PrPC → PrPSc conversion can occur.
Experimental Evidence for Disulfide-Bond Reactions During Conversion
The hypothesis of disulfide polymerization would account for the apparently critical role of thiol groups and disulfide bonds in the PrPC → PrPSc conversion. The pretreatment of PrPSc aggregates with the thiol-blocking agent N-ethyl maleimide inhibits the ability of such aggregates to convert added PrPC to a protease-resistant, scrapie-like form (17), which suggests an essential role for free thiols in the conversion reaction. The PrPC → PrPSc conversion is also blocked by the elimination of the intramolecular PrPC disulfide bond, whether by reduction (17) or by mutagenesis (23), suggesting that the intramolecular disulfide bond of PrPC must be intact for the successful conversion of PrPC to PrPSc. Reduction of the intramolecular PrPC disulfide bond has also been reported to induce a transition to a β-enriched scrapie-like conformation (24). This observation is consistent with our model, which postulates that the conformational transition to the scrapie isoform occurs after the thiolate attack on the intramolecular PrPC disulfide bond (Fig. 1b, second step). Finally, many of the mutations that provoke inherited prion diseases are clustered near the (intramolecular) PrPC disulfide bond (7). Because mutations that destabilize this region would likely facilitate the attack of a thiolate on the intramolecular PrPC disulfide bond, the disulfide-polymerization model can also account for the inherited and sporadic forms of prion diseases.
This hypothesis may account for other singular features of prion diseases. For example, scrapie infectivity seems to survive extremely high temperatures (25), which is more unusual for a polymer stabilized by only noncovalent interactions than for a polymer stabilized by both covalent bonds and (protective) noncovalent interactions. The cellular form PrPC also has a remarkable conformational stability in the vicinity of the disulfide bond (21) and an unusually fast folding time (26), which may derive from the need to stabilize the critical intracellular disulfide bond against thiolate attack. Finally, redox reactions with disulfide bonds are catalyzed by metal ions, which may be related to the experimental observation that the PrPC → PrPSc conversion is catalyzed by increasing the number of metal-ion-binding octapeptides in the unstructured N-terminal segment of the prion protein (27), although this N-terminal segment is not necessary for the conversion (28). Additional experiments have demonstrated directly that bound metal ions can influence the conversion (29, 30).
Kinetic Arguments Supporting the Hypothesis of Intermolecular Disulfide Bonds
Kinetic models have been developed to account for the exponential progression of prion diseases and for the fact that prion diseases may be infectious, sporadic, and inherited (3, 8, 9, 31, 32). However, these kinetic models have been criticized as not reproducing the observed behavior under realistic conditions (33); in particular, they reproduce the observed exponential progression only by requiring an as-yet-unidentified mechanism by which large oligomers of PrPSc can be broken into smaller oligomers (33).
However, the hypothesis of intermolecular disulfide bonds provides such a mechanism. The intermolecular PrPSc disulfide bonds are susceptible to occasional cleavage in the presence of a reducing agent (Fig. 1c), which generates new terminal thiolates at which polymerization can occur (Fig. 1b). This reduction mechanism produces exponential growth of the PrPSc polymers, provided that the rate of polymerization kpoly[PrPC] is greater than the rate of depolymerization kdepoly (see Appendix). It should be noted that the polymerization rate kpoly[PrPC] increases linearly with the PrPC concentration, which may account for the experimental observation that prion diseases can be provoked by high expression levels of PrPC (34). This reduction mechanism seems physiologically plausible, given that the cytoplasm is a reducing environment. However, the kinetics of prion diseases in vivo are difficult to model quantitatively with any confidence, because prion proteins are subject to anabolism and catabolism and are exposed to different pH and redox conditions over their life cycle.
Conclusions
In this article, we have proposed that the PrPC → PrPSc conversion involves not only a conformational transition but also a covalent thiol/disulfide exchange reaction between the terminal thiolate of an intermolecularly disulfide-bonded PrPSc polymer and the disulfide bond of a PrPC monomer, thus forming a new intermolecular disulfide bond (Fig. 1b). Contrary to the conclusions of earlier studies, the presence of intermolecular disulfide bonds in infectious PrPSc aggregates is not excluded by the available biochemical data. Indeed, the data suggest a critical role for the thiol groups and disulfide bonds in the PrPC → PrPSc conversion, and the hypothesis of intermolecular disulfide bonds accounts for several unusual features of prion diseases. Thus, infectious PrPSc aggregates may be stabilized by intermolecular disulfide bonds in addition to their noncovalent interactions, such as the “edge-to-edge” hydrogen bonding of β-sheets suggested by fiber diffraction data of the scrapie form (35).
Acknowledgments
This work was supported by National Institutes of Health Grant GM-24893. Support was also received from the National Foundation for Cancer Research.
Abbreviations
- PrP
the prion protein
- PrPC and PrPSc
the cellular and scrapie isoforms of the prion protein
Derivation of Exponential Kinetics
A simple kinetic model for the progression of prion diseases is presented in this Appendix to illustrate the point that the PrPC → PrPSc conversion is exponential, provided that the rate of polymerization 2kpoly[PrPC] at any individual terminal thiolate is greater than the rates of depolymerization 2kdepoly. Let the concentrations of intermolecular disulfide bonds and free thiolates be denoted as BSc and θSc, respectively. These quantities obey the kinetic equations
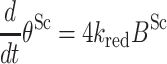 |
1 |
and
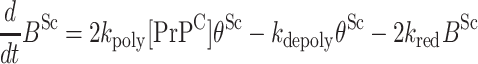 |
2 |
where kred, kpoly, and kdepoly represent the rate constants for reduction (Fig. 1c), polymerization (Fig. 1b), and depolymerization (Fig. 1a), respectively. The factors of two in these equations result from the facts (i) that the reduction of a single intermolecular disulfide bond results in two thiol groups and (ii) that a thiolate group may attack either of the two cysteines in a disulfide bond.
Strictly speaking, the θSc in the second term of the second equation should be corrected, because fully reduced PrP monomers have two free thiolates but cannot depolymerize. Thus, the second equation is equivalent to the assumption that the concentration of such fully reduced monomers is small compared with the total concentration of PrPSc polymers. Thus, our equations pertain to conditions under which the rate of reduction is relatively small compared with the net rate of growth of PrP polymers.
Differentiating the second equation and substituting the first equation yield the following equation.
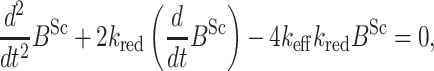 |
3 |
where keff 2kpoly[PrPC] − kdepoly is the effective rate of polymer growth at any individual terminal thiolate. The solution of this second-order differential equation indicates that the concentration of intermolecular disulfide bonds will grow exponentially if keff is a positive constant. Specifically, the exponential growth occurs at a rate given by
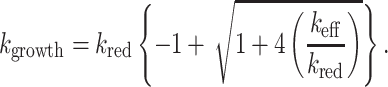 |
4 |
For simplicity, the process of cellular degradation of the scrapie isoform protein has been neglected in this derivation; nevertheless, this effect does not change the qualitative conclusion that exponential growth of the prion form is possible.
References
- 1.Horwich A L, Weissman J S. Cell. 1997;89:499–510. doi: 10.1016/s0092-8674(00)80232-9. [DOI] [PubMed] [Google Scholar]
- 2.Prusiner S B, Scott M R, DeArmond S J, Cohen F E. Cell. 1998;93:337–348. doi: 10.1016/s0092-8674(00)81163-0. [DOI] [PubMed] [Google Scholar]
- 3.Harris D A. Clin Microbiol Rev. 1999;12:429–444. doi: 10.1128/cmr.12.3.429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zahn R, Liu A, Lührs T, Riek R, von Schroetter C, Lopez García F, Billeter M, Calzolai L, Wider G, Wüthrich K. Proc Natl Acad Sci USA. 2000;97:145–150. doi: 10.1073/pnas.97.1.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lopez García F, Zahn R, Riek R, Wüthrich K. Proc Natl Acad Sci USA. 2000;97:8334–8339. doi: 10.1073/pnas.97.15.8334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stahl N, Baldwin M A, Teplow D B, Hood L, Gibson B W, Burlingame A L, Prusiner S B. Biochemistry. 1993;32:1991–2002. doi: 10.1021/bi00059a016. [DOI] [PubMed] [Google Scholar]
- 7.Wadsworth J D, Jackson G S, Hill A F, Collinge J. Curr Opin Genet Dev. 1999;9:338–345. doi: 10.1016/s0959-437x(99)80051-3. [DOI] [PubMed] [Google Scholar]
- 8.Horiuchi M, Caughey B. Structure Fold Des. 1999;7:R231–R240. doi: 10.1016/s0969-2126(00)80049-0. [DOI] [PubMed] [Google Scholar]
- 9.Jackson G S, Clarke A R. Curr Opin Struct Biol. 2000;10:69–74. doi: 10.1016/s0959-440x(99)00051-2. [DOI] [PubMed] [Google Scholar]
- 10.Caughey B W, Dong A, Bhat K S, Ernst D, Hayes S F, Caughey W S. Biochemistry. 1991;30:7672–7680. doi: 10.1021/bi00245a003. [DOI] [PubMed] [Google Scholar]
- 11.Pan K-M, Baldwin M, Nguyen J, Gasset M, Serban A, Groth D, Mehlhorn I, Huang Z, Fletterick R J, Cohen F E, et al. Proc Natl Acad Sci USA. 1993;90:10962–10966. doi: 10.1073/pnas.90.23.10962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Safar J, Roller P P, Gajdusek D C, Gibbs C J., Jr J Biol Chem. 1993;268:20276–20284. [PubMed] [Google Scholar]
- 13.Safar J, Roller P P, Gajdusek D C, Gibbs C J., Jr Biochemistry. 1994;33:8375–8383. doi: 10.1021/bi00193a027. [DOI] [PubMed] [Google Scholar]
- 14.Horiuchi M, Priola S A, Chabry J, Caughey B. Proc Natl Acad Sci USA. 2000;97:5836–5841. doi: 10.1073/pnas.110523897. . (First Published May 16, 2000; 10.1073/pnas.110523897) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kocisko D A, Lansbury P T, Jr, Caughey B. Biochemistry. 1996;35:13434–13442. doi: 10.1021/bi9610562. [DOI] [PubMed] [Google Scholar]
- 16.Turk E, Teplow D B, Hood L E, Prusiner S B. Eur J Biochem. 1988;176:21–30. doi: 10.1111/j.1432-1033.1988.tb14246.x. [DOI] [PubMed] [Google Scholar]
- 17.Herrmann L M, Caughey B. NeuroReport. 1998;9:2457–2461. doi: 10.1097/00001756-199808030-00006. [DOI] [PubMed] [Google Scholar]
- 18.Wedemeyer W J, Welker E, Narayan M, Scheraga H A. Biochemistry. 2000;39:4207–4216. doi: 10.1021/bi992922o. [DOI] [PubMed] [Google Scholar]
- 19.Narayan, M., Welker, E. & Scheraga, H. A. (2001) J. Am. Chem. Soc., in press. [DOI] [PubMed]
- 20.Welker E, Narayan M, Wedemeyer W J, Scheraga H A. Proc Natl Acad Sci USA. 2001;98:2312–2316. doi: 10.1073/pnas.041615798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hosszu L L, Baxter N J, Jackson G S, Power A, Clarke A R, Waltho J P, Craven C J, Collinge J. Nat Struct Biol. 1999;6:740–743. doi: 10.1038/11507. [DOI] [PubMed] [Google Scholar]
- 22.Horiuchi M, Caughey B. EMBO J. 1999;18:3193–3203. doi: 10.1093/emboj/18.12.3193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Muramoto T, Scott M, Cohen F E, Prusiner S B. Proc Natl Acad Sci USA. 1996;93:15457–15462. doi: 10.1073/pnas.93.26.15457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jackson G S, Hosszu L L P, Power A, Hill A F, Kenney J, Saibil H, Craven C J, Waltho J P, Clarke A R, Collinge J. Science. 1999;283:1935–1937. doi: 10.1126/science.283.5409.1935. [DOI] [PubMed] [Google Scholar]
- 25.Safar J, Roller P P, Gajdusek D C, Gibbs C J., Jr Protein Sci. 1993;2:2206–2216. doi: 10.1002/pro.5560021220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wildegger G, Liemann S, Glockshuber R. Nat Struct Biol. 1999;6:550–553. doi: 10.1038/9323. [DOI] [PubMed] [Google Scholar]
- 27.Vital C, Gray F, Vital A, Parchi P, Capellari S, Petersen R B, Ferrer X, Jarnier D, Julien J, Gambetti P. Neuropathol Appl Neurobiol. 1998;24:125–130. doi: 10.1046/j.1365-2990.1998.00098.x. [DOI] [PubMed] [Google Scholar]
- 28.Flechsig E, Shmerling D, Hegyi I, Raeber A J, Fischer M, Cozzio A, von Mering C, Aguzzi A, Weissmann C. Neuron. 2000;27:399–408. doi: 10.1016/s0896-6273(00)00046-5. [DOI] [PubMed] [Google Scholar]
- 29.McKenzie D, Bartz J, Mirwald J, Olander D, Marsh R, Aiken J. J Biol Chem. 1998;273:25545–25547. doi: 10.1074/jbc.273.40.25545. [DOI] [PubMed] [Google Scholar]
- 30.Brown D R, Hafiz F, Glasssmith L L, Wong B S, Jones I M, Clive C, Haswell S J. EMBO J. 2000;19:1180–1186. doi: 10.1093/emboj/19.6.1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harrison P M, Bamborough P, Daggett V, Prusiner S B, Cohen F E. Curr Opin Struct Biol. 1997;7:53–59. doi: 10.1016/s0959-440x(97)80007-3. [DOI] [PubMed] [Google Scholar]
- 32.Lansbury P T, Jr, Caughey B. Chem Biol. 1995;2:1–5. doi: 10.1016/1074-5521(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 33.Eigen M. Biophys Chem. 1996;63:A1–A18. doi: 10.1016/s0301-4622(96)02250-8. [DOI] [PubMed] [Google Scholar]
- 34.Prusiner S B, Scott M, Foster D, Pan K-M, Groth D, Mirenda C, Torchia M, Yang S-L, Serban D, Carlson G A, et al. Cell. 1990;63:673–686. doi: 10.1016/0092-8674(90)90134-z. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen J T, Inouye H, Baldwin M A, Fletterick R J, Cohen F E, Prusiner S B, Kirschner D A. J Mol Biol. 1995;252:412–422. doi: 10.1006/jmbi.1995.0507. [DOI] [PubMed] [Google Scholar]