

Abstract
HisH–hisF is a multidomain globular protein complex; hisH is a class I glutamine amidotransferase that hydrolyzes glutamine to form ammonia, and hisF is a (β/α)8 barrel cyclase that completes the ring formation of imidizole glycerol phosphate synthase. Together, hisH and hisF form a glutamine amidotransferase that carries out the fifth step of the histidine biosynthetic pathway. Recently, it has been suggested that the (β/α)8 barrel participates in a novel function: to channel ammonia from the active site of hisH to the active site of hisF. The present study presents a series of molecular dynamic simulations that investigate the channeling function of hisF. This article reconstructs potentials of mean force for the conduction of ammonia through the channel, and the entrance of ammonia through the strictly conserved channel gate, in both a closed and a hypothetical open conformation. The resulting energy landscape within the channel supports the idea that ammonia does indeed pass through the barrel, interacting with conserved hydrophilic residues along the way. The proposed open conformation, which involves an alternate rotamer state of one of the gate residues, presents only an ≈2.5-kcal energy barrier to ammonia entry. Another alternate open-gate conformation, which may play a role in non-nitrogen-fixing organisms, is deduced through bioinformatics.
The ability of proteins to direct ions and small molecules to active sites and across membranes is essential to many metabolic pathways, yet elucidating the mechanisms by which proteins control small molecule and substrate channeling presently remains a major challenge for scientists. Whether it is the ligand-induced gate opening of a calcium-gated potassium channel (1, 2), the dipole-orientation selectivity filter of an aquaporin channel (3), or the movement of a bulky residue in tryptophan synthase (4), revealing the mechanisms used by the various proteins to both regulate their conductance and expedite the delivery of a certain necessary molecule is central to understanding how these systems work. Metabolic channeling can be advantageous for many reasons and so it is not surprising that it is a mechanism used by both membrane and globular proteins. Substrate channeling in globular proteins is becoming more widely accepted as the number of enzymes demonstrating this core function and experimental evidence supporting these mechanisms continues to grow. Metabolic channeling allows directed transport of intermediates and therefore increases the overall rate of reaction. Simultaneously it may also ensure the integrity of the intermediate by providing a conduction path that is not accessible by other molecules (5).
HisH–hisF is a multidomain enzymatic complex regulating the fifth step of the histidine biosynthetic pathway. It belongs to the family of enzymes known as class I glutamine amidotransferases (GATases), which carry out two concerted reactions involving the production and subsequent use of ammonia. HisH comprises the glutaminase subunit that produces ammonia through glutamine hydrolysis (6, 7), and hisF, comprising the synthase subunit, uses the ammonia to complete a cyclase reaction (8). Recently, it has been suggested that the nascent ammonia is channeled through the (β/α)8 barrel of hisF (9, 10). The channeling of ammonia is common to GATases because they all are coupled to a second enzymatic reaction that requires unprotonated ammonia (5, 6, 11, 12). However, the mechanism involving ammonia conduction does not seem to be conserved among the GATases. In the GATase carbamoyl phosphate synthetase, experimental evidence has shown that ammonia does indeed traverse through a 96-Å intermolecular channel to get from one active site to the next without entering bulk solvent (13, 14). In another GATase, glutamine phosphoribosylpyrophosphate amidotransferase, the movement of a flexible loop forms a temporary channel for the ammonia to pass through to get to the next active site (11).
As of yet, no experimental evidence exists for hisH–hisF that conclusively shows that ammonia travels through the hisF barrel to complete the reaction. However, steady-state enzyme kinetics studies of the holoenzyme have shown that the binding of the hisF substrate to the active site of hisF stimulates enzymatic activity in hisH and that hisH shows no enzymatic activity in the absence of hisF (8, 15). This evidence shows strong coupling between the hisH and hisF reactions. Studies of both isolated hisF and the holoenzyme have also shown that glutamine is the preferred substrate as indicated by kcat/Km values that favor glutamine as the ammonia source by 3 orders of magnitude over ammonium/ammonia present in bulk solvent (8, 15). It has been suggested for other GATases (and experimentally shown for carbamoyl phosphate synthetase) that the ability to sequester ammonia from the bulk solvent is important to ensure that it can act as a nucleophile in the subsequent synthase reactions (11, 13, 16–18). Therefore the channeling in hisH–hisF of NH3 ≈25 Å from one active site to the next, to couple the two reactions and increase the enzymatic efficiency, seems to be a very plausible mechanism. This channeling presents a novel function for this ubiquitous enzymatic fold: hisF is the first (β/α)8 barrel suggested to use its barrel as an efficient channel for reaction intermediates.
Characteristic of the superfamily to which it belongs, hisH has a strictly conserved catalytic triad active site: Cys-84, His-178, and Glu-180 (19, 20). The active-site cysteine covalently binds glutamine, and the histidine, initially protonated, donates a proton to the amide group of glutamine to produce ammonia and glutamate. Subsequent steps allow the release of glutamate and reprotonation of histidine. Recently, high-resolution crystal structures of the hisH–hisF complex from Thermatoga maritima (9) and yeast (10) have been made available (Protein Data Bank codes 1GPW and 1JVN). Multiple sequence analysis and the available structures allow us to analyze the putative channel. HisH docks on the N-terminal side of hisF and forms 10 stabilizing hydrogen bonds between the subunits. The hisH active site is strategically positioned nearly directly above the mouth of the hisF barrel, and the number of crystallographically determined water molecules in the interface suggests that although it is partially solvated, it still remains a hydrophobic environment (Fig. 1).
Fig. 1.

HisH–hisF complex. Snapshot from one of the trajectories is shown, with NH3 within the barrel of hisF. Secondary structure motifs, hisH active site, strictly conserved Tyr-138 used in proposed open-gate conformation, and conserved residues in the hisF barrel are shown explicitly. NH3 is shown in van der Waals representation.
A multiple sequence alignment of all available hisF sequences is presented in Fig. 2, in which functionally relevant residues are highlighted. At the mouth of the barrel four strictly conserved charged residues (Arg-5, Glu-46, Lys-99, and Glu-167) form a gate (Figs. 1, 2, 3). It has been suggested that these groups control entrance to the barrel, although the recent crystal structures seem to only have the gate in a ``closed'' conformation that is not wide enough to allow the passage of ammonia (Fig. 3) (9, 10). Mutational studies involving Arg-5 and Glu-46 show that both residues are required for the proper function of the glutamine-dependent reaction (21). Once the ammonia enters the channel by a yet unknown mechanism, it continues its journey another 15 Å to the active site of hisF. On the C-terminal side of its barrel, hisF has two strictly conserved aspartate residues (Asp-11 and Asp-130) acting as the cyclase active site for the fifth step in histidine biosynthesis (9). The ammonia nitrogen is incorporated into the imidazole ring of imidizole glycerol phosphate, which eventually forms histidine (6, 7, 22).
Fig. 2.

Multiple sequence alignment: all three domains of life are shown as an excerpt from an alignment performed with 54 available sequences (yeast, Saccharomyces cerevisiae; THEMA, T. maritima; METTH, Methanobacterium thermoautotrophicum) BUCAP (Buchnera aphidicola) was chosen to represent nitrogen-fixing organisms. Strictly conserved residues across all sequences are shown in boldface, and residues corresponding to known and putative gating residues are marked with an asterisk. Numbering corresponds to THEMA. Note the strict conservation of gating residues Arg-5, Glu-46, Lys-99, and Glu-167. Aspartate residues suggested in possible gating mechanism (Asp-98, Asp-219) are conserved among non-nitrogen fixing organisms.
Fig. 3.

(a) Closed gate is present in five of six crystal structures, gate diameter 2.8 Å. (b) Chain C of 1GPW.pdb, partially closed, gate diameter 3.2 Å. (c) Top view of proposed open gate, Lys-99 within hydrogen-bonding distance of Tyr-138 of hisH, gate diameter 5.8 Å. (d) Side view of proposed open gate, 2.7-Å hydrogen bond indicated by gray line.
In the various crystal structures, Lys-99 has been reported in slightly different conformations. Already these different conformations suggest that movement of the lysine may be involved in the gating mechanism. Its strict conservation among the enzymes also suggests its functional significance; other residues of the channel gate may also play a role in the gating mechanism and the nearby conserved Asp-98 and Asp-219 (Fig. 2).
Although crystal structures shed invaluable light on many issues regarding protein function, they can only provide a snapshot of the protein at any time. Molecular dynamics (MD) simulations have been useful in elucidating function and exploring barriers to motion. Characterizing the energetics associated with select membrane proteins has been done with several different techniques. Umbrella sampling was used to determine the free energy surface associated with proton conduction in gramicidin A (23). An alternate method is the application of Jarzynski's identity (24, 25), which permits one to extract unperturbed free energy profiles from nonequilibrium force measurements. This technique was first demonstrated on repeated simulations of molecular pulling experiments on a model system (26). More recently, steered MD (SMD) simulations and single-molecule RNA atomic force microscopy experiments have been successfully used to reconstruct potentials of mean force (PMFs) through the use of this relatively new technique (27, 28). We have investigated the putative path of ammonia through the hisF barrel with MD and calculated the free energy landscape for this novel function of a (β/α)8 barrel. By characterizing the energetics and the conduction pathway, we attempt to answer fundamental questions regarding the function of the holoenzyme. In addition to generating the energy landscape for the conduction of ammonia through the (β/α)8 barrel, we investigate the energy barriers between the closed gate present in the crystal structures and a possible open-gate conformation.
Theory and Methods
Modeling. The crystal structure of the hisH–hisF complex from T. maritima was used in all simulations (Protein Data Bank code 1GPW; ref. 9). The original bacterial crystal structure had an active-site mutation (D11N) that we mutated back to its original, functional state. Of the three copies of the complex provided in the Protein Data Bank file, chains C and D were chosen because Lys-99 of the channel gate was in a partially closed conformation (Fig. 3). Active-site residues in both subunits were analyzed according to available biochemical information; as such, the hisH active site was neutralized corresponding to a postammonia release state (6, 22). The phosphate groups bound to hisF were removed from the crystal structure. As they are required to bind the hisF substrate at the side opposite to the interface, they should only minimally influence the conduction of ammonia as it nears the end of the channel.
After addition of the hydrogens, the complex was solvated by using the programs psfgen and solvate through vmd (29, 30). Crystallographic water molecules at the interface were augmented with additional water molecules suggested by the program dowser (31). Only those water molecules that favorably contributed (potential energy cutoff of –15 kcal/mol) and whose immediate environment seemed likely to accommodate a water molecule were included in the model (31). In total, 12 of the water molecules suggested by dowser were added at the interface. Eight sodium ions were added to neutralize the system. The composite system was comprised of 52,471 atoms; explicit solvent accounted for >40,000 of these atoms. The complex was then minimized for 10,000 steps with the MD program namd2 (29); the charmm27 parameter set was used throughout the simulations (32).
Ammonia was introduced into the system by replacing one crystallographic water molecule near the entrance of the channel. Parameters for ammonia were taken from the OPLS-AA force field (33–35). Ammonia and not ammonium was simulated for several reasons. First, ammonia is needed as a nucleophile to participate in the cyclase reaction at the hisF active site. Second, because the interface between the subunits is at least partially sequestered from solvent, as suggested by the limited number of water molecules present in the crystal structures, ammonia would probably remain unprotonated. Because of its pyramidal structure, ammonia has the ability to tunnel and perform an umbrella motion to change the plane of the hydrogens. The barrier associated with ammonia tunneling is ≈6 kcal/mol, and this inversion motion is on the gigahertz time scale. Because each trajectory was 1.2 ns and all of the energy barriers to ammonia's motion through the channel were <6 kcal/mol, we did not take the umbrella motion into account.
The starting configuration of the protein complex, all water molecules, and ammonia was equilibrated for 1 ns in the NPT ensemble by using periodic boundary conditions and the Langevin Piston method to control pressure at 1 atm. The Particle Mesh Ewald method was used to treat electrostatics without a cutoff (36). The system was equilibrated at 298.15 K with a time step of 1 fs. The nitrogen of ammonia was held fixed so it would stay close to the channel opening. The initial minimization and equilibration of the system revealed that to keep Lys-99 in the partially closed configuration, geometrical constraints were needed; otherwise after a few hundred femtoseconds of equilibration, the lysine would go back to the completely closed gate conformation. Protein equilibration was determined by monitoring the rms deviation of backbone atoms and fluctuations in volume. The rms deviation of the Cα atoms was ≈1 Å at the end of the equilibration.
The structure was examined for nearby charged and polar residues that could stabilize more open conformations of the gate. Using the molecular modeling program moe (version 2002.03, rotamer explorer) (37) energetically low-lying rotamer states of the gate residues were identified. The side-chain dihedral angles in the closed gate for Lys-99 (for 1GPW, chain C) are 175, –153, –72, and –57 degrees (χ1 through χ4, respectively). One of the low-lying rotamer states corresponded to a gate configuration that allowed hydrogen bonding between Lys-99 and a strictly conserved tyrosine from hisH at the interface (Tyr-138). We chose this Lys-99 rotamer to represent the open-gate configuration. In this rotamer, Lys-99 has angles of –66, 151, 167, and 55 degrees. This open gate corresponds to a distance between a Nz hydrogen of Lys-99 and the hydroxyl group of Tyr-138 of 2.5 Å. The structure with the new rotamer state for Lys-99 was equilibrated under identical simulation conditions for 150 ps before SMD simulations.
SMD Simulations. The center of mass of ammonia was subjected to a harmonic potential that moves along the channel axis with a constant velocity. The force vector was chosen along the channel axis (z axis), and the center of mass of ammonia was pulled through the channel until it reached the plane of Asp-11 and Asp-130 in the active site of hisF. A pulling speed of 15 Å/ns and a force constant k of 150 pN, which ensures minimal variance in the position of ammonia across the trajectories, were chosen. Each SMD trajectory was performed in the forward direction for 1.2 ns under simulation conditions similar to equilibration. SMD was also applied to conduct ammonia through the open-gate structure with Lys-99 in the alternate rotamer state. Also for this run we held the lysine fixed in the open position. Each run through the open-gate conformation was simulated for 1.2 ns; eight runs were performed to gather reasonable statistics. All runs were performed on either the National Center for Supercomputing Application's Platinum computer system (128 processors), the Pittsburgh Supercomputing Center's Terascale system (128 processors), or a local 32-processor Linux-i686-Scyld cluster. On these platforms each 1.2-ns trajectory took 20 h, 1 day, or 5 days, respectively. In total, >40 ns of simulation time were performed on the system.
PMF Reconstruction. The PMF is the average free energy potential experienced by ammonia as it moves through the channel. The system and SMD biasing potential are treated as a classical mechanical system with a time-dependent Hamiltonian H[x(t), t] = H0[x(t)] + 0.5k [z(x) – z0 – vt]2, where H0[x(t)] is the Hamiltonian of the unperturbed system at time t = 0, k is the spring constant of the harmonic constraint used to pull the ammonia, z(x) is the position of the ammonia center of mass initially positioned at z0, and v is the velocity at which the harmonic potential is moving. H0 is the generalized Hamiltonian: H0[x(t)] = H0[r(t), p(t)] for the canonical ensemble or H0[x(t)] = H0[r(t), p(t)] + PV for the isobaric-isothermal ensemble. The external work done by pulling ammonia through the protein from z0 to a point zM is
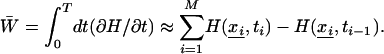 |
[1] |
Using the Jarzynski identity (24, 25) for systems perturbed by an external time-dependent force, we can relate the work along an ensemble of trajectories to the free energy difference
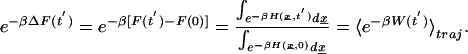 |
[2] |
By definition, the unperturbed free energy profile along the channel is
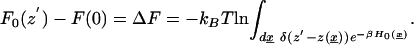 |
[3] |
Following Hummer and Szabo (26), we construct the unperturbed free energy profile by making use of a related form of the identity developed by Jarzynski (24, 25) and Crooks (38)
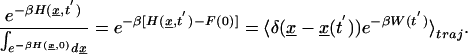 |
[4] |
After some manipulation and use of Eq. 3 this becomes
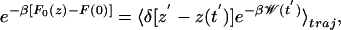 |
[5] |
where 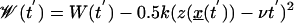 .
. 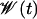 is the total external work performed on the system minus the instantaneous biasing potential.
is the total external work performed on the system minus the instantaneous biasing potential.
The unperturbed free energy profile following from Eq. 5 is 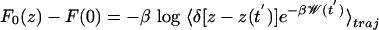 , and it is reconstructed by using the methods of Jensen et al. (27), where the exponential average is evaluated by using the second-order cumulant expansion and the ensemble average is restricted to trajectories satisfying z[x(t)] = z′.
, and it is reconstructed by using the methods of Jensen et al. (27), where the exponential average is evaluated by using the second-order cumulant expansion and the ensemble average is restricted to trajectories satisfying z[x(t)] = z′.
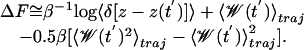 |
[6] |
The PMF force curves are shown in Fig. 4a. For each of the trajectories, one of every 100 data points is sampled (1 data point every 100 fs) and the total external work is numerically integrated with dt = 0.1 ps. The trajectories are broken up into j time slices of Δt = 10 ps each, and the mean position of ammonia in each time slice is determined. In any given trajectory, only a small area around the position z will be sampled. We average over the time slices and later, over multiple trajectories, to determine the most accurate free energy surface. Choosing a large spring constant reduces the fluctuations in position within a time window among trajectories, therefore z – z(t′) is minimized and the first term in Eq. 6 drops out. Ultimately, the free energy profile is calculated according to:
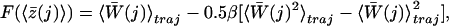 |
[7] |
where z̄(j) denotes the average position of ammonia in each time slice, 〈z̄(j)〉 is the average ammonia center of mass position in each time slice over all trajectories, and 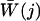 is the average work performed on ammonia in each time slice for each trajectory. The average total work over all trajectories in each jth time slice is:
is the average work performed on ammonia in each time slice for each trajectory. The average total work over all trajectories in each jth time slice is:
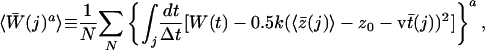 |
[8] |
where a is in our case either 1 or 2, N is the total number of trajectories, 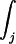 denotes integration over all of the j time slices, and t̄(j) is the average time that elapsed in the jth time slice over the trajectories.
denotes integration over all of the j time slices, and t̄(j) is the average time that elapsed in the jth time slice over the trajectories.
Fig. 4.

PMF reconstruction. (a) Force in kcal/mol per Å2 for five trajectories through the closed channel. (b) PMF shown for (i) closed- and (ii) open-gate conformations.
The simulations were carried out in both the NPT and NVT ensembles. Until now, most PMF calculations and derivations have been carried out in the NVT ensemble (24–27), although Liphardt et al. (28) reconstructed PMFs from atomic force microscopy experiments in the NPT ensemble for single-molecule RNA experiments. To mitigate any questions regarding the validity of the ensemble for PMF calculations, we ran 10 additional NVT ensemble runs under identical conditions. The force curves for both the NVT and NPT ensembles are nearly identical (data not shown).
Results and Discussion
Gating Mechanism. The gating mechanism of hisF is presently unknown. There is no experimental evidence to date that characterizes any gating mechanism. Although mutational studies of the complex have shown that Arg-5 and Glu-46 are indeed required for the glutamine-dependent reaction, no other significant evidence is available (8, 21).
The present simulated open-gate conformation of the hisF channel is consistent with a suggestion by Chaudhuri et al. (10) for the eukaryotic hisH–hisF complex. This low-lying alternate rotamer state of Lys-99 allows it to hydrogen-bond to the strictly conserved Tyr-138 from hisH, thereby increasing the gate diameter, defined by the distance between the nitrogen of Arg-5 to the next closest atom across the plane of the gate, from 3.72 to 5.84 Å (Fig. 3). To maintain the open-gate configuration throughout the entire simulation, the lysine needed to be held fixed in the alternate rotamer state. The fact that it closed after a few hundred femtoseconds of equilibration when no constraints were applied suggests that a force stronger than hydrogen bonding is needed to hold it in an open configuration.
In a parallel simulation of the hisF subunit alone (unpublished data), we used interactive MD (39) to form a stable open-gate configuration where Lys-99, instead of participating in a hydrogen bond with Tyr-138 of hisH, forms a salt bridge with the nearby and strictly conserved ASP98 of hisF. Although Douangamath et al. (9) suggest that this particular aspartate is involved in a hydrogen bond between the two subunits, it may also participate in the gating mechanism. Simulations on the unconstrained system showed that the salt bridge between these two residues remains intact for several hundred picoseconds. After ammonia passes the gate, the salt bridge breaks and a new ionic contact, with one of the glutamate residues of the gate, is formed. This rearrangement of salt bridges actually allows the gate to close once ammonia has passed. The stability of salt bridges in this region is increased by inaccessibility of the interface to bulk solvent; ionic contacts in a partially hydrophobic environment are even stronger than they would be in a completely solvated environment because of decreased coulombic screening. It is also interesting to note that in the simulations of the hisH–hisF complex, Asp-98 is involved with a salt bridge to Lys-181 of the hisH subunit.
Without knowing the energy barriers to reach the low-lying rotamer states, we cannot rationalize this as a conformation gating mechanism as seen in acetylcholinesterase (AChE) (40). In AChE, there are rapid conformational fluctuations between open and closed states. Because extended simulations do not show gate-opening fluctuations in the gate configuration, it is likely that the gating mechanism is part of a more activated event, such as a conformational change in the protein accompanying substrate binding. If this is the case, then the infrequency of this motion is better described by an intermittency model (41).
Another possible gating mechanism may involve Arg-5 of the gate and a nearby conserved Asp-219, which if both residues are in other rotamer states, may form another salt bridge. Interestingly, of 54 aligned sequences, Asp-219 is occupied by a negatively charged residue in 44 organisms (Asp or Glu), and only in 10 species, which are all nitrogen-fixing organisms, is it some other residue (Fig. 2). One of the unique properties of nitrogen-fixing organisms is that they are able to convert N2 into NH3, thus from an evolutionary standpoint it is possible that because these organisms can make their own ammonia, they don't need to use glutamine as the ammonia source. This idea is further corroborated by experimental evidence that shows the hisF reaction can still occur regardless of the source of ammonia; in fact, the reaction can proceed even when the hisH–hisF complex is not formed (8, 15). During the simulations, Asp-219 forms a salt bridge with a different arginine, Arg-191 of hisF. When both Lys-99 and Arg-5 are involved in these alternate salt bridges to Asp-98 and Asp-219, the diameter of the gate increases from 3.2 to 6.9 Å.
Conduction Mechanism, Energetics, and Dynamics. The conduction mechanism for ammonia through the channel can be deduced from the PMF reconstructed in Fig. 4b together with the examination of the interactions between ammonia and the channel. The small barriers of the PMF illustrate the network of hydrogen bonds formed and broken as ammonia progresses through the channel. In the trajectories, ammonia interacts with the hydrophilic residues Thr-78, Ser-101, and Ser-201 as it passes through the barrel center; the positions of these residues are shown in Fig. 4b. All three residues are conserved: Thr-78 is either a threonine or cysteine in 52 of 54 available sequences, Ser-101 is highly conserved in 51, and Ser-201 is invariant. The high conservation of these groups strongly suggests their vital functional role as an ``ammonia relay'' where ammonia is passed through the channel from one of these residues to the next, until it reaches its final destination at the active site of hisF. Also helping to break these hydrogen bonds in the channel is the constant presence of one or two water molecules. Although our simulations do not appear to suggest a constant ``proton wire'' (23), there is at least one water molecule within hydrogen-bonding proximity of ammonia for the duration of the simulation. The activity of the water molecules is not the same in different trajectories, and only some of the trajectories show a water conduction event through the predominantly hydrophobic tunnel along with ammonia. This surprising water passage indicates that it may indeed be possible for the predominantly hydrophobic channel to pass water and ammonia during the conduction process. In other trajectories, ammonia interacts with one crystallographically resolved water molecule when it enters the channel. As it proceeds through the channel this water molecule travels with the ammonia until after it passes Ser-101. At that point, a water molecule originally located in the bulk solvent near the end of the channel travels up into the barrel until it reaches the ammonia and then ``escorts'' the ammonia the rest of the way back out of the channel to the active site of hisF.
On average, it took the ammonia ≈400 ps to pass through the closed gate, as compared with 150 ps in the proposed open conformation. Fig. 4a shows the forces required to pull ammonia through the closed gate; the steep wall of the PMF (Fig. 4b) suggests that the free energy barrier for the passage of ammonia through the closed gate is at least 1 order of magnitude higher than the proposed open gate. We estimate the energy barrier for ammonia entry through the closed gate is between 25 and 40 kcal/mol. The PMF of the open gate is also shown in Fig. 4b and has a barrier of 2.8 kcal/mol. This significantly smaller barrier to ammonia entry makes physical sense because it should be easier for ammonia to pass through a larger opening. Because a 25– to 40-kcal/mol free energy barrier would be nearly impossible to be carried out physiologically, the PMF strongly suggests that the gate undergoes a conformational change that disrupts the gate salt bridges to expedite the passage of ammonia.
Accurate reconstruction of any PMF relies on minimal variance in the work applied to the system; it has been suggested that it is ideal if the standard deviation of work is on the order of kBT (26). A smooth trajectory was generated in every region of the channel except for the closed gate. The large forces required to pull ammonia through the closed gate caused a sudden large displacement of ammonia after passing through the plane of the gate residues. Consequently, these strong nonequilibrium effects caused the variance of the work to increase in this poorly sampled region. Fig. 4b shows the steep upward curve for the closed gate, after which the poor statistics destroy the accuracy of the PMF. For the conduction of ammonia through the channel, the PMF in Fig. 4b shows that it fluctuates between 3.8 and –0.5 kcal/mol depending on its position in the channel. The variance of the work in all of the segments for analysis was <0.4 kcal/mol (less than kBT).
In all simulations, at least one water molecule is within hydrogen-bonding distance of ammonia at all times. The interactions of the water molecule seem especially important when ammonia makes large translational moves as it switches its bonding from one hydrophilic residue to the next. The dipole orientation parameter of ammonia was averaged over all of the trajectories and it clearly shows that the dipole moment undergoes several transitions as the ammonia progresses through the channel (3) (Fig. 5). The various dipole orientations correspond to the positions of the highly conserved hydrophilic residues lining the channel. As ammonia passes through the gate, it has a clear preference for the dipole to be oriented so that its nitrogen can form a hydrogen bond with the hydrogen of Lys-99. However, once it is inside the channel, it flips its orientation several times as it optimizes its position to interact with the conserved hydrophilic residues and form stable hydrogen bonds. There are several places where its interaction with water also causes a directional dependence (Fig. 5).
Fig. 5.

Dipole orientation parameter of ammonia averaged over all eight open-gate trajectories shows a clear directional preference of ammonia at different positions in the channel corresponding to conserved hydrophilic residues in the channel. θ is the angle between the channel axis (z axis) and the normalized ammonia dipole vector (3).
Conclusion
Channeling enzymes are of intense interest as the number of enzymes demonstrating this type of function grows. The need for glutamine amidotransferases to use substrate channeling as a core enzymatic function has been identified with an increasing number of systems, although as of yet no similarities, other than the need to shuttle ammonia from one active site to the next, seem evident. The closed-gate energy barrier to entry of ammonia is very high and some motion at the interface is needed to bring the gate residues into one of their low-lying rotamer states. Although there are a number of energerically low-lying rotamer states, rotating Lys-99 to hydrogen-bond to Tyr-138 of hisH clearly opens the channel enough for the passage of ammonia. The small barriers as seen in our calculation of the open gate channel are consistent with suggestions of the bound complex providing a putative channel for ammonia passage (9, 10). The reconstructed PMFs and dipole analysis elucidate a full conduction event. Although the channel is predominantly hydrophobic, it is evident from the analysis that the conserved hydrophilic residues within the channel and the presence of a few water molecules inside the channel are essential to the conduction of ammonia. Mutations of the interacting residues should affect the free energy profile of ammonia in the channel and the overall reaction kinetics.
Although rotation of Lys-99 opens up the channel and significantly decreases the energy barrier for the entrance of ammonia into the channel, we believe this is only one part of the gating mechanism. For the gated membrane channel protein, OmpA, Bond et al. (42) suggest a gating mechanism involving an alternate rotamer state of an arginine in the channel. It is entirely possible that multiple residues are in alternate rotamer states to allow ammonia into the channel. Other rotamer states involving the salt bridges between gate residues and nearby charged residues, such as Asp-98 and Asp-219, might give rise to longer-lived open states. Preliminary simulations indicate that inclusion of the hisH and hisF substrates results in an increase in the fluctuations of the four gate residues and the backbone of hisF. This seems to support the idea the hisF substrate plays a key role in initiating the activated gating events.
Acknowledgments
We thank the Theoretical and Computational Biophysics Group at the Beckman Institute for their helpful discussions and generous use of computer time, especially Sanghyun Park for help with the PMF reconstruction. moe software was used at the University of Illinois Urbana–Champaign School of Chemical Sciences Computing Center. This work was funded by National Science Foundation Grant MCB02-35144.
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: GATase, glutamine amidotransferase; MD, molecular dynamics; SMD, steered MD; PMF, potential of mean force.
References
- 1.Jiang, Y., Lee, A., Chen, J., Cadene, M., Chait, B. T. & MacKinnon, R. (2002) Nature 417, 515–522. [DOI] [PubMed] [Google Scholar]
- 2.Jiang, Y., Lee, A., Chen, J., Cadene, M., Chait, B. T. & MacKinnon, R. (2002) Nature 417, 523–526. [DOI] [PubMed] [Google Scholar]
- 3.Tajkhorshid, E., Nollert, P., Jensen, M. O., Miercke, L. J., O'Connell, J., Stroud, R. M. & Schulten, K. (2002) Science 296, 525–530. [DOI] [PubMed] [Google Scholar]
- 4.Rhee, S., Parris, K., Ahmed, S. A., Miles, E. W. & Davies, D. R. (1996) Biochemistry 35, 4211–4221. [DOI] [PubMed] [Google Scholar]
- 5.Miles, E. W., Rhee, S. & Davies, D. R. (1999) J. Biol. Chem. 274, 12193–12196. [DOI] [PubMed] [Google Scholar]
- 6.Raushel, F. M., Thoden, J. B. & Holden, H. M. (1999) Biochemistry 38, 7891–7899. [DOI] [PubMed] [Google Scholar]
- 7.O'Donoghue, P., Amaro, R. E. & Luthey-Schulten, Z. (2001) J. Struct. Biol. 134, 257–268. [DOI] [PubMed] [Google Scholar]
- 8.Beismann-Driemeyer, S. & Sterner, R. (2001) J. Biol. Chem. 276, 20387–20396. [DOI] [PubMed] [Google Scholar]
- 9.Douangamath, A., Walker, M., Beismann-Driemeyer, S., Vega-Fernandez, M. C., Sterner, R. & Wilmanns, M. (2002) Structure (London) 10, 185–193. [DOI] [PubMed] [Google Scholar]
- 10.Chaudhuri, B. N., Lange, S. C., Myers, R. S., Chittur, S. V., Davisson, V. J. & Smith, J. L. (2001) Structure (London) 9, 987–997. [PubMed] [Google Scholar]
- 11.Bera, A. K., Smith, J. & Zalkin, H. (2000) J. Biol. Chem. 275, 7975–7979. [DOI] [PubMed] [Google Scholar]
- 12.Huang, X. & Raushel, F. (2000) Biochemistry 39, 3240–3247. [DOI] [PubMed] [Google Scholar]
- 13.Mullins, L. & Raushel, F. (1999) J. Am. Chem. Soc. 121, 3803–3804. [Google Scholar]
- 14.Thoden, J., Holden, H., Wesenberg, G., Raushel, F. & Rayment, I. (1997) Biochemistry 36, 6305–6316. [DOI] [PubMed] [Google Scholar]
- 15.Klem, T. J. & Davisson, V. J. (1993) Biochemistry 32, 5177–5186. [DOI] [PubMed] [Google Scholar]
- 16.Raushel, F. M., Mullins, L. & Gibson, G. (1998) Biochemistry 37, 10272–10278. [DOI] [PubMed] [Google Scholar]
- 17.Krahn, J., Kim, J. H., Burns, M., Parry, R., Zalkin, H. & Smith, J. (1997) Biochemistry 36, 11061–11068. [DOI] [PubMed] [Google Scholar]
- 18.Larsen, T., Boehlein, S., Schuster, S., Richards, N., Thoden, J., Holden, H. & Rayment, I. (1999) Biochemistry 38, 16146–16157. [DOI] [PubMed] [Google Scholar]
- 19.Tesmer, J., Klem, T., Deras, M., Davisson, V. J. & Smith, J. (1996) Nat. Struct. Biol 3, 74–86. [DOI] [PubMed] [Google Scholar]
- 20.Zalkin, H. & Smith, J. (1998) Adv. Enzymol. Relat. Areas Mol. Biol. 72, 87–144. [DOI] [PubMed] [Google Scholar]
- 21.Klem, T. J., Chen, Y. & Davisson, V. J. (2001) J. Bacteriol. 183, 989–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Thoden, J. B., Huang, X., Raushel, F. M. & Holden, H. M. (1999) Biochemistry 38, 16158–16166. [DOI] [PubMed] [Google Scholar]
- 23.Pomes, R. & Roux, B. (1998) Biophys. J. 75, 33–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jarzynski, C. (1997) Phys. Rev. Lett. 78, 2690–2693. [Google Scholar]
- 25.Jarzynski, C. (1997) Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Top. 56, 5018–5035. [Google Scholar]
- 26.Hummer, G. & Szabo, A. (2001) Proc. Natl. Acad. Sci. USA 98, 3658–3661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jensen, M. O., Park, S., Tajkhorshid, E. & Schulten, K. (2002) Proc. Natl. Acad. Sci. USA 99, 6731–6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liphardt, J., Dumont, S., Smith, S. B., Tinoco, I., Jr., & Bustamante, C. (2002) Science 296, 1832–1835. [DOI] [PubMed] [Google Scholar]
- 29.Kale, L., Skeel, R., Bhandarkar, M., Brunner, R., Gursoy, A., Krawetz, N., Phillips, J., Shinozaki, A., Varadarajan, K. & Schulten, K. (1999) J. Comp. Phys. 151, 283–312. [Google Scholar]
- 30.Grubmuller, H. (1996) solvate (Theoretical Biophysics Group, Institute for Medical Optics, Ludwig-Maximilians University, Munich), Version 1.0.
- 31.Zhang, L. & Hermans, J. (1996) Proteins Struct. Funct. Genet. 24, 433–438. [DOI] [PubMed] [Google Scholar]
- 32.MacKerrell, A. D., Jr., Bashford, D., Bellott, M., Dunbrack, R. L., Jr., Evansecl, J. D., Field, M. J., Fischer, S., Gao, J., Guo, H., Ha, S., et al. (1998) J. Phys. Chem. B 102, 3586–3616. [DOI] [PubMed] [Google Scholar]
- 33.Rizzo, R. C. & Jorgensen, W. L. (1999) J. Am. Chem. Soc. 121, 4827–4836. [Google Scholar]
- 34.Jorgensen, W. L., Maxwell, D. S. & Tirado-Rives, J. (1996) J. Am. Chem. Soc. 117, 11225–11236. [Google Scholar]
- 35.Maxwell, D. S., Tirado-Rives, J. & Jorgensen, W. L. (1995) J. Comput. Chem. 16, 984–1010. [Google Scholar]
- 36.Darden, T., York, D. & Pedersen, L. (1993) J. Chem. Phys. 98, 10089–10092. [Google Scholar]
- 37.Chemical Computing Group (2001) moe (Chemical Computing Group, Montreal).
- 38.Crooks, G. E. (2000) Phys. Rev. E Stat. Phys. Plasmas Fluids Relat. Interdiscip. Top. 61, 2361–2366. [Google Scholar]
- 39.Stone, J., Gullingsrud, J., Schulten, K. & Grayson, P. (2001) in 2001 ACM Symposium on Interactive 3D Graphics, ed. Sequin, J. H. A. C. (Association for Computing Machinery SIGGRAPH, New York), pp. 191–194.
- 40.Zhou, H., Wlodek, S. & McCammon, J. A. (1998) Proc. Natl. Acad. Sci. USA 95, 9280–9283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang, J. & Wolynes, P. (1999) J. Chem. Phys. 110, 4812–4819. [Google Scholar]
- 42.Bond, P. J., Faraldo-Gomez, J. D. & Sansom, M. S. (2002) Biophys. J. 83, 763–775. [DOI] [PMC free article] [PubMed] [Google Scholar]