

Abstract
The glyoxalase system, comprizing glyoxalase I and glyoxalase II, is a ubiquitous pathway that detoxifies highly reactive aldehydes, such as methylglyoxal, using glutathione as a cofactor. Recent studies of Leishmania major glyoxalase I and Trypanosoma brucei glyoxalase II have revealed a unique dependence upon the trypanosomatid thiol trypanothione as a cofactor. This difference suggests that the trypanothione-dependent glyoxalase system may be an attractive target for rational drug design against the trypanosomatid parasites. Here we describe the cloning, expression and kinetic characterization of glyoxalase I from Trypanosoma cruzi. Like L. major glyoxalase I, recombinant T. cruzi glyoxalase I showed a preference for nickel as its metal cofactor. In contrast with the L. major enzyme, T. cruzi glyoxalase I was far less fast-idious in its choice of metal cofactor efficiently utilizing cobalt, manganese and zinc. T. cruzi glyoxalase I isomerized hemithio-acetal adducts of trypanothione more than 2400 times more efficiently than glutathione adducts, with the methylglyoxal adducts 2–3-fold better substrates than the equivalent phenylglyoxal adducts. However, glutathionylspermidine hemithioacetal adducts were most efficiently isomerized and the glutathionylspermidine-based inhibitor S-4-bromobenzylglutathionylspermidine was found to be a potent linear competitive inhibitor of the T. cruzi enzyme with a Ki of 5.4±0.6 μM. Prediction algorithms, combined with subcellular fractionation, suggest that T. cruzi glyoxalase I localizes not only to the cytosol but also the mitochondria of T. cruzi epimastigotes. The contrasting substrate specificities of human and trypanosomatid glyoxalase enzymes, confirmed in the present study, suggest that the glyoxalase system may be an attractive target for anti-trypanosomal chemotherapy.
Keywords: drug discovery, glyoxalase, methylglyoxal, trypano-thione
Abbreviations: ECL, enhanced chemiluminesence; GLO1, glyoxalase I; TcGLO1, Trypanosoma cruzi GLO1; MALDI–TOF-MS, matrix-assisted laser-desorption ionization–time-of-flight MS; TCEP, tris(2-carboxyethyl)phosphine; T[SH]2, trypanothione, N1,N8-bis(glutathionyl)spermidine
INTRODUCTION
Chagas' disease, or American trypanosomiasis, is caused by the flagellated protozoan parasite Trypanosoma cruzi. Despite a marked reduction in rates of transmission in countries such as Brazil, Chile and Uruguay over the last 20 years, the disease re-mains a major health risk in many Latin American countries with more than 15 million infected people and an annual death toll of approx. 13000 [1]. The efficacy of the current nitro-drugs, nifurtimox and benznidazole, is poor, particularly in the chronic stage of infection. Both drugs have a high incidence of side effects [2] so that many patients are unable to complete a full course of treatment. Consequently, identification of novel drug targets and more effective replacement drug therapies has now become a matter of urgency.
In the search for antiparasitic drug targets, biochemical path-ways that are both essential for parasite survival and absent from the host are sought after [3]. One such pathway is the thiol meta-bolism of trypanosomes and Leishmania. These parasites are uniquely dependent upon T[SH]2 [trypanothione, N1,N8-bis(glu-tathionyl) spermidine] as their principal low-molecular-mass thiol, which, together with trypanothione reductase, assumes many of the antioxidant functions commonly undertaken by glutathione and glutathione reductase in their human hosts [4]. In related trypanosomatids, T[SH]2 has already been implicated in the mode of action as well as the mechanism of resistance of antimonial drugs in Leishmania spp. [5–7] and in defence against chemical and oxidant stress induced by arsenicals and nifurtimox in African trypanosomes [8–10]. Therefore, in an attempt to exploit this essential and unique metabolic pathway, trypanothione-dependent enzymes have become a major focus for drug discovery against these neglected tropical diseases.
In our previous study [11], we reported the characterization of the T[SH]2-dependent enzyme GLO1 (glyoxalase I) in L. major. This finding, combined with the identification of T[SH]2-dependent GLO2 in both Trypanosoma brucei [12] and L. donovani [13], provides compelling evidence for a unique T[SH]2-dependent glyoxalase system in certain trypanosomatids. The gly-oxalase system is a ubiquitous pathway for the detoxification of highly reactive oxoaldehydes. The metalloenzymes glyoxalase I (lactoylglutathione lyase) and glyoxalase II (hydroxyacylglu-tathione lyase) catalyse the step-wise dismutation of 2-oxoal-dehydes into the corresponding 2-hydroxyacids, using glutathione as a cofactor [14,15]. The principal role of the glyoxalase system is thought to be the detoxification of methylglyoxal (2-oxopro-panal), a highly toxic α-oxoaldehyde produced as a by-product of glycolysis and possessing cytotoxic, cytostatic and mutagenic properties [16]. In addition, methylglyoxal is also produced by the catabolism of threonine via aminoacetone or hydroxyacetone [17]. The inherent toxicity of this molecule stems from its pro-pensity to react with the nucleophilic centres of DNA, RNA and proteins. In particular, the oxoaldehyde reacts with the side chains of arginine, lysine and cysteine, and with the base guanine.
Inhibitors of GLO1 exhibit anticancer and antimalarial pro-perties and are selectively toxic for rapidly proliferating cells [18]. The T[SH]2-dependent glyoxalase system may provide an ideal target for Chagas' disease drug development. With this in mind, here we describe the expression, purification and kinetic characterization of GLO1 from T. cruzi.
EXPERIMENTAL
Materials
Methylglyoxal was prepared from methylglyoxal dimethylacetal and confirmed to be free of formaldehyde, as described previously [19]. All other reagents were standard commercial products of high purity.
Cloning and expression of TcGLO1 (T. cruzi GLO1)
A putative T. cruzi GLO1 gene (Tc00.1047053510659.240) was identified in the T. cruzi genome database (www.genedb.org). This gene was amplified by PCR from T. cruzi CL Brener genomic DNA using the sense primer: 5′ CATATGTCAACACGACGAC-TTATGCACACGATG 3′ with an additional NdeI site (underlined); and the antisense primer: 5′ GGATCCGGATCCTTAAG-CCGTTCCCTGTTC 3′ with an additional BamHI site (underlined). The PCR product was then cloned into the pCR®-Blunt II-TOPO® vector (Invitrogen) and sequenced. The pCR®-Blunt II-TOPO®-TcGLO1 constructs were then digested with NdeI and BamHI and the fragments cloned into the pET15b expression vector (Novagen) resulting in a pET15b-TcGLO1 construct.
Transformed BL21(DE3)pLysS Escherichia coli cells were grown in Luria–Bertani medium containing 100 μg/ml ampicillin and 12.5 μg/ml chloramphenicol to a D600 of ∼0.6. Expression was induced with 1 mM isopropyl-β-D-thiogalactoside for 4 h at 25 °C. Following expression, cells were pelleted (3000 g for 10 min at 4 °C), resuspended in lysis buffer [75 mM sodium phosphate, pH 7.5, 500 mM NaCl, 10 mM 2-mercaptoethanol and protease inhibitor cocktail (Roche)] and then lysed by sonication (four 30 s bursts at 15 microns amplitude), with cooling between pulses. The lysate was centrifuged (30000 g for 30 min) and the supernatant loaded at 2 ml/min onto a 5 ml HiTrap™ chelating Sepharose column (Amersham Pharmacia) in 50 mM sodium phosphate (pH 7.5) and 200 mM NaCl. TcGLO1 was eluted with a gradient of 0 to 500 mM imidazole in the same buffer. Eluted protein was then dialysed against 25 mM Bis-Tris, pH 6.5 and loaded onto a 5 ml HiTrap™ Q-Sepharose column (Amersham Pharmacia) at 2 ml/min and eluted with a gradient of 0–500 mM NaCl in the same buffer. Finally, the N-terminal His6-tag was removed by thrombin cleavage (30 units/mg for 12 h at 25 °C) and untagged TcGLO1 was recovered using a HiTrap™ Q-Sepharose column, as described previously [11].
Physical properties of TcGLO1
The native Mr of TcGLO1 was determined using Superdex 200 HR 10/30 size exclusion column chromatography (Amersham Pharmacia) against gel filtration standards (Bio-Rad). Molecular mass was determined by MALDI–TOF-MS (matrix-assisted laser-desorption ionization–time-of-flight MS) in linear mode using sinapinic acid as a matrix on a Voyager-DE STR mass spectro-meter (PerSeptive Biosystems).
Production of thiols
Trypanothione disulfide (Bachem) was reduced using TCEP [tris(2-carboxyethyl)phosphine] agarose, as described previously [11].
Apoenzyme production and metal reconstitution
Apoenzyme was prepared by dialysis of TcGLO1 for 24 h [11] against 1 litre of 20 mM imidazole, 1 mM EDTA, pH 7.0 contain-ing 5 g Chelex resin (2 volume changes). The enzyme was then reconstituted by incubation with a 10-fold molar excess of metal chloride in Chelex-treated 50 mM Hepes, pH 7.0, for 10 min at room temperature (25 °C).
Kinetic analysis of TcGLO1 activity
The pH optimum for the enzyme was determined over the pH range 4–10 using a mixed buffer system of 50 mM each of Ches [2-(N-cyclohexylamino)ethanesulfonic acid], Hepps and Mes (pH adjusted using KOH). The effect of ionic strength on enzyme activity was determined as above in 50 mM Hepes (pH 7.0) with various concentrations (0.025–0.4 M) of NaCl and (NH4)2SO4. To ensure that NiCl2 co-factor was not a limiting factor for enzyme activity in these assays, TcGLO1 was pre-incubated with a 10-fold molar excess of NiCl2.
Kinetic constants (Km and kcat) and pH optimization studies were determined by measuring the rate of isomerized hemithioacetal adduct formation at 240 nm using a continuous spectrophotometric assay (Shimadzu UV-2401 PC dual beam Spectrophotometer) [11]. The rate of phenylglyoxal adduct formation was measured at 263 nm. Hemithioacetal concentrations were calculated using the previously established Kd values of 3.0 mM for the methylglyoxal-thiol adduct and 0.6 mM for the phenylglyoxal-thiol adduct [20], in the presence of 0.1 M reduced thiol. Assays (0.5 ml) were performed in 50 mM Hepes/KOH (pH 7.0) with 25 mM NaCl at 27 °C. With the methylglyoxal and phenylglyoxal glutathione hemithioacetals, kcat/Km values were determined from the slope of rates plotted against enzyme concentration.
Inhibitor studies
Inhibition constants for TcGLO1 were determined over a range of substrate concentrations (0.5–2.5 times the Km with the hemithioacetal of trypanothione) at three fixed concentrations of S-4-bromobenzylglutathionylspermidine [21]. Linear Lineweaver–Burk transformations of each data set were inspected for the inhibition pattern and found to be competitive. Data sets were fitted by non-linear regression using the GraFit program and kinetic parameters determined.
Western analyses of cell extracts
Polyclonal antiserum against L. major GLO1 was raised in adult male Wistar rats. An initial injection of 100 μg of purified antigen [11], emulsified in complete Freund's adjuvant, was followed by two identical booster injections of antigen emulsified in Freund's incomplete adjuvant at 2-week intervals. L. major promastigotes (Friedlin strain; World Health Organization designation: MHOM/JL/81/Friedlin; 2×107 ml−1, 1 litre) and T. cruzi epimastigotes (CL Brener, genome project standard clone; 3×107 ml−1, 1 litre) were pelleted by centrifugation (1600 g for 10 min at 4 °C), washed twice in 20 mM Tris/HCl (pH 7.0) containing 0.1 mM sucrose and re-suspended in cell lysis buffer (10 mM Hepes, pH 7.0). For biological safety, parasites were in-activated by three cycles of freezing in liquid nitrogen and thawing in a 37 °C water bath, before lysis under pressure (30 klbf/in2; 1 lb/in2≡6.9 kPa) using a one shot cell disruptor (Constant Systems). Following centrifugation (20000 g for 40 min), cell supernatant was collected and protein concentration determined. Whole cell extracts (30 μg) were then separated by SDS/PAGE (12% gels) and subsequently transferred onto nitrocellulose. After blocking with 5% (w/v) skimmed milk in PBS at room temperature for 1 h, the blot was incubated with L. major GLO1 polyclonal antiserum (1:700 dilution) at room temperature for 1 h, washed in PBS containing 0.1% (v/v) Tween 20 and then incubated with a secondary rabbit anti-rat (IgG) antibody (1:10000 dilution; Dako). The immunoblot was subsequently developed using the ECL® (enhanced chemiluminescence) Plus system from Amersham Biosciences.
Subcellular fractionation
Subcellular fractions of L. major promastigotes and T. cruzi epimastigotes were prepared essentially as described previously [22]. Each fraction (30 μg) was then separated by SDS/PAGE (12% gels) and transferred onto nitrocellulose. After blocking, the blot was probed with L. major GLO1 polyclonal antiserum and detected by chemiluminescence, as described above. Following analysis, the immunoblot was stripped using Restore Western Blot Stripping Buffer (Pierce) and reprobed using the following primary antibodies in turn: anti-Hsp60 monoclonal antibody (1:5000 dilution; Stressgen); anti-(L. major trypanothione syn-thetase) antiserum (1:1000 dilution; [22]) and anti-(T. brucei BiP) antiserum (1:5000; [23]). Rabbit anti-rat (IgG) antibody (1:10000 dilution) and anti-(mouse IgG–horseradish peroxidase) antibody (1:10000 dilution; Sigma) were used as secondary antibodies where appropriate, and immunoblots were once again developed by ECL®.
RESULTS AND DISCUSSION
Identification of T. cruzi GLO1 sequences
The identification of a unique trypanothione-dependent glyoxal-ase system within Leishmania spp. and T. brucei [11–13] has raised hopes that the enzymes of this pathway may provide suitable targets for rational drug design against all trypanosomatids. However, despite having certain unique metabolic features in common [24], the evolutionary distance between Leishmania and Trypanosoma is large, approaching that of the fly and human [25]. With this in mind, we have determined the physicochemical and kinetic properties of GLO1 from T. cruzi.
BLAST searches of the T. cruzi genome database, using E. coli and L. major GLO1 sequences as templates, revealed a 426 bp sequence (Tc00.1047053510659.240) which shared 72% and 44% sequence identity to the L. major and E. coli GLO1 sequences res-pectively. Alignment of the putative TcGLO1 along with other trypanosomatid, eukaryotic and bacterial GLO1 amino acid sequences (Figure 1) illustrated a high degree of similarity be-tween the trypanosomatid and bacterial GLO1 proteins. Similarity with the eukaryotic GLO1 sequences was between 39.4 and 32.4%, with mammalian enzymes differing in that they have a number of unique insertions and an N-terminal extension. The conservation of a histidine residue near the N-terminus of the try-panosomatid proteins (residue 8 in T. cruzi) is suggestive of a nickel cofactor-binding site. However, in eukaryotic sequences a conserved glutamine in the same position is indicative of a zinc cofactor-binding site [26]. Conserved acidic residues (Asp100 and Tyr101) in the trypanosomatid enzymes are believed to be involved in the specific binding of T[SH]2 [21].
Figure 1. Alignment of the predicted amino acid sequences of glyoxalase I.

Gaps introduced into sequences to optimize alignments are represented by dashes. Conserved and similar residues are indicated by asterisks and dots respectively. The circles indicate conserved metal binding residues in the various sequences. Squares indicate conserved residues implicated in the binding of γ-glutamate moieties of thiol cofactors while triangles highlight conserved acidic residues (Asp100 and Tyr101) in the trypanosomatid enzymes thought to be involved in the binding of T[SH]2 [28]. Protein sequences are from T. cruzi (Tc00.1047053510659), L. major (LmjF35.3010), E. coli (Swiss-Prot accession no. P77036), Synechoccus sp. WH 8102 (TrEMBL accession no. Q7U3T2), human (Swiss-Prot accession no. P78375) and mouse (Swiss-Prot accession no. Q9CPU0).
Cloning and expression of TcGLO1 in E. coli
Sequence-specific sense and anti-sense primers were used to amplify the 426 bp TcGLO1 gene from T. cruzi CL Brener geno-mic DNA by PCR. The product was sub-cloned into the pCR®-Blunt II-TOPO® vector, sequenced to verify identity and cloned into pET15b giving the plasmid pET15b-TcGLO1. The resulting plasmid contains the entire open reading frame of TcGLO1 fused in frame with a cleavable His6-tag at the N-terminus. E. coli BL21(DE3)pLysS transformed with pET15b-TcGLO1 produced soluble and enzymatically active protein. TcGLO1 was purified to homogeneity via nickel affinity and anion exchange chromatography. The N-terminal His6-tag was then removed by thrombin cleavage followed by a second anion exchange chro-matography step (Figure 2). No residual His6-tagged enzyme could be detected by SDS/PAGE and Western analysis with an anti-His6 antibody. Both His6-tagged and untagged TcGLO1 readily formed dimers that were resistant to SDS denaturation and reduction with dithiotheitol, TCEP or 2-mercaptoethanol. Typical yields were between 5 and 7 mg/litre of starting culture.
Figure 2. Purification of recombinant TcGLO1.
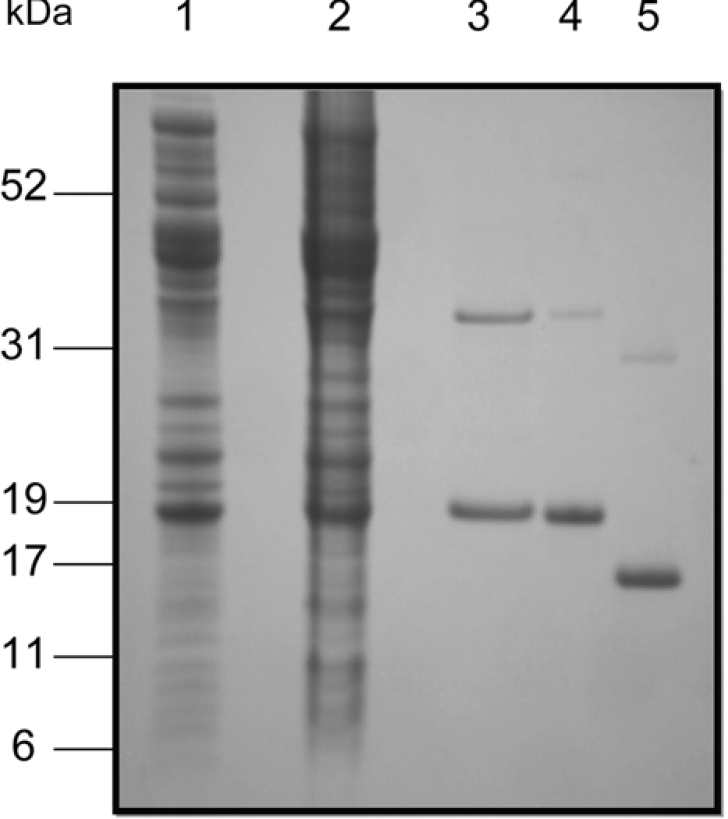
Lane 1, soluble fraction of BL21 (DE3)pLysS [pET15b-TcGLO1], uninduced cells; lane 2, soluble fraction of BL21 (DE3)pLysS [pET15b-TcGLO1], induced cells; lane 3, pooled fractions from nickel affinity chromatography; lane 4, pooled fractions from anion exchange chromatography (Resource-Q); lane 5, pooled fractions from anion exchange chromatography (Resource-Q) after removal of the His6-tag with thrombin protease.
Determination of TcGLO1 physical properties
SDS/PAGE analysis of TcGLO1 revealed a recombinant protein with an apparent molecular mass of 17 kDa (18.5 kDa with the His6-tag), in good agreement with the predicted size (16647 Da). The nominal molecular mass of recombinant TcGLO1 was determined to be 16650 Da by MALDI–TOF-MS analysis. Size exclusion chromatography and analytical ultracentrifugation showed native TcGLO1 to actively form dimers in solution with an Mr of 31300 (results not shown). These physiological properties are in good agreement with those for L. major GLO1 [11].
Metal specificity of TcGLO1
Sequence analyses suggested that nickel was likely to be the preferred metal cofactor of TcGLO1. To test this assumption, a range of bivalent cations were analysed for their ability to reactivate enzyme activity in the TcGLO1 apo-protein (Figure 3). Like the Leishmania enzyme, nickel was found to be the most effective in reactivating glyoxalase I enzyme activity. However, reactivation by cobalt was found to be only marginally less effect-ive than nickel. Indeed, in comparison with the L. major enzyme, TcGLO1 was found to be far less fastidious in its choice of metal cofactor, with both zinc and manganese capable of significant levels of reactivation. The versatility of TcGLO1 in utilizing a wider range of metal cofactors might be due to differing bioavail-ability in the mammalian host cells parasitized or in their res-pective insect vectors.
Figure 3. Metal reconstitution of TcGLO1 in various bivalent metal chlorides.

The apo form of TcGLO1 was incubated with a 10-fold molar excess of bivalent metal ions for 10 min at room temperature. Assays contained 25 μM trypanothione hemithioacetal and 0.1 mM free T[SH]2. L. major GLO1 reconstituted activity (black bars; [11]) is shown next to the TcGLO1 data (white bars). All assays were performed in triplicate, with the mean activity relative to the nickel-activated enzymes displayed.
Kinetic characterization
Using a mixed buffer system, the optimum for TcGLO1 activity was determined to be pH 7.0 (Figure 4A). Interestingly, a rapid decline in enzyme activity was noted in mildly alkaline conditions. This loss in TcGLO1 activity was subsequently shown to be reversible indicating that it is not due to denaturation at alkaline pH (results not shown). In 50 mM Hepes buffer at the optimum pH the optimum ionic conditions of the assay were found to be 25 mM NaCl (Figure 4B). L. major GLO1 has a high degree of substrate specificity for trypanothione hemithioacetals in preference to glutathione hemithioacetals [11], the substrate of choice for human GLO1 [16]. Similarly, TcGLO1 isomerized hemithioacetal adducts of trypanothione and glutathionylspermidine 2400 and 14000 times more efficiently than glutathione adducts respectively (Table 1). Of these two preferred substrates, TcGLO1 had a significantly higher affinity for the glutathionylspermidine adducts with either methylglyoxal or phenylglyoxal, as demonstrated by a markedly lower Km value, resulting in a 6-fold difference in kcat/Km. The T. cruzi enzyme displayed substrate specificity based not only upon thiol cofactor, but also on the oxoaldehyde component of the hemithioacetal. Methylglyoxal adducts were found to be between 2- and 3-fold better substrates than the equivalent phenylglyoxal adducts. The most striking difference between the L. major and T. cruzi enzymes is the 5-fold greater efficiency with which the L. major enzyme utilizes trypanothione hemithioacetals. This observation may be explained by the significantly lower intracellular levels of trypanothione in L. major promastigotes [27].
Figure 4. Enzymatic characterization of TcGLO1.

(A) Determination of assay pH optimum. Assays were performed with 50 μM trypanothione hemithioacetal in a mixed buffer system, as described in the Materials and methods section. Activity is expressed as a percentage relative to the maximum activity determined for TcGLO1. Results are presented as means±S.D. from three measurements. (B) Effect of buffer concentration and ionic strength on TcGLO1 activity. The assay mixtures contained either various amounts of Hepes buffer, pH 7.0 (closed circles), NaCl (open circles), or (NH4)2SO4 (open squares). Activity is expressed as a percentage of the activity determined in 50 mM Hepes buffer/25 mM NaCl. The inset shows the effect on activity as a function of buffer or salt concentration.
Table 1. Kinetic properties of T. cruzi GLO1.
Kinetic constants (Km and kcat) were determined as described previously [11]. Results are means±S.D. for triplicate measurements. ND, not determined.
| Hemithioacetal substrate | Km, μM | kcat, s−1 | kcat/Km×106·M−1·s−1 | kcat/Km(relative) |
|---|---|---|---|---|
| Glutathionylspermidine | ||||
| Methylglyoxal | 8.0±0.4 | 161±12 | 20 | 100 |
| Phenylglyoxal | 14±2.1 | 105±28 | 7.5 | 37.5 |
| Trypanothione | ||||
| Methylglyoxal | 109±10 | 363±33 | 3.3 | 16.5 |
| Phenylglyoxal | 207±23 | 265±31 | 1.5 | 7.5 |
| Glutathione | ||||
| Methylglyoxal | >1800 | ND | 0.0014 | 0.007 |
| Phenylglyoxal | >1800 | ND | 0.0005 | 0.0025 |
While glutathionylspermidine adducts appear to be the most efficient substrate for TcGLO1, 6-fold more efficient that trypanothione hemithioacetals, it should be noted that trypanothione levels are 8-fold higher than gluthionylspermidine in T. cruzi epimastigotes [27]. Based on their relevant ratios it would seem that both glutathionylspermidine and trypanothione hemithio-acetal adducts may act as the physiological substrates of the T. cruzi enzyme in vivo. The mutually exclusive substrate specifi-cities of the human and parasitic GLO1 enzymes have been directly related to major differences in active site architecture [28]. It is hoped that these differences can be exploited in the structure-based design of selective inhibitors against the trypanosomatids.
Inhibitor studies
The high affinity of the trypanosomatid GLO1 enzymes for glutathionylspermidine adducts may be useful in the future design of substrate-mimetic competitive inhibitors. (S)-4-Bromobenzyl-glutathionylspermidine was found to be a linear competitive inhibitor of GLO1 with respect to trypanothione hemithioacetal, as shown by diagnostic Lineweaver–Burk plots (Ki of 5.4±0.6 μM, Figure 5) which is 10-fold higher than for the L. major enzyme [28]. Comparison of L. major GLO1 with a homology model of TcGLO1 did not identify any striking differences between the active sites of both enzymes that could account for differences in inhibitor potency (T. Ariza and C. Bond, unpublished results). In contrast with the glutathionylspermidine analogue, S-4-bromobenzylglutathione is inactive as an inhibitor of both trypanosomatid enzymes (results not shown).
Figure 5. S-4-bromobenzylglutathionylspermidine inhibition of TcGLO1.

TcGLO1 inhibition constants were determined at three fixed concentrations of the inhibitor [(S-4-bromobenzylglutathionylspermidine] and over a range of substrate concentrations (25-125 μM trypanothione hemithioacetal). Data sets for each inhibitor concentration were fitted by non-linear regression and are presented as Lineweaver–Burk transformations. Trypanothione hemithioacetal was varied in the presence of 0 (○), 3.15 (■), 6.3 (□) and 12.6 μM (●) (S)-4-bromobenzylglutathionylspermidine.
Substrate analogues have been successfully used as inhibitors against GLO1 in several other organisms [29–31]. For example, (S)-(N-aryl-N-hydroxycarbamoyl)glutathione and (S)-(N-alkyl-N-hydroxycarbamoyl)glutathione compounds have been shown to inhibit human GLO1 with Ki values in the nanomolar range [32] and in the form of a diester to rapidly kill proliferating cancer cells [33]. It is tempting to suggest that the glutathionylspermidine version of these compounds may selectively inhibit trypanosomatid GLO1 activity and in their diester forms may prove to be toxic for parasites.
Western analyses of cell extracts and subcellular fractions
Immunoblots of L. major promastigote and T. cruzi epimastigote whole cell lysates were probed with a L. major GLO1-specific polyclonal antiserum (Figure 6A). As expected, a 17 kDa protein equivalent to the predicted molecular mass of the GLO1 enzymes reacted strongly with the antiserum in both the L. major and the T. cruzi lysates. Crude subcellular fractions of L. major promasti-gotes were also prepared and analysed by Western blot (Fig-ure 6B). The purity of each fraction was demonstrated using antibodies against marker proteins in each subcellular location. The endoplasmic reticulum-localized BiP protein was used as a marker for the microsomal fraction while Hsp60 and trypano-thione synthetase were used as marker proteins for the large granular and cytosolic fractions respectively. Interestingly, GLO1 was found to localize not only in the cytosol, but also in the large granular fraction from these parasites. Similar GLO1 localization was observed in lysates of T. cruzi epimastigotes (results not shown). Subsequent analysis of the TcGLO1 and L. major GLO1 primary sequences with MitoProt II [34], used to predict targeting of proteins to the mitochondria, suggested that both enzymes are likely to be exported to the mitochondria with a P=0.909 and 0.8704 respectively. However, no cleavage site was predicted for the mitochondrial targeting sequences of either protein.
Figure 6. Immunoblot analysis of cell lysates and cellular localization of TcGLO1.

(A) Blots of whole cell extracts (30 μg of protein in each lane) from T. cruzi epimastigotes (lane 1) and L. major promastigotes (lane 2) were probed with an anti-(L. major GLO1) antiserum. (B) Subcellular fractions of L. major promastigotes, containing the large granule (LG), cytosol (C), and microsomal fractions (MF), were prepared as described in the Materials and methods section. Western blots of these equally loaded fractions (30 μg of protein in each lane) were probed with anti-(L. major GLO1) antibody. In addition, blots were stripped and reprobed with antiserum to marker proteins for each subcellular fraction to demonstrate the purity of each fraction [LG, anti-Hsp60; C, anti-(L. major trypanothione synthetase); and MF, anti-BiP].
While comparatively little is known about the cellular location of GLO1 enzymes in other organisms, GLO2 has been extensively studied and detected in the cytoplasm and mitochondria of several higher eukaryotes including rat [35], human [36], yeast [37] and Arabidopsis thaliana [38]. In higher plants and yeast, distinct genes encode the alternate forms of GLO2, whereas analysis of GLO2 mRNA sequences in vertebrates indicates that dual initiation from alternative AUG codons is responsible [36]. In our future studies we hope to explore the targeting mechanism of GLO1 to trypanosomatid mitochondria.
In conclusion, the contrasting substrate specificities of human and trypanosomatid glyoxalase enzymes suggest that the glyoxalase system may be an attractive target for anti-trypanosomal chemotherapy. Gene knockout studies are underway to determine the role this pathway plays in parasite growth, survival or virulence.
Acknowledgments
We would like to thank Ian Eggleston and colleagues for providing inhibitors for this and previous studies, Charlie Bond and Tony Ariza for modelling studies, Irene Hallyburton for AUC and Douglas Lamont for performing mass spectrometry (all from the Division of Biological Chemistry and Molecular Microbiology, Wellcome Trust Biocentre, College of Life Sciences, University of Dundee, Dundee, DD1 5EH, Scotland, U.K.). This work was funded by the Wellcome Trust.
References
- 1.World Health Organization. promoting healthy life. Geneva: World Health Organization; 2002. The World health report 2002: reducing risks. [Google Scholar]
- 2.Castro J. A., Diaz-de-Toranzo E. G. Toxic effects of nifurtimox and benznidazole, two drugs used against American trypanosomiasis (Chagas' disease) Biomed. Environ. Sci. 1988;1:19–33. [PubMed] [Google Scholar]
- 3.El Sayed N. M., Myler P. J., Blandin G., Berriman M., Crabtree J., Aggarwal G., Caler E., Renauld H., Worthey E. A., Hertz-Fowler C., et al. Comparative genomics of trypanosomatid parasitic protozoa. Science. 2005;309:404–409. doi: 10.1126/science.1112181. [DOI] [PubMed] [Google Scholar]
- 4.Fairlamb A. H., Cerami A. Metabolism and functions of trypanothione in the Kinetoplastida. Annu. Rev. Microbiol. 1992;46:695–729. doi: 10.1146/annurev.mi.46.100192.003403. [DOI] [PubMed] [Google Scholar]
- 5.Wyllie S., Cunningham M. L., Fairlamb A. H. Dual action of antimonial drugs on thiol redox metabolism in the human pathogen Leishmania donovani. J. Biol. Chem. 2004;279:39925–39932. doi: 10.1074/jbc.M405635200. [DOI] [PubMed] [Google Scholar]
- 6.Croft S. L., Sundar S., Fairlamb A. H. Drug resistance in Leishmaniasis. Clin. Microbiol. Rev. 2006;19:111–126. doi: 10.1128/CMR.19.1.111-126.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mukhopadhyay R., Dey S., Xu N., Gage D., Lightbody J., Ouellette M., Rosen B. P. Trypanothione overproduction and resistance to antimonials and arsenicals in Leishmania. Proc. Natl. Acad. Sci. U.S.A. 1996;93:10383–10387. doi: 10.1073/pnas.93.19.10383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Alibu V. P., Richter C., Voncken F., Marti G., Shahi S., Renggli C. K., Seebeck T., Brun R., Clayton C. The role of Trypanosoma brucei MRPA in melarsoprol susceptibility. Mol. Biochem. Parasitol. 2006;146:38–44. doi: 10.1016/j.molbiopara.2005.10.016. [DOI] [PubMed] [Google Scholar]
- 9.Fairlamb A. H., Henderson G. B., Cerami A. Trypanothione is the primary target for arsenical drugs against African trypanosomes. Proc. Natl. Acad. Sci. U.S.A. 1989;86:2607–2611. doi: 10.1073/pnas.86.8.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ariyanayagam M. R., Oza S. L., Guther M. L., Fairlamb A. H. Phenotypic analysis of trypanothione synthetase knockdown in the African trypanosome. Biochem. J. 2005;391:425–432. doi: 10.1042/BJ20050911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vickers T. J., Greig N., Fairlamb A. H. A trypanothione-dependent glyoxalase I with a prokaryotic ancestry in Leishmania major. Proc. Natl. Acad. Sci. U.S.A. 2004;101:13186–13191. doi: 10.1073/pnas.0402918101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Irsch T., Krauth-Siegel R. L. Glyoxalase II of African trypanosomes is trypanothione-dependent. J. Biol. Chem. 2004;279:22209–22217. doi: 10.1074/jbc.M401240200. [DOI] [PubMed] [Google Scholar]
- 13.Padmanabhan P. K., Mukherjee A., Rentala M. Characterization of the gene encoding glyoxalase II from Leishmania donovani: a potential target for anti-parasite drug. Biochem. J. 2005;393:227–234. doi: 10.1042/BJ20050948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thornalley P. J. The glyoxalase system: new developments towards functional characterization of a metabolic pathway fundamental to biological life. Biochem. J. 1990;269:1–11. doi: 10.1042/bj2690001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vander Jagt D. L., Hunsaker L. A., Kibirige M., Campos N. M. NADPH production by the malarial parasite Plasmodium falciparum. Blood. 1989;74:471–474. [PubMed] [Google Scholar]
- 16.Thornalley P. J. Pharmacology of methylglyoxal: formation, modification of proteins and nucleic acids, and enzymatic detoxification – a role in pathogenesis and antiproliferative chemotherapy. Gen. Pharmacol. 1996;27:565–573. doi: 10.1016/0306-3623(95)02054-3. [DOI] [PubMed] [Google Scholar]
- 17.Phillips S. A., Thornalley P. J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993;212:101–105. doi: 10.1111/j.1432-1033.1993.tb17638.x. [DOI] [PubMed] [Google Scholar]
- 18.Creighton D. J., Zheng Z. B., Holewinski R., Hamilton D. S., Eiseman J. L. Glyoxalase I inhibitors in cancer chemotherapy. Biochem. Soc. Trans. 2003;31:1378–1382. doi: 10.1042/bst0311378. [DOI] [PubMed] [Google Scholar]
- 19.Vince R., Daluge S., Wadd W. B. Studies on the inhibition of glyoxalase I by S-substituted glutathiones. J. Med. Chem. 1971;14:402–404. doi: 10.1021/jm00287a006. [DOI] [PubMed] [Google Scholar]
- 20.Vander Jagt D. L., Han L. P., Lehman C. H. Kinetic evaluation of substrate specificity in the glyoxalase-I-catalysed disproportionation of ketoaldehydes. Biochemistry. 1972;11:3735–3740. doi: 10.1021/bi00770a011. [DOI] [PubMed] [Google Scholar]
- 21.Ariza A., Vickers T. J., Greig N., Fairlamb A. H., Bond C. S. Crystallization and preliminary X-ray analysis of Leishmania major glyoxalase I. Acta Crystallogr. Sect. F struct. Biol. Cryst. Commun. 2005;61:769–772. doi: 10.1107/S174430910502169X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oza S. L., Shaw M. P., Wyllie S., Fairlamb A. H. Trypanothione biosynthesis in Leishmania major. Mol. Biochem. Parasitol. 2005;139:107–116. doi: 10.1016/j.molbiopara.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 23.Bangs J. D., Uyetake L., Brickman M. J., Balber A. E., Boothroyd J. C. Molecular cloning and cellular localization of a BiP homologue in Trypanosoma brucei. Divergent ER retention signals in a lower eukaryote. J. Cell Sci. 1993;105:1101–1113. doi: 10.1242/jcs.105.4.1101. [DOI] [PubMed] [Google Scholar]
- 24.Berriman M., Ghedin E., Hertz-Fowler C., Blandin G., Renauld H., Bartholomeu D. C., Lennard N. J., Caler E., Hamlin N. E., Haas B., et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. doi: 10.1126/science.1112642. [DOI] [PubMed] [Google Scholar]
- 25.Fernandes A. P., Nelson K., Beverley S. M. Evolution of nuclear ribosomal RNAs in kinetoplastid protozoa: perspectives on the age and origins of parasitism. Proc. Natl. Acad. Sci. U.S.A. 1993;90:11608–11612. doi: 10.1073/pnas.90.24.11608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sukdeo N., Clugston S. L., Daub E., Honek J. F. Distinct classes of glyoxalase I: metal specificity of the Yersinia pestis, Pseudomonas aeruginosa and Neisseria meningitidis enzymes. Biochem. J. 2004;384:111–117. doi: 10.1042/BJ20041006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ariyanayagam M. R., Fairlamb A. H. Ovothiol and trypanothione as antioxidants in trypanosomatids. Mol. Biochem. Parasitol. 2001;115:189–198. doi: 10.1016/s0166-6851(01)00285-7. [DOI] [PubMed] [Google Scholar]
- 28.Ariza A., Vickers T. J., Greig N., Armour K. A., Dixon M. J., Eggleston I. M., Fairlamb A. H., Bond C. S. Specificity of the trypanothione-dependent Leishmania major glyoxalase I: structure and biochemical comparison with the human enzyme. Mol. Microbiol. 2006;59:1239–1248. doi: 10.1111/j.1365-2958.2006.05022.x. [DOI] [PubMed] [Google Scholar]
- 29.Kavarana M. J., Kovaleva E. G., Creighton D. J., Wollman M. B., Eiseman J. L. Mechanism-based competitive inhibitors of glyoxalase I: intracellular delivery, in vitro antitumor activities, and stabilities in human serum and mouse serum. J. Med. Chem. 1999;42:221–228. doi: 10.1021/jm9708036. [DOI] [PubMed] [Google Scholar]
- 30.Elia A. C., Chyan M. K., Principato G. B., Giovannini E., Rosi G., Norton S. J. N,S-bis-fluorenylmethoxycarbonylglutathione – a new, very potent inhibitor of mammalian glyoxalase-II. Biochem. Mol. Biol. Int. 1995;35:763–771. [PubMed] [Google Scholar]
- 31.Thornalley P. J., Strath M., Wilson R. J. M. Antimalarial activity in vitro of the glyoxalase I inhibitor diester, S-p-bromobenzylglutathione diethyl ester. Biochem. Pharmacol. 1994;47:418–420. doi: 10.1016/0006-2952(94)90035-3. [DOI] [PubMed] [Google Scholar]
- 32.Kalsi A., Kavarana M. J., Lu T. F., Whalen D. L., Hamilton D. S., Creighton D. J. Role of hydrophobic interactions in binding S-(N-aryl/alkyl-N- hydroxycarbamoyl)glutathiones to the active site of the antitumor target enzyme glyoxalase I. J. Med. Chem. 2000;43:3981–3986. doi: 10.1021/jm000160l. [DOI] [PubMed] [Google Scholar]
- 33.Sharkey E. M., O'Neill H. B., Kavarana M. J., Wang H. B., Creighton D. J., Sentz D. L., Eiseman J. L. Pharmacokinetics and antitumor properties in tumor-bearing mice of an enediol analogue inhibitor of glyoxalase I. Cancer Chemoth. Pharm. 2000;46:156–166. doi: 10.1007/s002800000130. [DOI] [PubMed] [Google Scholar]
- 34.Claros M. G., Vincens P. Computational method to predict mitochondrially imported proteins and their targeting sequences. Eur. J. Biochem. 1996;241:779–786. doi: 10.1111/j.1432-1033.1996.00779.x. [DOI] [PubMed] [Google Scholar]
- 35.Talesa V., Uotila L., Koivusalo M., Principato G., Giovannini E., Rosi G. Demonstration of glyoxalase-II in rat-liver mitochondria: partial-purification and occurrence in multiple forms. Biochim. Biophys. Acta. 1988;955:103–110. doi: 10.1016/0167-4838(88)90183-5. [DOI] [PubMed] [Google Scholar]
- 36.Cordell P. A., Futers T. S., Grant P. J., Pease R. J. The human hydroxyacylglutathione hydrolase (HAGH) gene encodes both cytosolic and mitochondrial forms of glyoxalase II. J. Biol. Chem. 2004;279:28653–28661. doi: 10.1074/jbc.M403470200. [DOI] [PubMed] [Google Scholar]
- 37.Bito A., Haider M., Hadler I., Breitenbach M. Identification and phenotypic analysis of two glyoxalase II encoding genes from Saccharomyces cerevisiae, GLO2 and GLO4, and intracellular localization of the corresponding proteins. J. Biol. Chem. 1997;272:21509–21519. doi: 10.1074/jbc.272.34.21509. [DOI] [PubMed] [Google Scholar]
- 38.Maiti M. K., Krishnasamy S., Owen H. A., Makaroff C. A. Molecular characterization of glyoxalase II from Arabidopsis thaliana. Plant Mol. Biol. 1997;35:471–481. doi: 10.1023/a:1005891123344. [DOI] [PubMed] [Google Scholar]