

Abstract
We consider a data set of DNA sequence variation at three Y chromosome genes (SMCY, DBY, and DFFRY) in a worldwide sample of human Y chromosomes. Between 53 and 70 chromosomes were fully screened for sequence variation at each locus by using the method of denaturing high-performance liquid chromatography. The sum of the lengths of the three genes is 64,120 bp. We have used these data to study the ancestral genealogy of human Y chromosomes. In particular, we focused on estimating the expected time to the most recent common ancestor and the expected ages of certain mutations with interesting geographic distributions. Although the geographic structure of the inferred haplotype tree is reminiscent of that obtained for other loci (the root is in Africa, and most of the oldest non-African lineages are Asian), the expected time to the most recent common ancestor is remarkably short, on the order of 50,000 years. Thus, although previous studies have noted that Y chromosome variation shows extreme geographic structure, we estimate that the spread of Y chromosomes out of Africa is much more recent than previously was thought. We also show that our data indicate substantial population growth in the effective number of human Y chromosomes.
Keywords: most recent common ancestorMRCA, population growth, human evolution, geographic pattern, genealogical analysis
Over the past 10 years, DNA polymorphisms have been widely used to reconstruct human evolutionary history (1–5). Mitochondrial DNA originally was used for this purpose, because the high mutation rate produced numerous polymorphisms and the absence of recombination facilitated their interpretation. In male lineages, the Y chromosome shares some of these properties, namely uniparental inheritance and absence of recombination in the nonrecombining part. Until recently, studies of the Y chromosome have been hampered by the scarcity of DNA sequence polymorphisms. Studies have been limited to a few segregating nucleotide sites (6–12) or to microsatellite polymorphisms whose mutation mechanisms are not well understood (13–16).
One of the main objectives of these studies is to estimate times of evolutionary events such as major migrations (9, 17). Vigilant et al. (1) argued that under the out-of-Africa model, the time of the most recent common ancestor (MRCA), which we write as TMRCA, is of particular interest, because it presumably precedes the departure of modern humans from Africa. Moreover, the ages of particular haplotypes or mutations have been used to estimate the ages of particular migration events (for examples, see refs. 4 and 18).
By using a worldwide sample of 445 Y chromosomes typed at eight microsatellite loci, Pritchard et al. (15) estimated the expected time to the MRCA, denoted by E[TMRCA], under a set of different mutation models. Their estimates ranged from 46,000 to 91,000 years B.P. under the different models, considerably less than those obtained by previous authors whose estimates were based on very small numbers of segregating sites (5–9, 19), but consistent with the microsatellite-based estimates of Wilson and Balding (14). The estimates of Pritchard et al. (15) of E[TMRCA] are also much younger than those obtained at other loci, which include 143,000 years for mtDNA (20), 535,000 years for a noncoding region at Xq13.3 (21), 800,000 years for β-globin (4), and 1,860,000 years for PDHA1 (22).
A second objective of the studies of DNA polymorphisms is to make inferences about demographic history, including population bottlenecks and expansions (4, 15, 23–25). The results of these studies often are conflicting, with evidence for expansions at some loci [e.g., mtDNA (24)] but not at others [e.g., β-globin (4)]. In this paper, we test for evidence of growth in the effective number of Y chromosomes.
In the companion paper, Shen et al. (26) report on the use of denaturing high-performance liquid chromatography (DHPLC) to reveal single-nucleotide polymorphisms in a worldwide sample of Y chromosomes. The sample of chromosomes analyzed in the present paper was completely screened by DHPLC‡‡ in the regions of the three genes SMCY, DFFRY, and DBY. Shen et al. report a total of 78 polymorphisms across the three genomic regions, a much larger number than found in previous studies of Y chromosomes. For SMCY, 53 chromosomes were typed, whereas 70 were typed for the other two genes. These data are summarized in Table 1. In addition, the same regions were sequenced in a chimpanzee and, where possible, in a gorilla, orangutan, and old- and new world monkeys. The chimpanzee sequences were used to infer the roots and the ancestral genotypes of the human Y chromosome trees.
Table 1.
Data and estimated mutation rate per site per year
| Gene | Sequence length | Sample size | No. polymorphisms | No. substitutions | Mutation rate |
|---|---|---|---|---|---|
| SMCY | 39,931 | 53 | 47 (41) | 528 | 1.32 × 10−9 |
| DBY | 8547 | 70 | 14 (12) | 107 | 1.25 × 10−9 |
| DFFRY | 15,642 | 70 | 17 (15) | 159 | 1.02 × 10−9 |
| Three genes | 64,120 | 43 | 65 (56) | 794 | 1.24 × 10−9 |
For each gene, we give the total number of polymorphisms and, in parentheses, the number of polymorphisms after the removal of length variants, repeat sequences, and indels.
In this paper, we use the data of Shen et al. (26) to build Y chromosome trees, to estimate E[TMRCA] for each gene as well as all three together, and to estimate the expected times of mutations as well as the growth rate of the worldwide population represented by the sample. Our estimate of E[TMRCA] for the sampled Y chromosomes is of the order of 59,000 years which agrees closely with the estimate of Pritchard et al. (15) from Y chromosome microsatellite polymorphisms. We also consider the geographic distribution of the observed haplotypes and estimate the expected ages of two mutations that are of particular interest.
Mutation Rate
For the ages of major events in these trees, an estimate for the mutation (single-nucleotide substitution) rate was needed. To obtain this rate, the number of substitutions was found between a chimpanzee sequence and a human sequence for the genomic region in question. From this information, the mutation rates per site per year for the three genes were estimated by:
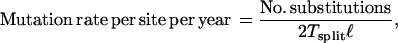 |
1 |
where ℓ is the sequence length (number of base pairs) and Tsplit is the time in years since the human and chimpanzee split, assumed here to be 5 million years ago. The mutation rate estimates for the three genes are given in Table 1. This method of estimation of the mutation rate assumes selective neutrality of all changes on the Y chromosome since divergence.
Geographic Structure of the Tree
The tree for the three genes combined shown in Fig. 1 reveals considerable geographic structure. Most of the shared haplotypes are within geographic groups, as seen previously with Y chromosome microsatellites (13, 15). Furthermore, presence or absence of certain mutations is strongly correlated with geography.
Figure 1.

A rendition of the three gene trees, where the mutations (56 single-nucleotide substitutions) are represented by circles, and those mutations that distinguish important geographic clades are numbered 1 and 2 (see Geographic Structure of the Tree).
In the tree of Fig. 1, the chromosomes that branch into the tree at the deepest points are African. Mutation 1 defines a clade, separate from the deep African lineages. Within this clade, a younger clade, consisting of 21 lineages of which only one is African, is defined by mutation 2. Eight of the 11 lineages that are not in this younger clade are African. Consequently, the tree is fairly consistent with the hypothesis developed from mtDNA (1) that the genetic diversity seen outside Africa is derived from a small number of African ancestors. It is interesting that the four deepest non-African lineages consist of three Asians and one Oceanian. A similar pattern of archaic African and Asian lineages was observed by Harding et al. (4) at the β-globin locus. However, as we shall show, the ages of the oldest Y chromosome clades are far younger than in that study.
It is of interest to estimate the expected age of mutation 1, which presumably preceded any movement out of Africa, and of 2, which would have been present in any hypothetical bottleneck before the global expansion.
Analysis Using genetree
genetree is a program written by R. C. Griffiths that estimates the probability of obtaining a given set of sequences from a sample of individuals randomly chosen from a large population. The method requires the input of θ = 2Neμ and other parameters, such as the population growth rate. Ne is defined as the effective number of Y chromosomes in the population and μ as the mutation rate per gene per generation. By varying θ until the probability of the data is maximized for a given data set, it is possible to obtain θ̂, the maximum likelihood estimate of θ.
The main applications of the program are to estimate θ and Ne as well as the expected ages of mutations and the expected time since the MRCA (E[TMRCA]). The theory behind genetree, developed by Griffiths and Tavaré (27), utilizes an importance sampling approach to estimate the above-mentioned probability. The program itself can be downloaded from www.stats.ox. ac.uk/≈stephens/group/software.html.
The assumptions made for this methodology include the coalescent process (28, 29) and an infinitely-many-sites mutation model (30). The infinitely-many-sites mutation model assumes that the mutation rate is low enough that each mutation occurs at a new site.
Analyses of the Y chromosome samples were carried out by using genetree. Four insertions, three deletions, and two repeat mutations were omitted from the analyses, as they mutate at different rates from single-nucleotide substitutions. Of all of the segregating sites, only one (in SMCY) appeared to have mutated more than once. Thus, except for this site, the data appear to fit the infinitely-many-sites model. Rather than removing the offending site, it was included in the analysis as two segregating sites. There were a total of 47 polymorphic sites at SMCY, 17 at DFFRY, and 14 at DBY. Forty-three individuals were sequenced for all three genes, resulting in 65 polymorphic sites.
Each simulation run carried out by genetree produces a possible ordering of coalescent and mutation events for a given data set. Because the distributions of the times between coalescent and mutation events are known, it is possible to simulate the age of pertinent mutations and of the MRCA of the tree for a given run. These simulated ages then are weighted by the probability of that run. An estimate of the expected age in question is obtained from the weighted average of the simulated ages over a large number of independent runs. This estimate is denoted by E[TMRCA].
Constant Population Size Model
The maximum likelihood estimates θ̂ of θ were found for the three genes, under the assumption of a constant population size over time. From the formula θ = 2Neμ and the mutation rates shown in Table 1, the effective population size, Ne, was estimated. To convert μ into a rate per gene per generation, it was assumed that the generation length of humans has been 25 years. [Note that this value is slightly less than the 27 years suggested by Weiss (31).] The estimates are presented in Table 2. The distribution of θ̂ is asymptotically normal. Because this distribution is proportional to the likelihood curve, these curves were generated by using an error function to join points from genetree.
Table 2.
Parameter estimates for a constant population model
| Gene | θ̂ | L1 | L2 | P (data|θ̂) | N̂e |
|---|---|---|---|---|---|
| SMCY | 16 | 10.5 | 22.3 | 5.7 × 10−25 | 6,100 |
| DBY | 4 | 2.5 | 6.5 | 2.0 × 10−6 | 7,500 |
| DFFRY | 4 | 2.7 | 7.7 | 9.0 × 10−13 | 5,000 |
| Three genes | 24 | 6.7 | 33.9 | 1.3 × 10−34 | 6,000 |
L1 and L2 are the lower and upper central 95% probability limits for the estimate of θ.
By using θ̂ as the input for genetree, the expected ages of the MRCA (E[TMRCA]) for the three samples were estimated. It is possible to use genetree to compute the sample distribution of TMRCA. This distribution assumes that θ and other parameters are already known and, hence, does not represent the uncertainty that the lack of knowledge about these parameters creates. To include this uncertainty, prior distributions could be placed on the parameters. This facility is not yet available in genetree.
Fig. 2 represents the effect of θ on E[TMRCA] by plots of the likelihood curves and the estimated E[TMRCA]s for various values of θ. As an alternative to placing a prior distribution on θ, E(θ̂) and Var(θ̂) were used to estimate E[TMRCA] by the delta method. Var(θ̂) was estimated in two ways: (i) from the second derivative with respect to θ of the logarithm of the likelihoods [ℓ′′(θ)], by using a cubic spline for ℓ(θ); and (ii) from the curves in Fig. 2. The two methods gave similar results. To be conservative, we report the consistently larger values from the second method.
Figure 2.

The likelihood curve and expected age of the MRCA in units of N generations, given θ under the model of constant population size. A number of points (nine for the three genes combined) were obtained by using genetree, and an error function (cubic spline) was fitted between the points for the likelihood curve (expected age of the MRCA).
Ninety-five percent probability intervals for E[TMRCA] were estimated by using the probability intervals for θ̂. This method used the Taylor expansion, as would be required for the delta method of approximating Var[TMRCA].
genetree estimates the ages in units of Ne generations. To obtain the estimates in terms of years, these values were multiplied by the generation time of humans (25 years) and the effective population size. The resulting probability intervals are reported in Table 3.
Table 3.
Estimated distribution of the MRCA (TMRCA) with constant population
| Gene | T̂MRCA* | T1* | T2* | T̂MRCA** | T1** | T2** |
|---|---|---|---|---|---|---|
| SMCY | 0.56 | 0.40 | 0.82 | 85,000 | 61,000 | 125,000 |
| DBY | 0.83 | 0.60 | 1.10 | 154,000 | 112,000 | 206,000 |
| DFFRY | 0.96 | 0.55 | 1.21 | 120,000 | 69,000 | 152,000 |
| Three genes | 0.55 | 0.36 | 0.98 | 84,000 | 55,000 | 149,000 |
T̂MRCA* is expected age in units of Ne generations. T1* and T2* are the central 95% probability intervals for the expected age of MRCA. T̂MRCA**, T1**, and T2** are the corresponding values in years.
Exponential Growth Model
Several estimators of θ have been suggested for the neutral, constant-sized, random-mating population model. These include estimators based on the number of segregating sites (30), the average number of pairwise differences (32), and the maximum likelihood estimator based on the full data (computed here with genetree). When the population model is correct, these estimators should produce similar estimates of θ.
We estimate θ at SMCY as 16.0, 9.0, and 2.87, by using the maximum likelihood estimate, the number of segregating sites, and the number of pairwise differences, respectively. At DFFRY, the estimates are 4.0, 2.5, and 0.68; and at DBY, the estimates are 4.0, 3.1, and 1.19. For the three genes combined, the corresponding estimates of θ are 24, 12.9, and 4.73. Although these estimates have large variances, the disparity among them suggests that the data may not be drawn from the assumed population model. As a simple test of the model, we can make use of Tajima's D. Tajima (33) suggested using the difference between estimates based on k and S as a test of neutrality. The values of Tajima's test statistic, D, are −2.31, −2.04, and −1.79 for the SMCY, DBY, and DFFRY genes, respectively. These statistics are all significant at the 5% level. The value of Tajima's D for the combination is −2.25, also significant at the 5% level. Negative values of D can indicate selection, but also population growth or population subdivision.
We also have computed Tajima's D within continents for the 43 chromosomes for which all three genes were typed, to account for the effect of population subdivision. D was significant at the 5% level in Asia and close to the 5% cutoff in Africa (despite the small sample size of just 18 chromosomes).
It is possible to incorporate a model of exponential population growth into the analysis conducted by genetree. The population size is modeled by:
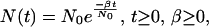 |
2 |
where N0 is the present-day effective population, N(t) is the effective population size t generations in the past, and β/N0 is the exponential growth rate per generation. For the theory behind the inclusion of a varying population size into a coalescent model, see Slatkin and Hudson (23) and Griffiths and Tavaré (34).
The inclusion of exponential population growth into the model results in a model with two parameters (θ, β). Table 4 reports the maximum likelihood estimates (θ̂, β̂) for (θ, β). These values were obtained by estimating the likelihood for a large number of (θ, β) pairs with genetree. The estimate of N0 derived from (θ̂, β̂) also is reported in Table 4, as well as the growth rate per generation. By obtaining the probability of the data for various pairs of values of θ and β around θ̂ and β̂, and by applying an error function between points, likelihood surfaces were estimated. Probability intervals were estimated from these likelihood surfaces, taking into account uncertainty in θ and β. These intervals were obtained from the approximate area underneath a curve that follows θ̂ for a given β.
Table 4.
The present day effective population size, N0, and maximum likelihood estimates of the population growth rate, β and θ, with central 95% probability intervals for each
| Gene | β̂ | β1 | β2 | θ̂ | θ1 | θ2 | P (data) | N̂0 | β̂/N̂0 |
|---|---|---|---|---|---|---|---|---|---|
| SMCY | 70 | 47.6 | 79.9 | 70 | 36.5 | 85.0 | 3.5 × 10−20 | 27,000 | 0.0026 |
| DBY | 110 | 33.8 | 142.8 | 22 | 11.7 | 26.3 | 9.4 × 10−4 | 41,000 | 0.0027 |
| DFFRY | 100 | 65.9 | 119.7 | 29 | 21.1 | 31.3 | 2.3 × 10−10 | 36,000 | 0.0027 |
| Three genes | 70 | 6.0 | 103.4 | 110 | 37.5 | 139.6 | 1.8 × 10−30 | 28,000 | 0.0025 |
θ is the rate for the entire gene. β̂/N̂0 is the estimated growth rate per generation. The 95% central probability intervals for β and θ are (β1, β2) and (θ1, θ2) respectively.
A cubic spline was fitted between points to estimate surfaces for E[TMRCA] over a range of θ and β values. These surfaces are presented in Fig. 3. Table 5 gives the mean and 95% probability intervals for E[TMRCA], which were obtained according to the methods described above for the constant population case but using the two dimensions θ and β.
Figure 3.

The likelihood surfaces and E[TMRCA] surfaces in units of N0 generations, given θ and β, under the exponential growth model. The three single gene surfaces used an error function to connect nine points on the likelihood curve. For the three genes combined, an error function was used where possible to connect 37 points. When the error function did not fit, linear interpolation was used. A cubic spline was used for all E[TMRCA] surfaces.
Table 5.
Estimated expected age of the MRCA (T̂MRCA) under a model of exponential population growth
| Gene | T̂MRCA, in N0 generations | T1g | T2g | T̂MRCA, years | T1y | T2y |
|---|---|---|---|---|---|---|
| SMCY | 0.0731 | 0.0618 | 0.1030 | 48,000 | 41,000 | 68,000 |
| DBY | 0.0538 | 0.0382 | 0.0975 | 55,000 | 39,000 | 100,000 |
| DFFRY | 0.0582 | 0.0440 | 0.0720 | 53,000 | 40,000 | 65,000 |
| Three genes | 0.0853 | 0.0580 | 0.2070 | 59,000 | 40,000 | 140,000 |
The 95% central probability intervals (T1g, T2g) and (T1y, T2y) are for time in units of N0 generations and years, respectively, using 25 years per generation.
Estimating the Expected Age of Mutations
As stated in Geographic Structure of the Tree, it is likely that migration first occurred from Africa at some time between the occurrences of mutations 1 and 2 on the tree of Fig. 1. The expected times at which these mutations occurred were estimated by using genetree, with the inclusion of a model of population growth. We used the symbol Tm to indicate these expectations with T̂m as their estimates. They also were estimated by using the number of segregating sites, S, that were found within the B sequences that contained the mutation in question. The theory behind this estimation uses the age of a mutation given its frequency within a random sample (35) and a rejection technique similar to the one described by Tavaré et al. (19). The theory can be found in Griffiths and Tavaré (R. C. Griffiths and S. Tavaré, unpublished work) and is written up in Thomson (36).
Both estimates are given in Table 6. Probability intervals were found from the likelihood surfaces used in the estimation of θ and β as shown in Fig. 3. These results indicate that male movement out of Africa first occurred around 47,000 years ago. The age of mutation 2, at around 40,000 years ago, represents an estimate of the time of the beginning of global expansion.
Table 6.
Estimated expected ages of mutations in the tree of Fig. 1
| Mutation | T̂m using genetree | B | S | T̂m using (B, S) |
|---|---|---|---|---|
| 1 | 47,000 (35,000; 89,000) | 42 | 51 | 43,000 (37,000; 111,000) |
| 2 | 40,000 (31,000; 79,000) | 38 | 45 | 42,000 (36,000; 109,000) |
B is the number of individuals found in the sample that contained the mutation in question, and S is the number of segregating sites found within those individuals. The 95% probability intervals are obtained by using likelihood surfaces found in Fig. 3.
A Simple Estimate of the Time Since the MRCA
The time estimates given above are based on a specific population model and use all of the information in the data. Although these estimates make full use of the data, they are not necessarily robust to departures from the model. As a simple alternative, to complement our model-based estimates, we suggest the following estimator, which does not assume a specific population genetic model.
Let T be the time since the MRCA. Also, let xi be the number of mutational differences between the ith sequence and the MRCA. The distribution of xi is Poisson with mean μT. Then an estimator of the time to the MRCA is
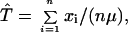 |
3 |
where n is the total number of sequences in the sample.
Under the infinitely-many-sites assumption (so that the tree can be determined unambiguously), T̂ is an unbiased estimator of T. However, the observations of xi are correlated among lineages, so it is not straightforward to estimate the variance of T̂. To get an upper bound on the variance, notice that Var(T̂) will be less than (probably much less than!) the variance of the estimate that we would obtain by picking a random sequence and simply using that sequence to estimate T̂ (i.e., drawing i at random from {1, . . . , n} and setting T̂* = xi/μ). We can get an upper bound on the variance by noting that Var(T̂) < Var(T̂*) = T/μ. Then, because we don't know T, we might estimate the variance and SE of T̂ by (T̂/μ) and (T̂/μ)1/2, respectively, noting that these values will usually be overestimates.
The ages of the MRCAs of the three Y chromosome genes and their SEs were estimated by using this method; the results are presented in Table 7.
Table 7.
Estimates of TMRCA using the average number of differences between each sequence and the root
| Gene | ∑i=1nxi/n | T̂, years |
|---|---|---|
| SMCY | 3.83 | 73,000 (37,000) |
| DBY | 0.357 | 33,000 (56,000) |
| DFFRY | 0.629 | 39,000 (50,000) |
| Three genes | 5.56 | 70,000 (30,000) |
Numbers in parentheses are SE.
Conclusions
Our estimate for the expected age of the Y chromosome root of human males was substantially smaller than has been found in previous studies using sequence data. A major difference between this study and previous studies is the greater size of the sample and the length of sequence examined. Previous studies that used much smaller data sets have reported an age that is much greater. With a smaller data set, the resulting age estimates were more influenced by the coalescent model than by the data themselves.
Another difference between this study and most previous studies is that we have included variable population size. Under a model of exponential population growth, the age of the MRCA is expected to be substantially smaller than that for the constant population model. However, this is not the only cause of the lower age estimates in this study, because our age estimates under a constant population model are also smaller than those found in previous studies.
The age estimates of this study were very close to the estimates found recently in a study of the human Y chromosome using microsatellites (15). That study used a population size model that was exponential in the recent past and constant in the distant past.
Under a neutral, constant-sized population model, the expected time to the Y chromosome common ancestor is a quarter of that for autosomal regions. In view of recent results for autosomal genes, it seems that this simple-minded prediction may be roughly accurate (4). However, as found previously using microsatellites, the current data are not consistent with a neutral constant-sized population model (recall the strongly significant Tajima test result). In view of the fact that for much of the last 50,000 years humans have been widely dispersed around the globe, with rapid population growth for a significant fraction of that time, it is striking that the estimated time to the MRCA is so short. From the Y chromosome, one would conclude that the ancestral population size 50,000 years ago was very small indeed. Yet this view is at odds with the results from other loci such as β-globin, which have very ancient MRCA times.
One solution to this apparent discrepancy is the possibility that the Y chromosome is subject to fairly strong selection, either in the form of positive selection for advantageous mutations (hitchhiking) or negative selection against mildly deleterious mutations (background selection). The possible role of selection seems quite plausible in the light of results from Drosophila [reviewed by Pritchard et al. (15)].
In this study, we found evidence for growth in the effective number of Y chromosomes, as observed previously for mtDNA (24). However, evidence for population growth has been absent at autosomal loci, such as β-globin (4) and PDHA1 (22). It is possible that this discrepancy reflects recent population growth from a population of fixed size [cf. Pritchard et al. (15)]. The much deeper ancestral trees of autosomal loci such as β-globin and PDHA1 would be affected less by recent population growth than would the relatively short genealogies of the Y chromosome and mtDNA.
The Y chromosome tree (Fig. 1) reveals substantial continental structure in the data, with the older clade primarily representing Africa and the younger representing non-African populations. Previous studies of Y chromosome microsatellite polymorphisms (13) also revealed substantial continental structure. It is remarkable that although the sequence data for β-globin (an autosomal locus) revealed similar tree topology, the estimated E[TMRCA] for Y variation is an order of magnitude less than that for β-globin (4).
Acknowledgments
J.K.P. is supported by a Hitchings-Elion Fellowship from the Burroughs-Wellcome Fund. This research was supported in part by National Institutes of Health Grants GM28016 and GM28428.
Abbreviation
- MRCA
most recent common ancestor
Footnotes
See commentary on page 6927.
Oefner, P. J. & Underhill, P. A. (1995) Am. J. Hum. Genet. 57, A266 (abstr.).
References
- 1.Vigilant L, Stoneking M, Harpending H, Hawkes K, Wilson A C. Science. 1991;253:1503–1507. doi: 10.1126/science.1840702. [DOI] [PubMed] [Google Scholar]
- 2.Bowcock A M, Ruiz-Linares A, Tomfohrde J, Minch E, Kidd J R, Cavalli-Sforza L L. Nature (London) 1994;368:455–457. doi: 10.1038/368455a0. [DOI] [PubMed] [Google Scholar]
- 3.Goldstein D B, Ruiz-Linares A, Cavalli-Sforza L L, Feldman M W. Proc Natl Acad Sci USA. 1995;92:6723–6727. doi: 10.1073/pnas.92.15.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Harding R M, Fullerton S M, Griffiths R C, Bond J, Cox M J, Schneider J A, Moulin D S, Clegg J B. Am J Hum Genet. 1997;60:772–789. [PMC free article] [PubMed] [Google Scholar]
- 5.Underhill P A, Jin L, Lin A A, Mehdi S Q, Jenkins T, Vollrath D, Davis R W, Cavalli-Sforza L L, Oefner P J. Genome Res. 1997;7:996–1005. doi: 10.1101/gr.7.10.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dorit R L, Akashi H, Gilbert W. Science. 1995;268:1183–1185. doi: 10.1126/science.7761836. [DOI] [PubMed] [Google Scholar]
- 7.Whitfield L S, Sulston J E, Goodfellow P N. Nature (London) 1995;378:379–380. doi: 10.1038/378379a0. [DOI] [PubMed] [Google Scholar]
- 8.Hammer M F. Nature (London) 1995;378:376–378. doi: 10.1038/378376a0. [DOI] [PubMed] [Google Scholar]
- 9.Hammer M F, Karafet T, Rasanayagam A, Wood E T, Altheide T K, Jenkins T, Griffiths R C, Templeton A R, Zegura S L. Mol Biol Evol. 1998;15:427–441. doi: 10.1093/oxfordjournals.molbev.a025939. [DOI] [PubMed] [Google Scholar]
- 10.Underhill P A, Jin L, Zemans R, Oefner P J, Cavalli-Sforza L L. Proc Natl Acad Sci USA. 1996;93:196–200. doi: 10.1073/pnas.93.1.196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jaruzelska J, Zietkiewicz E, Labuda D. Mol Biol Evol. 1999;16:1633–1640. doi: 10.1093/oxfordjournals.molbev.a026076. [DOI] [PubMed] [Google Scholar]
- 12.Jaruzelska J, Zietkiewicz E, Batzer M, Cole D E C, Moisan J-P, Scozzari R, Tavaré S, Labuda D. Genetics. 1999;152:1091–1101. doi: 10.1093/genetics/152.3.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruiz-Linares A, Nayar K, Goldstein D B, Seielstad M, Lin A, Herbert J, Feldman M W, Cavalli-Sforza L L. Ann Hum Genet. 1996;60:401–408. doi: 10.1111/j.1469-1809.1996.tb00438.x. [DOI] [PubMed] [Google Scholar]
- 14.Wilson I J, Balding D J. Genetics. 1998;150:499–510. doi: 10.1093/genetics/150.1.499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pritchard J K, Seielstad M T, Perez-Lezaun A, Feldman M W. Mol Biol Evol. 1999;16:1791–1798. doi: 10.1093/oxfordjournals.molbev.a026091. [DOI] [PubMed] [Google Scholar]
- 16.Ruiz-Linares A, Ortíz-Barrientos D, Figueroa M, Mesa N, Múnera J G, Bedoya G, Vélez I D, García L F, Pérez-Lezaun A, Bertranpetit J, et al. Proc Natl Acad Sci USA. 1999;96:6312–6317. doi: 10.1073/pnas.96.11.6312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Seielstad M T, Minch E, Cavalli-Sforza L L. Nat Genet. 1998;20:278–280. doi: 10.1038/3088. [DOI] [PubMed] [Google Scholar]
- 18.Watson E, Forster P, Richards M, Bandelt H-J. Am J Hum Genet. 1997;61:691–704. doi: 10.1086/515503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tavaré S, Balding D J, Griffiths R C, Donnelly P. Genetics. 1997;145:505–518. doi: 10.1093/genetics/145.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Horai S, Hayasaka K, Kondo R, Tsugane K, Takahata N. Proc Natl Acad Sci USA. 1995;92:532–536. doi: 10.1073/pnas.92.2.532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kaessmann H, Heissig F, von Haeseler A, Paabo S. Nat Genet. 1999;22:78–81. doi: 10.1038/8785. [DOI] [PubMed] [Google Scholar]
- 22.Harris E E, Hey J. Proc Natl Acad Sci USA. 1999;96:3320–3324. doi: 10.1073/pnas.96.6.3320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Slatkin M, Hudson R R. Genetics. 1991;129:555–562. doi: 10.1093/genetics/129.2.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rogers A R, Harpending H. Mol Biol Evol. 1992;9:552–569. doi: 10.1093/oxfordjournals.molbev.a040727. [DOI] [PubMed] [Google Scholar]
- 25.Sherry S T, Rogers A R, Harpending H, Soodyall H, Jenkins T, Stoneking M. Hum Biol. 1994;66:761–775. [PubMed] [Google Scholar]
- 26.Shen P, Wang F, Underhill P A, Franco C, Yang W-H, Roxas A, Sun R, Lin A A, Hyman R W, Vollrath D, et al. Proc Natl Acad Sci USA. 2000;97:7354–7359. doi: 10.1073/pnas.97.13.7354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Griffiths R C, Tavaré S. Stat Sci. 1994;9:307–319. [Google Scholar]
- 28.Kingman J F C. J Appl Prob. 1982;19A:27–43. [Google Scholar]
- 29.Hudson R R. In: Oxford Surveys in Evolutionary Biology. Futuyma D J, Antonovics J, editors. Oxford: Oxford Univ. Press; 1990. pp. pp.1–44. [Google Scholar]
- 30.Watterson G A. Theor Popul Biol. 1975;7:256–276. doi: 10.1016/0040-5809(75)90020-9. [DOI] [PubMed] [Google Scholar]
- 31.Weiss K. Am Antiquity. 1973;38:1–86. [Google Scholar]
- 32.Tajima F. Genetics. 1983;105:437–460. doi: 10.1093/genetics/105.2.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tajima F. Genetics. 1989;123:585–595. doi: 10.1093/genetics/123.3.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Griffiths R C, Tavaré S. Philos Trans R Soc London B. 1994;344:403–410. doi: 10.1098/rstb.1994.0079. [DOI] [PubMed] [Google Scholar]
- 35.Griffiths R C, Tavaré S. Stochastic Models. 1998;14:273–295. [Google Scholar]
- 36.Thomson R. The Shape of a Coalescent Tree, Ph.D. thesis. Clayton, Australia: Monash University; 1998. [Google Scholar]