

Abstract
The RecBCD enzyme is required for homologous recombination and DNA repair in Escherichia coli. The structure and function of RecBCD enzyme is altered on its interaction with the recombination hotspot Chi (5′-GCTGGTGG-3′). It has been hypothesized that the RecD subunit plays a role in Chi-dependent regulation of enzyme activity [Thaler, D. S., Sampson, E., Siddiqi, I., Rosenberg, S. M., Stahl, F. W. & Stahl, M. (1988) in Mechanisms and Consequences of DNA Damage Processing, eds. Friedberg, E. & Hanawalt, P. (Liss, New York), pp. 413–422; Churchill, J. J., Anderson, D. G. & Kowalczykowski, S. C. (1999) Genes Dev. 13, 901–911]. We tested the hypothesis that the RecD subunit inhibits recombination by deleting recD from the nuclease- and recombination-deficient mutant recBD1080ACD. We report here that the resulting strain, recBD1080AC, was proficient for recombination and DNA repair. Recombination proficiency was accompanied by a change in enzyme activity: RecBD1080AC enzyme loaded RecA protein onto DNA during DNA unwinding whereas RecBD1080ACD enzyme did not. Together, these genetic and biochemical results demonstrate that RecA loading by RecBCD enzyme is required for recombination in E. coli cells and suggest that RecD interferes with the enzyme domain required for its loading. A nuclease-dependent signal appears to be required for a change in RecD that allows RecA loading. Because RecA loading is not observed with wild-type RecBCD enzyme until it acts at a Chi site, our observations support the view that RecD inhibits recombination until the enzyme acts at Chi.
RecBCD enzyme plays a central role in the major pathway of genetic exchange and DNA repair in Escherichia coli (1–3). The degradative and recombinational activities of RecBCD enzyme, as well as its structure, are regulated by a specific DNA sequence called Chi (5′-GCTGGTGG-3′). As a consequence of the RecBCD enzyme-Chi interaction, both the DNA substrate (4–7) and enzyme (8–10) are changed. Genetic and biochemical experiments have suggested that one or another RecBCD enzyme subunit directs this regulation (8, 11–16). We show here that the RecD subunit inhibits E. coli recombination by blocking RecBCD enzyme-facilitated loading of RecA protein onto single-stranded (ss) DNA.
The structure of the RecBCD enzyme and the activities it promotes are complex. RecBCD enzyme is a heterotrimer composed of one copy of each of the products of the recB, recC, and recD genes (17). The enzyme is an ATP-dependent double-stranded (ds) and ss exonuclease, a ss endonuclease, and a DNA helicase (1). RecBCD enzyme interacts with Chi sites, which stimulate recombination in E. coli and bacteriophage lambda (18). Null mutations in recB and recC result in recombination deficiency and sensitivity to DNA damaging agents (19–21). Cultures of such strains contain many inviable cells (22), reflecting their inability to repair DNA damage by homologous recombination. In contrast, null mutations in recD leave strains highly viable and proficient in recombination and DNA repair (refs. 11 and 23; see below).
The role of RecBCD enzyme in homologous recombination begins when it binds to the end of a dsDNA substrate and initiates unwinding. Further reactions of RecBCD enzyme with DNA occur in a manner dependent on the presence of Chi sites and the concentrations of Mg2+ and ATP. When the concentration of ATP exceeds that of Mg2+, RecBCD enzyme makes a ss endonucleolytic cut a few nt to the 3′ side of Chi (4, 5). When the concentration of Mg2+ exceeds that of ATP, RecBCD enzyme degrades the 3′ terminated strand during DNA unwinding; the degradation is reduced when the enzyme reaches Chi (10) and the new 3′ ssDNA end is loaded with RecA by RecBCD enzyme (6). RecA protein stimulates pairing and strand exchange with a homolog (24), and other activities including RuvABC and RecG convert heteroduplex DNA to the final recombinant or repaired products (24, 25).
The individual activities of RecBCD enzyme depend on complex interactions between the three subunits, but at least part of the nuclease domain has been located in the C-terminal portion of the RecB polypeptide (26). A single bp change in recB resulting in the substitution of alanine (A) for aspartic acid (D) at position 1080 eliminates the nuclease activity and some of the Chi-dependent activities of the holoenzyme (27). The mutant enzyme retains DNA unwinding activity (27) but fails to load RecA protein (28). As expected from the supposition that RecA loading by RecBCD enzyme is essential for recombination, strains carrying the recBD1080A mutation are recombination-deficient, as we report here.
It is clear that RecBCD enzyme alters a DNA substrate in several ways during a reaction. Other evidence shows that RecBCD enzyme is changed by its interaction with Chi in a two-step process (8, 9). First, a RecBCD enzyme molecule that makes an endonucleolytic cut at Chi loses the ability to nick at a second Chi site on the same DNA, although it continues to unwind this DNA (9). The second change in the enzyme follows unwinding of the DNA and results in the disassembly of RecBCD enzyme into its three subunits (8). These changes reflect a mechanism of enzyme regulation that may allow only one recombination event per enzyme molecule (ref. 8; see Discussion).
The complex structure and activities of RecBCD enzyme have led to models that account for regulation of the enzyme's degradative and recombinogenic activities. Genetic analysis of recD null mutants and examination of purified enzyme led to the hypothesis that RecD acts as an inhibitor of recombination (11) until it is modified or ejected after a RecBCD enzyme-Chi interaction (12, 16). This model is supported by the fact that recD mutants are recombination-proficient (11, 23, 29) and hyperrecombination-proficient in the absence of Chi sites (11), and they cluster recombination events at the ends of DNA in lambda replication-blocked crosses (30). Purified RecBC enzyme (i.e., lacking RecD) has a low affinity for dsDNA (unpublished data, see Table 5) ends but can unwind DNA (16, 31, 32) and facilitates loading of RecA protein at the 3′ termini of DNA during unwinding (16). This is in contrast to RecBCD enzyme, which requires a Chi site for significant loading of RecA protein (6), joint molecule formation (7), and recombination (18). The constitutive loading of RecA protein by RecBC enzyme suggested that RecD inhibits RecA loading by the holoenzyme until an interaction with Chi (16).
Table 5.
Summary of recBCD phenotypes
| Enzyme | DNA damage*
|
Recombination†
|
Enzyme activity‡
|
||||||
|---|---|---|---|---|---|---|---|---|---|
| UV | Mit. C | Hfr | λ×λ | Chi | Exo | Unwinding | Chi | RecA loading | |
| RecBCD | R | R | + | + | + | + | + | + | +¶ |
| RecBC | R | R | + | + | − | − | +§ | − | +‖ |
| RecBD1080ACD | S | S | − | − | − | − | + | − | − |
| RecBD1080AC | R | R | + | + | − | − | +§ | − | +‖ |
S, sensitive to agent; R, resistant to agent.
Recombination proficiency and Chi activity in Hfr and lambda red− gam− crosses. +, recombination-proficient or Chi active; −, recombination-deficient or Chi inactive.
Activity in enzyme assays. +, activity present; −, activity absent.
Unwinding by RecBC or RecBD1080AC enzyme requires higher protein concentrations because of their lower affinity for DNA than RecBCD or RecBD1080ACD enzyme (>200-fold for RecBC enzyme compared to RecBCD enzyme, unpublished observations).
RecA loaded on 3′ Chi tail.
RecA loaded constitutively on 3′ end.
If RecD is an inhibitor of recombination, there might be a class of mutants in which this inhibition cannot be alleviated by Chi. In this case a derivative lacking RecD would be recombination-proficient, and assays of enzymatic activity would indicate the mechanism of inhibition. In this paper we demonstrate such an inhibition: recBD1080ACD was recombination-deficient, but recBD1080AC was recombination-proficient. Purified RecBD1080AC enzyme facilitated loading of RecA protein during DNA unwinding (see Results) whereas RecBD1080ACD did not (28). The correlation between genetic assays of recombination proficiency and enzymatic assays of RecA loading demonstrates that E. coli recombination requires loading of RecA by RecBCD enzyme and that the RecD subunit inhibits this reaction.
Materials and Methods
Bacterial Strains, Phage, and Plasmids.
All are listed in Table 1 with their genotypes and sources. The genotype recBCD indicates recB+ recC+ recD+; recBC indicates recB+ recC+; recD− indicates the absence of the RecD subunit and is used for clarity. The genotype recBD1080ACD indicates that amino acid 1080 of the RecB polypeptide has been changed from aspartic acid to alanine by mutation (27).
Table 1.
Bacterial strains and plasmids used
| Strain/plasmid number | Genotype | Ref. or source* |
|---|---|---|
| V66 | argA21 hisG4 recF143 met rpsL31 galK2 xyl-5 λ−F− | Ref. 33 |
| V67 | As V66, plus recB21∷IS186† | Ref. 23 |
| V330 | Δ (recC- argA)234 λ−F− | AC111, ref. 34 |
| V1306 | thi-1 relA1 λ− (Hfr PO44) | Ref. 33 |
| V2333 | As V66 but hisG+ hisD∷kanR | P. Dabert, ref. 35 |
| V2570 | Δ (recC- argA)234 hisD∷kanRrpsL31 λ−F− | V66 × V2571 |
| V2571 | Δ (recC- argA)234 hisD∷kanR λ−F− | V2333 × V330 |
| 594 | lac-3350 galK2 galT22 rpsL179 λ−F− | Ref. 33 |
| C600 | thr-1 leuB6 thi-1 lacY1 tonA21 supE44 rfbD1 λ−F− | Ref. 33 |
| pBR322χ+F225‡ | None | Ref. 36 |
| pSA114§ | recBCD | This work |
| pSA122§ | recBC | This work |
| pABD1080ACD§ | recBD1080ACD | Ref. 27 |
| pSA123§ | recBD1080AC | This work |
| pDWS2‡ | recBCD | Ref. 4 |
| pSA21‡ | recB21CD | Ref. 23 |
A × B indicates a P1 transduction in which A is the donor, and B is the recipient.
The recB21 mutation is an IS186 insertion at nucleotide 913 of the recB coding sequence (ref. 23; data not shown).
Vector is pBR322.
Vector is pACYC184.
Tryptone broth and agar, LB broth and agar, minimal medium, and suspension medium have been described (37). BBL agar contained trypticase (Baltimore Biological Laboratory) instead of tryptone and 0.2% yeast extract. Minimal medium contained the required amino acids at 20 μg/ml. Transformants were grown on agar or in broth containing chloramphenicol (40 μg/ml).
λ Crosses, Chi Activity Measurement, and High-Frequency Recombination (Hfr) Conjugation.
Recombination proficiency was measured in λ crosses and Hfr conjugation as described (33). Hfr conjugational crosses were performed in a recB21 strain with the recBCD alleles present on plasmid derivatives. The recB21 mutation is an IS186 insertion mutation (ref. 23; Table 1; data not shown) that is polar on recD. Polarity was demonstrated by the lack of complementing activity in genetic assays (11, 23) and by the lack of RecD expression in cell extracts as detected by Western blot analysis with mAbs as probes (data not shown).
Cloning the recBC Genes.
BseRI fragments (11.7 kb) containing either the wild-type recB and recC genes from pDWS2 (4) or the recBD1080Aand recC genes from pABD1080ACD (27) were blunt-end ligated into the BamHI site of pACYC184 (38) after removal of 2 nt overhangs by T4 DNA polymerase and the Klenow fragment of DNA polymerase I (New England Biolabs). BseRI cuts 8 nt to the 3′ side of the recB coding sequence and thus effectively removes the adjacent recD gene.
Enzyme Purification and Detection.
We purified the RecBCD, RecBD1080ACD, and RecBD1080AC enzymes as described below. RecBC enzyme was produced by mixing purified RecB and RecC polypeptides as described (39). RecBCD enzyme had 2.3 × 105 units of dsDNA exonuclease activity per mg of protein, whereas RecBD1080ACD and RecBD1080AC enzymes had <25. The unwinding specific activities of RecBD1080ACD enzyme relative to RecBCD enzyme or RecBD1080AC enzyme relative to RecBC enzyme were within a factor of 2 (see Fig. 2 and data not shown).
Figure 2.

RecD inhibits RecA loading during DNA unwinding by RecBD1080ACD enzyme. RecBCD, RecBC, RecBD1080ACD, and RecBD1080AC enzymes were assayed by using 5′-32P-labeled (*) pBR322 χ+F225 DNA (see diagram) as described in Materials and Methods. The DNA substrate (4.7 nM) and indicated amount of mutant or wild-type RecBCD enzyme were incubated at 37o for 2 min in a reaction mix with RecA protein (lanes 3–6, 11–14, 19–22, and 27–30) or without RecA protein (lanes 7–10, 15–18, 23–26, and 31–34). An aliquot was removed for analysis (0 min Exo I). Exonuclease I was added to the remaining sample, and incubation was continued for the times indicated. The products of reaction were analyzed by electrophoresis in a 1% agarose gel (Materials and Methods). The position of ds substrate DNA (DS; lane 1), unwound ssDNA (SS; boiled, in lane 2), and the major product of Chi-dependent nuclease attenuation (Chi) are shown.
RecBD1080ACD and RecBD1080AC enzymes were purified from 3-liter cultures of V330 (pABD1080ACD) and V330 (pSA123), respectively, as described (8). The concentrations of RecBD1080ACD and RecBD1080AC enzymes in unfractionated extracts were estimated from Western blots of native gels. Both enzymes were present at approximately 80% the concentration of wild-type RecBCD enzyme carried on a similar (pACYC184-based) plasmid (data not shown). Purification was through HiTrap Q, Sephacryl S-300, and HiTrap Heparin columns (all from Amersham Pharmacia), followed, for the RecBD1080AC enzyme purification, by a hydroxyapatite column (Bio-Rad CHT-II cartridge). Purification was monitored by native and SDS gel electrophoresis with proteins detected by Coomassie staining or (for the HiTrap Q column eluate) by Western blot analysis (8).
RecBCD Enzyme Reaction Conditions and DNA Substrates.
Plasmid pBR322 χ+F225 DNA was digested with HindIII (New England Biolabs), treated with shrimp alkaline phosphatase (United States Biochemical) and labeled at the 5′ end with [γ-32P] ATP (7,000 Ci/mmol; ICN) using T4 polynucleotide kinase (New England Biolabs). Unincorporated nucleotides were removed from the substrate by passage through an SR200 minicolumn (Amersham Pharmacia Biotech). The substrate then was digested with PstI (New England Biolabs) to remove the 5′ label from the non-Chi-containing strand and to provide a 3′ overhang to facilitate RecBC enzyme binding to DNA (ref. 31; data not shown). The 3,578-bp fragment was purified with a QIAquick column (Qiagen, Chatsworth, CA) from a 1% agarose gel and used in RecBCD enzyme reactions.
RecBCD enzyme Chi cutting, DNA unwinding, and RecA loading assays were performed as described (6). 5′-32P-labeled pBR322 χ+F225 substrate DNA (4.7 nM molecules) was reacted with the amount of enzyme indicated in Fig. 2 at 37o for 2 min in 40 μl of buffer containing 25 mM Tris acetate (pH 7.5), 8 mM magnesium acetate, 5 mM ATP, 1 mM DTT, 1 mM phosphoenol pyruvate, 4 units/ml pyruvate kinase, 20 μM RecA protein (Promega), and 8 μM SS DNA-binding-protein (Promega). After 2 min ATP-γS was added to 5 mM and ss M13 DNA to 40 nM. One minute later a 5-μl sample was removed and added to 5 μl of stop buffer containing 0.125 M EDTA, 2.5% SDS, 10% sucrose, 0.125% bromophenol blue, and 0.125% xylene cyanol. Exonuclease I (100 units/ml; United States Biochemicals) was added to the remaining sample and incubation continued for 6, 12, or 18 min. At each time point a 5-μl sample was removed to stop buffer. Reaction products were separated as described (40, 41) and analyzed by PhosphorImager and imagequant software (version 3.2, Molecular Dynamics).
Results
RecD Inhibits DNA Repair in the recBD1080AMutant.
To test the hypothesis that the RecD subunit of RecBCD enzyme acts as an inhibitor of recombination, we compared the phenotypes of E. coli strains carrying plasmid-borne recBC or recBCD alleles. The recB alleles tested were wild-type or recBD1080A, which contains a mutation in the RecB nuclease domain that eliminates nuclease activity (27).
Strains with and without recD were first tested for phenotypes associated with the absence of RecBCD enzyme, including sensitivity to DNA damaging agents. The recBCD wild-type strain was resistant to mitomycin C, but the ΔrecBCD and recBD1080ACD mutant strains were equally sensitive (Table 2). In accordance with the hypothesis that RecD is an inhibitor, the recD− derivative, recBD1080AC, was resistant to mitomycin C (Table 2).
Table 2.
RecD makes recBD1080A sensitive to mitomycin C
| rec alleles* | E.O.P. on mitomycin C |
|---|---|
| None | <0.01, 0.007 |
| recBCD | 0.8, 0.8 |
| recBC | 0.9, 0.7 |
| recBD1080ACD | <0.01, 0.006 |
| recBD1080AC | 0.8, 0.7 |
Cultures were grown to mid-log phase in LB broth with 40 μg/ml chloramphenicol, diluted, and plated on LB agar plates with or without mitomycin C (0.25 μg/ml). The efficiency of plating (E.O.P.) is the titer on plates with mitomycin C divided by that on plates without mitomycin C. The results of two experiments in which 75–300 colonies were counted for each determination are shown.
Strains are transformants of strain V2570 [Δ(argA-recC)234 hisD∷kan rpsL31] with the indicated rec alleles present on derivatives of plasmid pACYC184.
Similar results were obtained when strains were tested for the ability to repair DNA damage induced by exposure to UV light. The recB21 and recBD1080ACD strains were equally sensitive to UV light (Fig. 1). The recD− derivative, recBD1080AC, and the recBCD strains were equally resistant to UV light. These data show that RecD inhibits DNA repair in the presence of RecBD1080AC.
Figure 1.

RecD inhibits survival of recBD1080A strains after exposure to UV light. Strains are transformants of V67 (recB21∷IS186) with the indicated rec alleles present on a plasmid. Cultures were grown to mid-log phase at 37°C in LB broth with 40 μg/ml chloramphenicol, harvested by centrifugation, resuspended in 10 mM MgSO4, exposed to UV light, and plated on LB agar with chloramphenicol. Survival is the fraction of initial colony-forming units surviving after exposure to the indicated amount of UV light.
RecD Inhibits Recombination in the recBD1080AMutant.
To determine whether RecD inhibits recombination in the recBD1080A strain, we measured recombination proficiency during Hfr conjugation and in mixed phage lambda infections (Table 3). Similar results were obtained from both types of crosses.
Table 3.
RecD inhibits recombination in the recBD1080A mutant
| rec alleles* | Hfr recombination (%His+ [Strr])†
|
Phage λ recombination (% J+R+)‡
|
Chi activity§
|
|||
|---|---|---|---|---|---|---|
| Mean | Range | Mean | Range | Mean | Range | |
| None | 0.001 | 0.0009–0.001 | 0.6 | 0.5–0.8 | 1.1 | 0.9–1.3 |
| recBCD | 1.4 | 0.9–1.8 | 4.5 | 4.1–5.1 | 6.0 | 5.9–6.1 |
| recBC | 0.8 | 0.7–0.9 | 4.9 | 4.8–5.3 | 0.9 | 0.7–1.1 |
| recBD1080ACD | 0.005 | 0.004–0.007 | 0.9 | 0.8–1.1 | 1.0 | 0.7–1.2 |
| recBD1080AC | 0.9 | 0.6–1.1 | 5.2 | 4.8–5.8 | 0.9 | 0.8–1.0 |
Strains are transformants of strain V67 with the indicated rec alleles present on derivatives of pACYC184.
The number of His+ (StrR) recombinants per Hfr donor cell, corrected for the viability of the recipient. The mean and range are shown for three independent matings in which 75–600 His+ colonies were counted for each determination. For each recipient, the frequency of F' his+ transfer from V156 (33) was 0.12–0.24.
The frequency of J+R+ recombinants in cross lysates of phages 1081 and 1082 or 1083 and 1084 (33, 45) was determined by plating on strain 594 (sup+) for recombinants and on strain C600 (supE) for total phage titer. The mean and range of the frequencies are reported for three independent crosses.
The Chi activity for each set of crosses (phage 1081 × 1082 and 1083 × 1084) was determined as described (33). Chi activity = 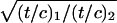 where (t/c) is the ratio of turbid to clear plaques from cross 1 (phage 1081 × phage 1082) or cross 2 (phage 1083 × 1084) among J+R+ recombinants.
where (t/c) is the ratio of turbid to clear plaques from cross 1 (phage 1081 × phage 1082) or cross 2 (phage 1083 × 1084) among J+R+ recombinants.
We first tested recombination proficiency by using Hfr conjugation, a sensitive measure of recombination by the RecBCD pathway (20, 42, 43). The recBD1080ACD strain was as recombination-deficient as the ΔrecBCD strain (Table 3). The frequency of His+ recombinants in the recD− derivative, recBD1080AC, increased approximately 1,000-fold to the level for recBCD and recBC. These results show that RecD is an inhibitor of RecBD1080AC recombination as measured in Hfr conjugation.
Recombination in a mixed lambda infection was measured in strains carrying mutant or wild-type recBCD alleles. The phage were red− to inactivate lambda's own recombination pathway and gam− to inactivate a phage-encoded inhibitor of the RecBCD enzyme (44). The ΔrecBCD and the recBD1080ACD strains were recombination-deficient (Table 3). In contrast, recBD1080AC was as recombination-proficient as recBCD and recBC, confirming that RecD inhibits RecBD1080AC enzyme-mediated recombination.
Chi hotspot activity was measured in lambda vegetative crosses to determine whether the mutant and wild-type RecBCD enzymes interacted with Chi (Table 3). Chi activity is detected when more recombination events occur in an interval with Chi than in the same interval without Chi (45). Wild-type recBCD cells had high Chi activity (6.0), indicating that exchanges were clustered in the interval with Chi. In all of the other strains Chi had no significant effect on the distribution of exchanges, as reflected by a Chi activity of approximately 1 (Table 3). This result shows that recBD1080A with or without recD had no Chi activity and is consistent with the lack of Chi nicking activity in purified enzyme (ref. 27; data not shown). Taken together, these results show that RecD is an inhibitor of RecBD1080AC recombination as measured in Hfr conjugation and lambda vegetative crosses.
RecD Inhibits RecA Loading in the recBD1080AMutant.
Because recBD1080AC was recombination-proficient and recBD1080ACD was not, we expected that enzyme purified from the two strains would differ in one or more activities. We compared enzyme activities to identify a difference that would account for the change in recombination proficiency.
We simultaneously monitored the DNA unwinding, Chi cleavage, and RecA loading activities of RecBCD enzyme by reaction with singly labeled ds pBR322 χ+F225 DNA (Fig. 2). As shown previously (10) under the conditions of these reactions (Mg2+>ATP), wild-type RecBCD enzyme entering from the right (Fig. 2) degrades the upper (3′ terminated) strand of DNA until it reaches Chi, so little or no full-length ssDNA is observed; the subsequent Chi-dependent reduction of exonuclease activity results in the preservation of a fragment extending from the 5′-P32 label to the Chi site (Fig. 2, lane 3). RecBCD enzyme entering from the left (Fig. 2) degrades the bottom, unlabeled (3′-terminated) strand, producing full-length ssDNA. The resistance of RecA-coated ssDNA to exonuclease I distinguished ss reaction products bearing RecA protein from those coated solely with ssDNA-binding protein(6). In reactions containing RecA protein, approximately 70% of the Chi-dependent product was resistant to exonuclease I (Fig. 2, Chi, lanes 4–6; Table 4), showing it to be coated with RecA by RecBCD enzyme. In a similar reaction lacking RecA protein, the ssDNA extending from Chi was observed (Fig. 2, lane 7) but failed to survive exonuclease I digestion (Fig. 2, lanes 8–10, Table 4).
Table 4.
RecD inhibits RecA loading by RecBD1080AC enzyme
| Enzyme | Product* | RecA† | % product surviving exonuclease I digestion after:‡
|
||
|---|---|---|---|---|---|
| 6 min | 12 min | 18 min | |||
| RecBCD | Chi | + | 79, 66 | 69, 67 | 54 |
| − | <1, <1 | <1, <1 | <1 | ||
| RecBC | Unw | + | 53, 60 | 49, 60 | 52 |
| − | 1.7, 2.1 | 1.4, 1.3 | 2.1 | ||
| RecBD1080ACD | Unw | + | 5.0, 3.2, 2.2 | 7.5, 3.1, 1.8 | 2.8 |
| − | <1, <1, 1.2 | <1, <1, 1.3 | <1 | ||
| RecBD1080AC | Unw | + | 44, 76, 41 | 46, 72, 47 | 58 |
| − | 2.4, 3.6, 1.9 | 3.6, 2.9, 1.4 | 3.4 | ||
The major product from RecBCD wild-type or mutant enzyme reaction with linear pBR322 χ+F225 as described in the text. Chi is ssDNA labeled at the 5′ end and extending to the Chi site. Unw is full-length ssDNA labeled at the 5′ end.
Reactions were conducted in the absence (−) or presence (+) of 20 μM RecA protein by using the indicated enzyme. Multiple values are from independent experiments in which samples were taken at the indicated times. The reaction with RecBCD mutant or wild-type enzyme was for 2 min under conditions described in Materials and Methods. Exonuclease I was added and samples were removed at 6, 12, and 18 min (experiment 1) or at 6 and 12 min (experiments 2 and 3). Experiment 1 is shown in Fig. 2.
The percent of product present at the time listed after the addition of exonuclease I relative to the amount present at the end of the RecBCD enzyme reaction was determined by PhosphorImage analysis as described in Materials and Methods.
Mutant RecBCD enzymes that have unwinding activity but lack nuclease activity produce a full-length ss product and may load RecA constitutively on the 3′ end (16). Thus, RecBC enzyme, which lacks nuclease activity (11, 23), produced full-length ssDNA during DNA unwinding but showed no fragment extending from Chi (Fig. 2, lane 11). As shown previously (16), RecA protein is loaded on the 3′ termini where the RecBC enzyme enters the DNA (Fig. 2, lanes 12–14). Approximately 50% of the unwound DNA was protected from exonuclease I digestion in a RecA-dependent manner (Fig. 2, compare lanes 11–14 to lanes 15–18; Table 4). Only 50% of the ssDNA was protected because RecBC enzyme entering dsDNA from the labeled end would load RecA on the unlabeled strand but leave the labeled strand sensitive to exonuclease I digestion.
We compared RecBD1080ACD and RecBD1080AC enzyme in the DNA unwinding and RecA loading assay to see whether the change in DNA repair and recombination phenotypes correlated with a change in enzymatic activity. RecBD1080ACD enzyme has unwinding activity but no detectable nuclease activity (ref. 27; Materials and Methods) and hence produced full-length ssDNA products by unwinding (ref. 27; Fig. 2, lane 19). As shown previously (28), this product was sensitive to exonuclease I, as the enzyme cannot load RecA onto ssDNA (Fig. 2, lanes 20–22; Table 4). Less than 5% of the unwound DNA was protected from exonuclease I digestion in the presence or absence (Fig. 2, lanes 19–26; Table 4) of RecA protein. This result, the failure to load RecA protein on ssDNA, is consistent with the recombination and DNA repair deficiency of recBD1080ACD.
The activity of RecBD1080AC enzyme in this assay was unlike that of RecBD1080ACD enzyme. RecBD1080AC enzyme unwound DNA (Fig. 2, lane 27) and loaded RecA on the 3′ termini as demonstrated by the survival of approximately 55% of the unwound DNA during exonuclease I digestion (Fig. 2, lanes 28–30; Table 4). Protection of the ssDNA depended on RecA (Fig. 2, lanes 31–34; Table 4). Taken together, these results show that the RecD subunit inhibited the RecA loading activity of the RecBD1080AC enzyme and suggest that this activity was needed for recombination proficiency.
Discussion
We have used genetic and enzymatic assays of RecBCD enzyme and a mutant derivative to show that the RecD subunit inhibits recombination in the presence of Chi sites and that RecA loading by RecBCD enzyme is essential for homologous recombination. Analysis of the nuclease-deficient RecBD1080ACD enzyme further elucidated the changes that occur when wild-type enzyme interacts with a Chi site and how these changes contribute to the regulation of recombination events.
The inhibitory role of the RecD subunit was demonstrated by comparing the phenotypes and enzymatic activities of recBD1080ACD and recBD1080AC (Table 5). A recBD1080ACD strain was recombination-deficient (Table 3) and sensitive to DNA damaging agents (Table 2 and Fig. 1), and the purified enzyme failed to load RecA during DNA unwinding (Fig. 2; ref. 28). In contrast, we found that the recD− derivative, recBD1080AC, was recombination-proficient (Table 3) and resistant to DNA damaging agents (Table 2 and Fig. 1), and the RecBD1080AC enzyme was active in the RecA loading assay (Fig. 2). This result indicates that the recBD1080Amutation does not affect RecA loading directly but rather that the RecD subunit inhibits recombination by blocking RecA loading, one of two alternative models suggested previously (28). The mutant enzyme was unable to overcome the inhibitory activity of the RecD subunit (see below).
During DNA unwinding RecBCD enzyme loads RecA on the 3′ end of ssDNA (6), allowing the essential steps of pairing and strand exchange in recombination (24). The observed correlation between genetic measures of recombination proficiency and enzymatic assays of RecA loading by wild-type and mutant RecBCD enzymes indicates that this activity is required for homologous recombination. Although purified RecA can bind to ssDNA and facilitate pairing and strand exchange with dsDNA (24), in cells this activity is apparently not sufficient to support recombination proficiency. Rather, our results indicate that RecBCD enzyme must actively load RecA protein to promote recombination.
The interaction between RecBCD enzyme and Chi results in the stimulation of recombination and regulation of enzyme activity. RecBCD enzyme recognizes Chi (4) and produces a 3′ end for RecA loading by nicking the DNA (4, 5) or reducing its degradative activity (10). Additional features of a RecBCD enzyme-Chi interaction can be inferred from the analysis of RecBD1080ACD. We have shown that RecD inhibited RecA loading by the nuclease-deficient RecBD1080ACD enzyme. Anderson et al. (28) demonstrated that RecBD1080ACD enzyme is able to recognize Chi because this mutant enzyme, like wild-type enzyme, is inactivated during the unwinding of DNA containing Chi; inactivation does not occur with Chio DNA (8, 9, 28, 46). This result indicates that RecBD1080ACD enzyme recognizes Chi; lacking nuclease activity, however, the enzyme fails to cleave the DNA (27) or load RecA (28). The absence of RecA loading activity indicates that a second step other than Chi recognition is required for RecA loading. Our results suggest that this step, which apparently requires or is coordinated with nuclease activity, involves as-yet-undocumented changes in RecD, such as ejection or a conformational change in the enzyme that exposes a domain for RecA loading as hypothesized previously (12, 16). The nuclease activity of RecBCD enzyme may play a role in the signaling mechanism that results in the release or modification of RecD.
Inactivation of RecBCD enzyme by Chi is believed to occur in two steps. The first step includes an alteration of enzyme activity at Chi (see above) and limits RecBCD enzyme to interaction with a single Chi site on the DNA substrate (9). The second step occurs after continued DNA unwinding beyond Chi and results in enzyme inactivation by disassembly of the enzyme's subunits (8). The first step in enzyme regulation appears to require Chi recognition, the nuclease-dependent signal, and alleviation of RecD inhibition. The second step, with accompanying enzyme inactivation, appears to require Chi recognition but not the nuclease-dependent signal or the RecD associated change.
This regulatory mechanism suggests that one RecBCD enzyme molecule facilitates only a single genetic exchange, near a Chi site, at either end of a linear fragment (47). For example, in conjugation or transduction a linear fragment of DNA enters E. coli. The circularity of the chromosome is maintained only if an even number of genetic exchanges occurs between such DNA and the chromosome (8). By limiting the number of recombination events to two, one event at each end of the linear fragment, the integrity of the circular chromosome would be maintained.
When coupled with the recBD1080A mutation, RecD inhibits RecBCD enzyme-mediated recombination (Table 3). The observation that recD null mutants are hyperrecombination-proficient in the absence of Chi sites suggested that RecD is an inhibitor of recombination in the absence of Chi (11). Chaudhury and Smith (11) suggested that Chi sites might reduce a recombination-inhibiting function of RecBCD enzyme and that recD mutants lack the inhibitory function. Stahl and colleagues (12, 13) suggested that the degradative activity of RecBCD enzyme blocks recombination and that ejection of the RecD subunit abolishes nuclease activity and sets the enzyme in a recombinogenic mode. Current information about RecBCD enzymology and genetics supports these proposals in several ways. Interaction with a Chi site activates RecBCD enzyme to nick the DNA (or reduce its degradative activity), RecD is modified or released, and RecA is loaded on the ssDNA with a 3′ terminus near Chi. Subsequent unwinding, joint molecule formation, and resolution leads to recombination. In the absence of Chi (or Chi-like sequences) or in enzymes lacking nuclease activity, we propose that RecD is not modified or released and that there is little RecA loading or recombination. In the mutant strains studied here, RecD is an inhibitor of recombination even in the presence of Chi and in the absence of nuclease activity, because, although the RecBD1080ACD enzyme recognizes Chi (28), it does not carry out the change in RecD that allows RecA loading. The simple absence of RecBCD enzyme's nuclease activity is not sufficient to permit recombination.
Additional work may identify the change in RecD that allows RecA loading and identify the subunit(s) responsible for this essential activity.
Acknowledgments
We thank Doug Julin and colleagues for plasmid pABD1080ACD and helpful discussions, Patrick Dabert for strain V2333, and Luther Davis, Joseph Farah, and Walter Steiner for comments on the manuscript. This work was supported by Public Health Service Grant GM31693 from the National Institute of General Medical Sciences.
Abbreviations
- ss
single-stranded
- ds
double-stranded
- Hfr
high-frequency recombination
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.130192397.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.130192397
References
- 1.Taylor A F. In: Genetic Recombination. Kucherlapati R, Smith G R, editors. Washington, DC: Am. Soc. Microbiol.; 1988. pp. 231–263. [Google Scholar]
- 2.Kowalczykowski S C, Dixon D A, Eggleston A K, Lauder S D, Rehrauer W M. Microbiol Rev. 1994;58:401–465. doi: 10.1128/mr.58.3.401-465.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith G R. In: DNA Damage and Repair, Vol. I: DNA Repair in Prokaryotes and Lower Eukaryotes. Nickoloff J A, Hoekstra M F, editors. Totowa, NJ: Humana; 1998. pp. 135–162. [Google Scholar]
- 4.Ponticelli A S, Schultz D W, Taylor A F, Smith G R. Cell. 1985;41:145–151. doi: 10.1016/0092-8674(85)90069-8. [DOI] [PubMed] [Google Scholar]
- 5.Taylor A F, Schultz D W, Ponticelli A S, Smith G R. Cell. 1985;41:153–163. doi: 10.1016/0092-8674(85)90070-4. [DOI] [PubMed] [Google Scholar]
- 6.Anderson D G, Kowalczykowski S C. Cell. 1997;90:77–86. doi: 10.1016/s0092-8674(00)80315-3. [DOI] [PubMed] [Google Scholar]
- 7.Dixon D A, Kowalczykowski S C. Cell. 1991;66:361–371. doi: 10.1016/0092-8674(91)90625-9. [DOI] [PubMed] [Google Scholar]
- 8.Taylor A F, Smith G R. Genes Dev. 1999;13:890–900. doi: 10.1101/gad.13.7.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Taylor A F, Smith G R. Proc Natl Acad Sci USA. 1992;89:5226–5230. doi: 10.1073/pnas.89.12.5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dixon D A, Kowalczykowski S C. Cell. 1993;73:87–96. doi: 10.1016/0092-8674(93)90162-j. [DOI] [PubMed] [Google Scholar]
- 11.Chaudhury A M, Smith G R. Proc Natl Acad Sci USA. 1984;81:7850–7854. doi: 10.1073/pnas.81.24.7850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thaler D S, Sampson E, Siddiqi I, Rosenberg S M, Stahl F W, Stahl M. In: Mechanisms and Consequences of DNA Damage Processing. Friedberg E, Hanawalt P, editors. New York: Liss; 1988. pp. 413–422. [Google Scholar]
- 13.Stahl F W, Thomason L C, Siddiqi I, Stahl M M. Genetics. 1990;126:519–533. doi: 10.1093/genetics/126.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myers R S, Kuzminov A, Stahl F W. Proc Natl Acad Sci USA. 1995;92:6244–6248. doi: 10.1073/pnas.92.14.6244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Köppen A, Krobitsch S, Thoms B, Wackernagel W. Proc Natl Acad Sci USA. 1995;92:6249–6253. doi: 10.1073/pnas.92.14.6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Churchill J J, Anderson D G, Kowalczykowski S C. Genes Dev. 1999;13:901–911. doi: 10.1101/gad.13.7.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Taylor A F, Smith G R. J Biol Chem. 1995;270:24451–24458. doi: 10.1074/jbc.270.41.24451. [DOI] [PubMed] [Google Scholar]
- 18.Smith G R, Stahl F W. BioEssays. 1985;2:244–249. [Google Scholar]
- 19.Willetts N S, Clark A J, Low B. J Bacteriol. 1969;97:244–249. doi: 10.1128/jb.97.1.244-249.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Willetts N S, Mount D W. J Bacteriol. 1969;100:923–934. doi: 10.1128/jb.100.2.923-934.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howard-Flanders P, Theriot L. Genetics. 1966;53:1137–1150. doi: 10.1093/genetics/53.6.1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Capaldo-Kimball F, Barbour S D. J Bacteriol. 1971;106:204–212. doi: 10.1128/jb.106.1.204-212.1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Amundsen S K, Taylor A F, Chaudhury A M, Smith G R. Proc Natl Acad Sci USA. 1986;83:5558–5562. doi: 10.1073/pnas.83.15.5558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.West S C. Annu Rev Biochem. 1992;61:603–640. doi: 10.1146/annurev.bi.61.070192.003131. [DOI] [PubMed] [Google Scholar]
- 25.West S C. J Bacteriol. 1996;178:1237–1241. doi: 10.1128/jb.178.5.1237-1241.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yu M, Souaya J, Julin D A. Proc Natl Acad Sci USA. 1998;95:981–986. doi: 10.1073/pnas.95.3.981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yu M, Souaya J, Julin D A. J Mol Biol. 1998;283:797–808. doi: 10.1006/jmbi.1998.2127. [DOI] [PubMed] [Google Scholar]
- 28.Anderson D G, Churchill J J, Kowalczykowski S K. J Biol Chem. 1999;274:27139–27144. doi: 10.1074/jbc.274.38.27139. [DOI] [PubMed] [Google Scholar]
- 29.Lovett S T, Luisi-DeLuca C, Kolodner R D. Genetics. 1988;120:37–45. doi: 10.1093/genetics/120.1.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thaler D S, Sampson E, Siddiqi I, Rosenberg S M, Thomason L C, Stahl F W, Stahl M M. Genome. 1989;31:53–67. doi: 10.1139/g89-013. [DOI] [PubMed] [Google Scholar]
- 31.Korangy F, Julin D A. Biochemistry. 1993;32:4873–4880. doi: 10.1021/bi00069a024. [DOI] [PubMed] [Google Scholar]
- 32.Korangy F, Julin D A. Biochemistry. 1994;33:9552–9560. doi: 10.1021/bi00198a022. [DOI] [PubMed] [Google Scholar]
- 33.Schultz D W, Taylor A F, Smith G R. J Bacteriol. 1983;155:664–680. doi: 10.1128/jb.155.2.664-680.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chaudhury A M, Smith G R. J Bacteriol. 1984;160:788–791. doi: 10.1128/jb.160.2.788-791.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dabert P, Smith G R. Genetics. 1997;145:877–889. doi: 10.1093/genetics/145.4.877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Smith G R, Kunes S M, Schultz D W, Taylor A, Triman K L. Cell. 1981;24:429–436. doi: 10.1016/0092-8674(81)90333-0. [DOI] [PubMed] [Google Scholar]
- 37.Cheng K C, Smith G R. Genetics. 1989;123:5–17. doi: 10.1093/genetics/123.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chang A C Y, Cohen S N. J Bacteriol. 1978;134:1141–1156. doi: 10.1128/jb.134.3.1141-1156.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Masterson C, Boehmer P E, McDonald F, Chaudhuri S, Hickson I D, Emmerson P T. J Biol Chem. 1992;267:13564–13572. [PubMed] [Google Scholar]
- 40.Amundsen S K, Taylor A F, Smith G R. Nucleic Acids Res. 1998;26:2125–2131. doi: 10.1093/nar/26.9.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sambrook J, Fritsch E F, Maniatis T. Molecular Cloning: A Laboratory Manual. 2nd Ed. Plainview, NY: Cold Spring Harbor Lab. Press; 1989. [Google Scholar]
- 42.Clark A J, Margulies A. Proc Natl Acad Sci USA. 1965;53:451–459. doi: 10.1073/pnas.53.2.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Emmerson P T. Genetics. 1968;60:19–30. doi: 10.1093/genetics/60.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smith G R. In: Lambda II. Hendrix R W, Roberts J W, Stahl F W, Weisberg R A, editors. Plainview, NY: Cold Spring Harbor Lab. Press; 1983. pp. 175–209. [Google Scholar]
- 45.Stahl F W, Stahl M M. Genetics. 1977;86:715–725. doi: 10.1093/genetics/86.4.715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dixon D A, Churchill J J, Kowalczykowski S C. Proc Natl Acad Sci USA. 1994;91:2980–2984. doi: 10.1073/pnas.91.8.2980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Smith G R. Cell. 1991;64:19–27. doi: 10.1016/0092-8674(91)90205-d. [DOI] [PubMed] [Google Scholar]