

Abstract
Beta-cell replacement is considered to be the most promising approach for treatment of type 1 diabetes. Its application on a large scale is hindered by a shortage of cells for transplantation. Activation of insulin expression, storage, and regulated secretion in stem/progenitor cells offers novel ways to overcome this shortage. We explored whether fetal human progenitor liver cells (FH) could be induced to differentiate into insulin-producing cells after expression of the pancreatic duodenal homeobox 1 (Pdx1) gene, which is a key regulator of pancreatic development and insulin expression in beta cells. FH cells possess a considerable replication capacity, and this was further extended by introduction of the gene for the catalytic subunit of human telomerase. Immortalized FH cells expressing Pdx1 activated multiple beta-cell genes, produced and stored considerable amounts of insulin, and released insulin in a regulated manner in response to glucose. When transplanted into hyperglycemic immunodeficient mice, the cells restored and maintained euglycemia for prolonged periods. Quantitation of human C-peptide in the mouse serum confirmed that the glycemia was normalized by the transplanted human cells. This approach offers the potential of a novel source of cells for transplantation into patients with type 1 diabetes.
Type 1 diabetes mellitus is characterized by a progressive loss of pancreatic beta cells, leading to insufficient insulin production. Beta-cell replacement is considered the optimal treatment for type 1 diabetes. However, the availability of human organs for transplantation is limited. An effective cell replacement strategy depends on the development of an abundant supply of beta cells and their protection from immune destruction. Reversible immortalization of differentiated beta cells by conditional oncogene expression allowed the controlled expansion of murine cells in tissue culture, which were capable of replacing beta-cell function in vivo (1–3). However, this approach has not been successful yet with beta cells from human islets, which tend to lose insulin expression during forced replication (4). An alternative approach has used the natural proliferative capacity of stem cells, both embryonic and from adult tissues, as a source of cells which can be induced to differentiate into insulin-producing cells. Recent studies showed that cultured mouse (5) and human (6) embryonic stem (ES) cells can differentiate at low frequencies into insulin-producing cells. By enrichment and expansion of nestin-positive cells, Lumelsky et al. (7) significantly enhanced the generation of insulin-positive cells from mouse ES cell cultures.
Several groups demonstrated the ability of stem/progenitor cells isolated from adult organs to differentiate along additional cell lineages (8, 9). These findings may reflect the presence of pluripotent cells in adult tissues, or the ability of committed stem/ progenitor cells to transdifferentiate under suitable conditions. Lineage switching in stem/progenitor cells may be stimulated by soluble factors, as well as master regulatory genes, which can activate hierarchically determined cascades of gene expression in a dominant fashion.
The homeodomain transcription factor pancreatic duodenal homeobox 1 (Pdx1) plays key roles in pancreas development (10) and is expressed in mature beta cells as well, where it regulates expression of multiple genes, including insulin and the glucose transporter GLUT2 (11). Moreover, forced Pdx1 expression in mouse (12) and Xenopus (13) parenchymal liver cells, and in rat enterocytes in vitro (14), was shown to activate beta-cell genes, including insulin.
Study of pancreatic injury in animal models revealed that pancreatic duct epithelial cells are capable of islet neogenesis in adult animals. These cells may also be involved in pancreatic islet renewal in the absence of injury. Although epithelial cells isolated from pancreatic ducts were shown to differentiate into beta cells in culture (15, 16), human progenitor cells capable of differentiation into pancreatic beta-cells have not yet been isolated.
Stem/progenitor cells isolated from the human liver may offer an alternative source for generating insulin-producing cells. The parenchymal cells in liver and pancreas share embryological origin from the primitive foregut. Furthermore, mature hepatocytes and pancreatic beta cells manifest similarities in gene expression profiles, including genes encoding transcription factors, the glucose transporter GLUT2, and the glucose phosphorylating enzyme glucokinase (GK). Pluripotent progenitor cells cultured from mouse fetal liver were shown to differentiate in vivo into a number of hepatic, pancreatic, and intestinal cell types (17). In addition, adult rat hepatic stem cells termed oval cells were shown to give rise to pancreatic endocrine cells in vitro (18). Recently, procedures were developed for isolating and culturing unique populations of epithelial progenitor cells from human fetal liver (FH) (19). These cells express markers of hepatocytes, bile duct cells, and oval cells, and are capable of differentiating into mature hepatocytes in vivo (19). Moreover, FH cells were amenable to immortalization after retroviral introduction of the catalytic subunit of the human telomerase gene (FH-hTERT) (20). The replication potential of FH-hTERT cells was greatly enhanced, without evidence for neoplastic cell transformation, in either in vitro or in vivo assays. Here we explored the possibility of directing FH-hTERT cells along the pancreatic beta-cell lineage under Pdx1 regulation. Our findings demonstrated that Pdx1 activated numerous beta-cell genes in FH-hTERT cells. These cells produced, stored, and released insulin in response to glucose. They were able to restore and maintain euglycemia in hyperglycemic immunodeficient mice. This approach offers a novel source of cells for transplantation into patients with type 1 diabetes.
Methods
Cell Culture. FH cells were isolated and cultured as described (19), in DMEM containing 25 mM glucose and supplemented with 10% FBS, 100 units/ml penicillin, 100 μg/ml streptomycin, 5 μM hydrocortisone, and 5 μg/ml insulin. A subclone of FH cells that was stably transduced with hTERT, designated FH-B-hTERT (FH-B), was cultured in the same medium, as described (20). FH-B cells transduced with the Pdx1 gene (see below), designated FH-B-TPN cells, were maintained in the same culture medium, with the exception of hydrocortisone and insulin. For incubation in serum-free medium, the cells were placed in DMEM containing antibiotics in the presence of 100 ng/ml insulin, 55 ng/ml transferrin, and 50 pg/ml selenium (ITS, Sigma).
Construction of Pdx1 Lentivirus. A Pdx1 lentivirus was generated by using an equine infectious anemia virus (EIAV)-based vector (21, 22). A fragment including rat Pdx1 cDNA (23) under control of the mouse phosphoglycerate kinase 1 promoter (PGKP; ref. 24), placed upstream of an encephalomyocarditis virus internal ribosomal entry site (25), a neomycin resistance gene, and the posttranscriptional regulatory element of woodchuck hepatitis virus (WHV), was inserted between the XbaI and KpnI sites of the pONY4G SIN-MunI vector (Oxford Biomedica, Oxford) (26). The long terminal repeats (LTR) of this vector are mutated by removal of the U3 element to prevent LTR-mediated transactivation of gene expression and to limit recombination into replication-competent virus. The WHV element increases nuclear RNA stability and the efficiency of its transport out of the nucleus (27). The pONY4-Pdx1 construct (Fig. 1a) was cotransfected with the pONY3.1 plasmid (26) encoding viral gag/pol, and the pMD.G plasmid (28) encoding pseudotyped vesicular stomatitis virus envelope protein, into 293T cells as described (28). The culture medium was collected 36 h later and titrated in COS7 cells by counting geneticin (G418)-resistant colonies. FH-B cells were incubated overnight with Pdx1 lentivirus under 5:1 multiplicity of infection at 37°C in medium containing 5 mM glucose. After 2 days, the medium was switched to 25 mM glucose, and FH-B-TPN cells were selected with 200 μg/ml G418 for 16 days.
Fig. 1.

Pdx1 induces insulin expression in FH-B-TPN cells. (a) Diagram of the pONY4-Pdx1 viral vector. Transcription of the vector genome is driven by the human cytomegalovirus immediate-early enhancer/promoter (CMVP) fused to the R and U5 regions of the 5′ long terminal repeat of equine infectious anemia virus (EIAV). PGKP, phosphoglycerate kinase 1 promoter; IRES, internal ribosomal entry site; Neo, neomycin resistance gene; WHV posttranscriptional regulatory element of woodchuck hepatitis virus; rev, EIAV gene encoding the Rev protein; Δenv, mutated EIAV env gene. (b–e) Histological analyses of FH-B-TPN cells. (b) Nuclear PDX1 rhodamine-immunofluorescence. (c) Phase contrast image of a ball-shaped cluster formed in a confluent culture. (d) Cytoplasmic insulin Cy2-immunofluorescence. (e) Phase contrast image of the same field shown in d. b and d do not show the same field. FH-B cells were negative for staining for both proteins (data not shown). (Scale bar, 10 μm.)
RT-PCR Analyses. Total RNA was extracted from cultured cells, mature human hepatocytes (obtained from Incara Cell Technologies, Research Triangle Park, NC), and human pancreatic islets (obtained through the Juvenile Diabetes Research Foundation Islet Distribution Program), by using commercial kits (High Pure RNA isolation kit, Roche Molecular Biochemicals, Mannhein, Germany; Trizol Reagent, Invitrogen Life Technologies, Carlsbad, CA). Specific transcripts were analyzed with Promega RT-PCR kit or GeneAmp Gold RNA PCR kit (Perkin–Elmer), according to the manufacturers. The absence of DNA contamination in RNA samples was confirmed with PCR primers flanking an intron. cDNA was amplified for 40 cycles (94°C for 45 sec; annealing for 45 sec; 72°C for 40 sec), by using the primer pairs and annealing temperatures listed in Table 1, which is published as supporting information on the PNAS web site, www.pnas.org. PCR products were separated by electrophoresis in 1.5–2.5% agarose gels and visualized by ethidium bromide staining.
Insulin Secretion and Content. Insulin secretion from FH-B-TPN cells was measured by static incubation as described (2). Cells were plated in 24-well plates at 105 cells per well. The cells were preincubated for 1 h in Krebs–Ringer buffer (KRB), followed by incubation for the indicated period in KRB containing 0.5 mM 1-isobutyl 3-methylxanthine and glucose at various concentrations. The cells were then extracted in acetic acid, and the amount of insulin in the buffer and cell extract was determined by RIA using the INSIK-5 kit (DiaSorin, Vercelli, Italy) according to the manufacturer. This assay has <20% crossreactivity with proinsulin. In addition, insulin content was determined by using an ELISA kit (Diagnostic Systems Laboratories, Webster, TX), which recognizes only mature insulin. Insulin content was normalized to total cellular protein, measured by the Bio-Rad Protein Assay kit.
Cell Proliferation Assay. [3H]thymidine incorporation was measured in 104 cells during a 16-h pulse, as described (1).
Cell Transplantation. Six-week-old nonobese diabetic severe combined immunodeficient (NOD-scid) female mice (Harlan, Jerusalem) were made hyperglycemic by i.p. injection of streptozotocin (STZ) at 180 μg per g of body weight. When blood glucose reached levels >300 mg/dl, mice were transplanted on the same day with 107 FH-B-TPN cells in 0.5 ml of PBS i.p. Blood glucose levels were monitored twice a week in samples obtained from the tail vein of fed mice by using Accutrend strips (Roche Diagnostics). Serum insulin and human C-peptide levels were determined by RIA in blood samples obtained from the orbital plexus of fed mice, by using the INSIK-5 and Double Antibody C-Peptide (EURO/DPC, Llanberis, U.K.) kits, respectively, according to the manufacturers. The human C-peptide kit had 0% cross reactivity with mouse C-peptide. For transplantation under the renal capsule, 3 × 106 cells preincubated for 6 days in serum-free medium were placed in 50 μl of PBS and injected in the left kidney by using a 30-gauge needle. At the indicated time point, the mice were anesthetized and subjected to left kidney nephrectomy. Mice were monitored 1 day later for changes in blood glucose levels.
Glucose Tolerance Test. Mice fasted for 6 h were injected i.p. with glucose in saline at 1 mg per g of body weight. Blood glucose levels were monitored at the indicated time points in samples obtained from the tail vein.
Histological Assays. FH-B-TPN cells plated in six-well plates on sterilized coverslips were fixed in 4% paraformaldehyde. For insulin immunostaining, cells were blocked for 10 min at room temperature in 5% BSA, 5% FBS, and 0.1% Triton X-100, and incubated with guinea pig anti-insulin serum (Linco Research, St. Charles, MO) diluted 1:1,000 in blocking solution for 1 h at room temperature. The bound antibody was visualized with Cy2-conjugated donkey anti-guinea pig serum (Jackson ImmunoResearch; 1:400) under a Zeiss confocal microscope. For Pdx1 immunostaining, cells were incubated in 0.25% Nonidet P-40 for 10 min at room temperature before blocking, followed by incubation in rabbit-anti-Pdx1 serum diluted 1:5,000. The bound antibody was visualized with rhodamine-conjugated donkey anti-rabbit serum (Jackson ImmunoResearch; 1:600).
Histochemical Studies of Liver Gene Expression. Cells were stained for glycogen, dipeptidyl peptidase IV (DPPIV), and γ-glutamyl transpeptidase activities as described (29).
Statistical Analysis. Variance analysis was performed by using ANOVA.
Results
To induce differentiation of FH-hTERT cells into insulin-producing cells, a subclone, designated FH-B, was transduced with a lentivirus vector containing the Pdx1 and neomycin resistance genes, both expressed from a common promoter using an internal ribosome entry site (Fig. 1a). Cells surviving 16 days of selection in G418 were termed FH-B-TPN (for Telomerase, Pdx1, and Neo). FH-B-TPN cells continued to grow as monolayers on cell culture plastic, which was similar to the parental FH-B cells (Fig. 1e). In confluent cultures, ball-shaped cell clusters could be observed (Fig. 1c). However, the rate of cell proliferation in FH-B-TPN cells declined, compared with FH-B cells. [3H]thymidine incorporation in DNA was reduced 3-fold in FH-B-TPN cells, compared with FH-B cells (2,367 ± 394 cpm vs. 7,381 ± 668 cpm, mean ± SEM, n = 12, P < 0.001), and the population doubling time increased from 40 to 84 h.
The pattern of gene expression in FH-B-TPN cells was analyzed with a representative panel of liver genes expressed in parental FH cells and FH-B cells. These studies included RT-PCR as well as histochemical stainings (Fig. 2). Despite transduction with Pdx1 lentivirus and G418 selection, FH-B-TPN cells continued to express multiple liver genes, including glycogen (a hepatocyte marker), and DPPIV and γ-glutamyl transpeptidase (biliary markers), which was similar to the parental FH and FH-B cells. FH-B-TPN cells showed differences in transcription factor expression compared with FH and FH-B cells. Some transcription factors expressed in FH cells were extinguished in FH-B and FH-B-TPN cells, e.g., hepatocyte nuclear factor (HNF)-4, GATA-4, and CCAAT-enhancer binding protein (C/EBP)α, whereas C/EBPγ continued to be expressed. The expression of hepatocyte growth factor was unchanged in FH-B-TPN cells, whereas expression of HNF-1β, transforming growth factor (TGF)α, TGFβ1, and TGFβ1R mRNAs, and to a lesser extent GATA-1 mRNA, was down-regulated in FH-B-TPN, compared with FH-B cells. Moreover, FH-B-TPN cells expressed GATA-6, which was similar to FH cells. This multilineage gene expression pattern, with expression of hepatocyte markers, biliary markers and GATA-1, which is characteristic of hematopoietic cells, indicated that FH-B-TPN cells retained a stem/progenitor cell phenotype.
Fig. 2.

Expression of liver genes in mature adult hepatocytes, primary fetal liver cells, FH-B cells, and FH-B-TPN cells. (a) Genes analyzed by RT-PCR. Primers for human GAPDH gene were used to monitor mRNA and cDNA quality. (b) Histochemical studies of cultured FH-B-TPN cells showing flat epithelial cell morphology (phase contrast), and expression of glycogen (hepatocyte marker), DPPIV (bile canalicular marker), and γ-glutamyl transpeptidase (biliary marker). Magnification, ×90.
Analysis of Pdx1 expression showed that FH-B-TPN cells expressed both rat Pdx1, which was exogenously introduced, and endogenous human Pdx1, which was likely activated by the rat Pdx1 transgene (30, 31) (Fig. 3a). Rat Pdx1 shares 88% amino acid homology with the human Pdx1 protein, and was therefore expected to be active in human cells.
Fig. 3.

Expression of transcripts of pancreatic genes in FH-B-TPN cells. mRNA extracted from FH-B-TPN and FH-B cells was subjected to RT-PCR analysis with primers for the indicated genes. (a) Analysis of expression of rat Pdx1, with plasmid DNA as positive control. (b) Analysis of expression of human pancreatic genes, with human islets as positive control. Primers for myosin 6 (MYO6) were used to monitor mRNA and cDNA quality. The amount of human islet cDNA used in the analysis with insulin primers was 1/10th of the amount used for the other genes. Mix, PCR without cDNA.
Expression of beta-cell and pancreatic genes was evaluated in FH-B-TPN cells by RT-PCR analysis (Fig. 3b). Expression of genes encoding two transcription factors found in mature beta cells, BETA2 and NKX6.1, as well as neurogenin 3 (NGN3), a transcription factor found in fetal islet cells, was observed. In contrast, genes for three other beta-cell transcription factors, NKX2.2, ISL1, and PAX6, were not expressed in FH-B-TPN cells. PAX4, a transcription factor found in embryonic beta cells, was also not expressed in FH-B-TPN cells.
FH-B-TPN cells expressed insulin mRNA, along with transcripts for 2 proinsulin processing enzymes, prohormone convertase (PC) 1/3 and PC2, indicating that the Pdx1-modified cells acquired the ability to synthesize and process proinsulin to mature insulin. Transcripts encoding the beta-cell protein islet amyloid polypeptide were also detected. Moreover, expression of a major component of dense-core secretory granules, chromogranin A, was observed, suggesting induction of a regulated secretory pathway in the FH-B-TPN cells, which is not normally present in hepatocytes. Transcripts for another component of the secretory vesicle, synaptogyrin 3 (SYNG3), were present in both FH-B and FH-B-TPN cells. Of the two components of the 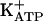 channel, SUR1 and KIR6.2, only the latter was expressed in FH-B-TPN cells. In contrast, expression of GLUT2 and GK, which participate in signal-secretion coupling in beta cells, and are also present in mature hepatocytes, could not be detected in FH-B-TPN cells.
channel, SUR1 and KIR6.2, only the latter was expressed in FH-B-TPN cells. In contrast, expression of GLUT2 and GK, which participate in signal-secretion coupling in beta cells, and are also present in mature hepatocytes, could not be detected in FH-B-TPN cells.
Additional transcripts for proteins found in non-beta islet cells, as well as in exocrine pancreas, were detected in FH-B-TPN cells. These included glucagon, pancreatic polypeptide (PP), and elastase. Transcripts for somatostatin were not detected. Notably, FH-B-TPN cells expressed glucagon mRNA in the absence of detectable PAX6, which is needed in pancreatic islets for alpha-cell development and gene expression (32).
To determine the proportion of insulin-positive cells within the FH-B-TPN cell population, PDX1 and insulin expression was analyzed by immunostaining (Fig. 1). Pdx1 staining was visible in all of the cells, which is expected because the cells underwent selection for neomycin resistance expressed from the same transcript. Insulin staining was also detected in all of the cells, as seen in Fig. 1 by comparing immunofluorescence and phase contrast images of the same field.
Presence of immunostainable insulin demonstrated that FH-B-TPN cells were capable of storing insulin. The ability to store insulin was further established by RIA. The insulin content of FH-B-TPN cells was found to be 185 ± 38 ng per 1 × 106 cells (or 100 μg of protein). The cells were monitored for insulin content during 38 population doublings after G418 selection without a notable change.
Insulin secretion in response to glucose was determined by static incubations. Most insulin was released from FH-B-TPN cells in response to stimulation with glucose concentrations between 8 and 20 mM (Fig. 4). This phenotype of regulated insulin secretion after glucose stimulation was maintained in multiple experiments using FH-B-TPN cells after 4–30 population doublings following G418 selection.
Fig. 4.

Glucose-induced insulin secretion from FH-B-TPN cells during a 2-h static incubation. Values are mean ± SEM of nine replicate wells from three independent experiments.
To determine the ability of FH-B-TPN cells to replace beta-cell function, they were transplanted into NOD-scid mice, which were treated with STZ to eliminate their beta cells. Starting from 2 weeks after transplantation, blood glucose levels in the transplanted mice decreased and were stabilized around 160 mg/dl for the remainder of the experiment, during ≈8 weeks (Fig. 5a), whereas untransplanted mice remained hyperglycemic. At the end of the experiment, serum was taken from the mice for insulin and human C-peptide RIA. Serum insulin levels measured in the STZ-treated transplanted mice averaged 0.98 ± 0.16 ng/ml, compared with 0.30 ± 0.04 ng per ml in the control group of untransplanted mice (P < 0.025). Serum human C-peptide levels averaged 1.18 ± 0.26 ng per ml, compared with undetectable levels in both normal NOD-scid and the STZ-treated untransplanted mice. The low insulin levels in the untransplanted mice, which could reflect residual beta-cell survival and/or limited regeneration, allowed survival of these mice with hyperglycemia until the end of the experiment, though they were insufficient for achieving euglycemia. Taken together, these findings indicate that the glycemia in the transplanted mice was normalized by insulin secretion from the human cells. An i.p. glucose tolerance test performed in the transplanted mice showed a clearance rate similar to that of normal NOD-scid mice (Fig. 5b), suggesting that insulin secretion from the cells in vivo was regulated by glucose as observed in the cultured cells. Analysis of the mice killed after completion of the experiment, close to 3 months after cell transplantation, did not demonstrate tumors in the peritoneal cavity.
Fig. 5.

FH-B-TPN cells can replace beta-cell function in NOD-scid STZ-induced diabetic mice. (a) Fed blood glucose levels. Filled circles, transplanted mice (n = 3); open circles, untransplanted mice (n = 4). Values are mean ± SEM. The differences between the two groups were significant on days 25–70 after transplantation (P < 0.0001). (b) Glucose tolerance test performed on the mice in a 70 days after transplantation. Circles, transplanted diabetic mice; triangles, nondiabetic NOD-scid mice. Values are mean ± SEM. The differences between transplanted and nondiabetic mice were not significant at the 30- to 120-min time points (P > 0.2). Untransplanted diabetic mice were hyperglycemic (>350 mg/dl) at all time points (data not shown).
To promote further differentiation of FH-B-TPN cells, they were incubated in serum-free medium supplemented with ITS. After a 6-day incubation, insulin content increased 15-fold, to 2,766 ± 232 ng per 1 × 106 cells. Quantitation of cellular insulin content using an ELISA kit that detects only mature insulin revealed a content of 2,580 ± 77 ng per 1 × 106 cells, indicating that most of the insulin was stored in the cells in a processed form. No insulin was detected in control FH-B cells incubated in the same conditions. Assay of glucose-induced insulin secretion during a 30-min static incubation demonstrated the same dose-response observed in cells grown in regular medium (Fig. 6a). RT-PCR analysis revealed a large increase in insulin mRNA levels, as well as induction of expression of NKX2.2 (Fig. 6b). However, transcripts for GLUT2, GK, and SUR1 were still absent after this treatment (data not shown). Transplantation of cells after a 6-day incubation in serum-free medium under the renal capsule of STZ-diabetic NOD-scid mice resulted in a rapid restoration of euglycemia (Fig. 6c). Fed serum insulin and human C-peptide levels 7 days after transplantation were 2.18 ± 0.32 and 3.48 ± 0.37 ng per ml, respectively. Removal of the transplanted cells by nephrectomy caused a rapid increase in blood glucose levels, demonstrating that the correction of blood glucose was caused by the transplanted cells. These results establish the capacity of FH-B-TPN cells to function as surrogate beta cells in vivo.
Fig. 6.

Characterization of FH-B-TPN cells after a 6-day incubation in serum-free medium. (a) Glucose-induced insulin secretion during a 30-min incubation. Values are mean ± SEM of three replicate wells. (b) Expression of transcripts of pancreatic genes analyzed by RT-PCR. 1, serum-free medium; 2, regular medium; 3, mix; 4, human islets. (c) Cell transplantation under the renal capsule of NOD-scid STZ-induced diabetic mice. Each line represents fed blood glucose levels of a single mouse. The arrow marks left-kidney nephrectomy of the mice labeled by circles and squares, leading to hyperglycemia.
Discussion
These results demonstrate that Pdx1 activates the expression of insulin, as well as other beta-cell genes, in human fetal liver progenitor cells. Induction of insulin expression in these cells was likely caused by the rat Pdx1 transgene, as well as by activation of the endogenous human Pdx1 gene by rat Pdx1, as indicated by our RT-PCR analysis, though we do not know whether both proteins were produced in FH-B-TPN cells.
In expressing Pdx1 in FH-B-TPN cells, our goal was not only to activate insulin expression, but rather to induce a cellular phenotype along the mature pancreatic beta-cell lineage, as manifested by insulin production in normal amounts, insulin processing and storage, and appropriate insulin release in response to physiological signals. Our findings suggest that FH-B-TPN cells have undergone profound changes as a result of Pdx1 expression. These cells acquired the ability to produce proinsulin, as shown by insulin mRNA and protein analyses. Expression of PC1/3 and PC2 in FH-B-TPN cells, as well as insulin ELISA, which recognizes only mature insulin, suggest that the cells possess the ability to process proinsulin to mature insulin, though direct verification of this possibility will require HPLC analyses. Analysis of insulin content in FH-B-TPN cells by immunofluorescence, RIA, and ELISA assays demonstrated an ability of these cells to store significant amounts of insulin. Cell incubation in serum-free medium resulted in a 15-fold increase in insulin content, up to ≈2.8% of cellular protein. This amount represents a significant fraction of the insulin found in normal pancreatic islets, and to our knowledge is the highest amount reported for insulin-producing cells differentiated from non-beta cells. Further investigation will be required to determine the precise subcellular compartment(s) in which insulin is stored in FH-B-TPN cells, because mature liver cells lack a regulated secretory pathway. Expression of mRNAs for proteins found in secretory vesicles, such as chromogranin A and SYNG3, suggests that insulin may be stored in vesicles. However, confirmation of this possibility will require ultrastructural studies. Release of the stored insulin in response to glucose may reflect the induction of a signal-secretion coupling apparatus in FH-B-TPN cells. However, most of the response occurred above 8 mM, which is somewhat shifted to the right, compared with normal islets that respond to 5 mM glucose. In addition, we could not demonstrate expression of GLUT2, GK, and the sulfonylurea receptor SUR1, a component of the 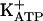 channel, in FH-B-TPN cells, all of which are required for glucose-regulated insulin secretion in beta cells. Further investigation will be required to reconcile these findings.
channel, in FH-B-TPN cells, all of which are required for glucose-regulated insulin secretion in beta cells. Further investigation will be required to reconcile these findings.
Expression of other endocrine and exocrine pancreatic genes, such as glucagon, PP, and elastase, was revealed by RT-PCR analysis. This could reflect the emergence of several distinct cell phenotypes, or coexpression of multiple gene products in some or all FH-B-TPN cells. However, although the negative RT-PCR results can be trusted to reflect a lack of expression of specific genes, the weak positive results should be confirmed by other methods, such as immunostaining, to determine the fraction of cells that express each gene. Presence of transcripts of Ngn3, a transcription factor that, in mice, is found in embryonic but not in mature pancreatic islets (33), indicates that phenotypically FH-B-TPN cells may resemble immature islet precursor cells more closely than mature beta cells.
Pdx1 expression did not extinguish liver gene expression in FH-B-TPN cells, as shown by the presence of glycogen, DPPIV, and γ-glutamyl transpeptidase, as well as expression of several liver transcription factor and growth factor genes. However, some differences in expression of these genes after Pdx1 expression were obvious in FH-B-TPN cells, including extinction of HNF-1β and reactivation of the GATA-6 transcription factor. In addition, FH-B-TPN cells lost expression of multiple growth factors, including TGFα and TGFβ, as well as TGFβ receptor. The precise significance of these changes is unclear at present. Nonetheless, proliferative activity was decreased in FH-B-TPN cells, compared with FH-B cells, and this could well be due to perturbed growth factor regulation after Pdx1 expression.
The clearest demonstration of the differentiation of FH-B-TPN cells along the beta-cell lineage is their ability to replace beta-cell function for long periods of time in vivo. The lag period of >2 weeks observed in the transplanted mice between the time of transplantation and normalization of blood glucose levels may reflect a need for further cell differentiation in vivo. This possibility is supported by the faster rate of glycemia correction with more differentiated cells generated by incubation in serum-free medium. We do not know whether FH-B-TPN cells can proliferate in vivo. It has been established that FH-B cells had no tumorigenic potential in NOD-scid mice (20). Indeed, no evidence for neoplasia was revealed in mice transplanted with FH-B-TPN cells 3 months after transplantation, though excluding this possibility will require in situ analyses of the transplanted cells after long-term studies.
In conclusion, our studies establish the potential of human liver progenitor cells in expressing insulin, releasing it in response to glucose, and replacing beta-cell function in vivo. Further manipulations will be needed to promote differentiation of FH-B-TPN cells along the beta-cell phenotype, aimed at activation of additional beta-cell genes, decreasing expression of non-beta-cell pancreas and liver genes, and increasing the amounts of insulin produced and stored in the cells. One manipulation presented here, cell incubation in serum-free medium, resulted in a large increase in insulin mRNA and protein levels, the induction of the beta-cell transcription factor NKX2.2, and a much faster correction of glycemia, as compared with untreated cells.
The use of progenitor cells derived from fetal human liver cells for developing a universal donor cell source for beta-cell replacement offers several advantages, compared with efforts to obtain human insulin-producing cells from other sources, such as ES cells. Liver-derived cells express a number of transcription factors, the HNFs, which are needed in addition to Pdx1 for beta-cell development and for maintaining the mature beta-cell phenotype. In addition, HLA-matching of banked fetal stem/progenitor liver cells could be achieved far more readily than with the limited number of human ES cell lines, thereby increasing the likelihood of allograft survival.
Supplementary Material
Acknowledgments
We thank Philippe Dupraz and Bernard Thorens for Pdx1 cassette and guidance with lentivirus construction, Chris Wright for Pdx1 antibody, and the Juvenile Diabetes Research Foundation Islet Distribution Program for human islets. This work was funded in part by the Israel Science Foundation (S.E.) and National Institutes of Health Grants DK52956 (to S.E. and N.F.), DK20541 (to N.F.), DK46952 (to S.G.), and DK41296 (to S.G.). This work was performed in partial fulfillment of the requirements for a Ph.D. degree of M.Z.
Abbreviations: Pdx1, pancreatic duodenal homeobox 1; FH, fetal hepatocyte; ES, embryonic stem; TGF, transforming growth factor; DPPIV, dipeptidyl peptidase IV; STZ, streptozotocin; HNF, hepatocyte nuclear factor; PC, prohormone convertase; PP, pancreatic polypeptide; GK, glucokinase.
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Efrat, S., Fusco-DeMane, D., Lemberg, H., Emran, O. A. & Wang, S. (1995) Proc. Natl. Acad. Sci. USA 92, 3576-3580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Fleischer, N., Chen, C., Surana, M., Leiser, M., Rossetti, L., Pralong, W. & Efrat, S. (1998) Diabetes 47, 1419-1425. [DOI] [PubMed] [Google Scholar]
- 3.Milo-Landesman, D., Berkovich, I., Surana, M., Compagni, A., Christofori, G., Fleischer, N. & Efrat, S. (2001) Cell Transplant. 10, 645-650. [PubMed] [Google Scholar]
- 4.de la Tour, D., Halvorsen, T., Demeterco, C., Tyrberg, B., Itkin-Ansari, P., Loy, M., Yoo, S. J., Hao, E., Bossie, S. & Levine, F. (2001) Mol. Endocrinol. 15, 476-483. [DOI] [PubMed] [Google Scholar]
- 5.Soria, B., Roche, E., Berna, G., Leon-Quinto, T., Reig, J. A. & Martin, F. (2000) Diabetes 49, 157-162. [DOI] [PubMed] [Google Scholar]
- 6.Assady, S., Maor, G., Amit, M., Itskovitz-Eldor, J., Skorecki, K. L. & Tzukerman, M. (2001) Diabetes 50, 1691-1697. [DOI] [PubMed] [Google Scholar]
- 7.Lumelsky, N., Blondel, O., Laeng, P., Velasco, I., Ravin, R. & McKay, R. (2001) Science 292, 1389-1394. [DOI] [PubMed] [Google Scholar]
- 8.Clarke, D. L., Johansson, C. B., Wilbertz, J., Veress, B., Nilsson, E., Karlstrom, H., Lendahl, U. & Frisen, J. (2000) Science 288, 1660-1663. [DOI] [PubMed] [Google Scholar]
- 9.Jiang, Y., Jahagirdar, B. N., Reinhardt, R. L., Schwartz, R. E., Keene, C. D., Ortiz-Gonzalez, X. R., Reyes, M., Lenvik, T., Lund, T., Blackstad, M., et al. (2002) Nature 418, 41-49. [DOI] [PubMed] [Google Scholar]
- 10.Jonsson, J., Carlsson, L., Edlund, T. & Edlund, H. (1994) Nature 371, 606-609. [DOI] [PubMed] [Google Scholar]
- 11.Ahlgren, U., Jonsson, J., Jonsson, L., Simu, K. & Edlund, H. (1998) Genes Dev. 12, 1763-1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ferber, S., Halkin, A., Cohen, H., Ber, I., Einav, Y., Goldberg, I., Barshack, I., Seijffers, R., Kopolovic, J., Kaiser, N. & Karasik, A. (2000) Nat. Med. 6, 568-572. [DOI] [PubMed] [Google Scholar]
- 13.Horb, M. E., Shen, C. N., Tosh, D. & Slack, J. M. (2003) Curr. Biol. 13, 105-115. [DOI] [PubMed] [Google Scholar]
- 14.Kojima, H., Nakamura, T., Fujita, Y., Kishi, A., Fujimiya, M., Yamada, S., Kudo, M., Nishio, Y., Maegawa, H., Haneda, M., et al. (2002) Diabetes 51, 1398-1408. [DOI] [PubMed] [Google Scholar]
- 15.Ramiya, V. K., Maraist, M., Arfors, K. E., Schatz, D. A., Peck, A. B. & Cornelius, J. G. (2000) Nat. Med. 6, 278-282. [DOI] [PubMed] [Google Scholar]
- 16.Bonner-Weir, S., Taneja, M., Weir, G. C., Tatarkiewic, K., Song, K. H., Sharma, A. & O'Neil, J. J. (2000) Proc. Natl. Acad. Sci. USA 97, 7999-8004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Suzuki, A., Zheng, Y.-W., Kaneko, S., Onodera, M., Fukao, K., Nakauchi, H. & Taniguchi, H. (2002) J. Cell Biol. 156, 173-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang, L., Li, S., Hatch, H., Ahrens, K., Cornelius, J. G., Petersen, B. E. & Peck, A. B. (2002) Proc. Natl. Acad. Sci. USA 99, 8078-8083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Malhi, H., Irani, A. N., Gagandeep, S. & Gupta, S. (2002) J. Cell. Sci. 115, 2679-2688. [DOI] [PubMed] [Google Scholar]
- 20.Wege, H., Le, H. T., Chui, M. S., Liu, L., Wu, J., Giri, R. K., Malhi, H., Sappal, B. S., Kumaran, V., Gupta, S. & Zern, M. A. (2003) Gastroenterology 124, 432-444. [DOI] [PubMed] [Google Scholar]
- 21.Olsen, J. C. (1998) Gene Ther. 5, 1481-1487. [DOI] [PubMed] [Google Scholar]
- 22.Rohll, J. B., Mitrophanous, K. A., Martin-Rendon, E., Ellard, F. M., Radcliffe, P. A., Mazarakis, N. D. & Kingsman, S. M. (2002) Methods Enzymol. 346, 466-500. [DOI] [PubMed] [Google Scholar]
- 23.Miller, C. P., McGehee, R. E., Jr., & Habener, J. F. (1994) EMBO J. 13, 1145-1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adra, C. N., Boer, P. H. & McBurney, M. W. (1987) Gene 60, 65-74. [DOI] [PubMed] [Google Scholar]
- 25.Jang, S. K., Krausslich, H. G., Nicklin, M. J., Duke, G. M., Palmenberg, A. C. & Wimmer, E. (1988) J. Virol. 62, 2636-2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mitrophanous, K., Yoon, S., Rohll, J., Patil, D., Wilkes, F., Kim, V., Kingsman, S., Kingsman, A. & Mazarakis, N. (1999) Gene Ther. 6, 1808-1818. [DOI] [PubMed] [Google Scholar]
- 27.Zufferey, R., Donello, J. E, Trono, D. & Hope, T. J. (1999) J. Virol. 73, 2886-2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Naldini, L., Blomer, U., Gage, F. H., Trono, D. & Verma, I. M. (1996) Proc. Natl. Acad. Sci. USA 93, 11382-11388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ott, M., Rajvanshi, P., Sokhi, R., Alpini, G., Aragona, E., Dabeva, M., Shafritz, D. A. & Gupta, S. (1999) J. Pathol. 187, 365-373. [DOI] [PubMed] [Google Scholar]
- 30.McKinnon, C. M. & Docherty, K. (2001) Diabetologia 44, 1203-1214. [DOI] [PubMed] [Google Scholar]
- 31.Marshak, S., Ben-Shushan, E., Shoshkes, M., Havin, L., Cerasi, E. & Melloul, D. (2001) Diabetes 50 (Suppl. 1), S37-S38. [DOI] [PubMed] [Google Scholar]
- 32.Dohrmann, C., Gruss, P. & Lemaire, L. (2000) Mech. Dev. 92, 47-54. [DOI] [PubMed] [Google Scholar]
- 33.Gradwohl, G., Dierich, A., LeMeur, M. & Guillemot, F. (2000) Proc. Natl. Acad. Sci. USA 97, 1607-1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.