

Abstract
Pheromones are water-soluble chemicals released and sensed by individuals of the same species to elicit social and reproductive behaviors or physiological changes; they are perceived primarily by the vomeronasal organ (VNO) in terrestrial vertebrates. Humans and some related primates possess only vestigial VNOs and have no or significantly reduced ability to detect pheromones, a phenomenon not well understood at the molecular level. Here we show that genes encoding the TRP2 ion channel and V1R pheromone receptors, two components of the vomeronasal pheromone signal transduction pathway, have been impaired and removed from functional constraints since shortly before the separation of hominoids and Old World monkeys ≈23 million years ago, and that the random inactivation of pheromone receptor genes is an ongoing process even in present-day humans. The phylogenetic distribution of vomeronasal pheromone insensitivity is concordant with those of conspicuous female sexual swelling and male trichromatic color vision, suggesting that a vision-based signaling-sensory mechanism may have in part replaced the VNO-mediated chemical-based system in the social/reproductive activities of hominoids and Old World monkeys (catarrhines).
Pheromones are water-soluble chemicals used in intraspecific communications to elicit social and reproductive behaviors or physiological changes such as male–male aggression, onset of puberty, estrus, and induction of mating. Pheromones are perceived primarily by the vomeronasal organ (VNO), which is at the base of the nasal cavity and separated from the main olfactory epithelium that senses thousands of volatile odorants (1). It has been known for decades that some primate species, including humans, do not possess functional VNOs, and these organisms lack vomeronasal chemoreception to pheromones (1–3). This insensitivity has likely had important impacts on the sexual and social behaviors of many primates. On the other hand, behavioral changes may have also altered natural selection on vomeronasal chemoreception. It is therefore of interest to find when the vomeronasal pheromone insensitivity occurred in evolution, how it occurred, and why it occurred.
Vomeronasal pheromone perception begins by the binding of pheromones to pheromone receptors located on the cell membrane of sensory neurons of the VNO, which triggers a signal transduction pathway that ultimately leads to the activation of the hypothalamus. Several components in the pathway have been identified, including GTP-binding proteins, phospholipase C, inositol 1,4,5-trisphosphate (IP3), and an ion channel of the transient receptor potential family named TRP2 (also known as TRPC2) (4). However, among members of this pathway, only TRP2 (5) and pheromone receptors of the V1R and V2R families (6–9) are unique to pheromone transduction and are not known to be used in other physiological processes. Disruption of either of these two components in mice hampers pheromone perception and causes dramatic changes in sexual and social behaviors (10–12). They thus represent the best genetic markers for a study of the evolution of VNO pheromone perception. We here use both of these markers to demonstrate that the vomeronasal pheromone transduction pathway was impaired in the common ancestor of hominoids and Old World (OW) monkeys (catarrhines), and we suggest that a vision-based sensory system might have in part replaced the chemical-based vomeronasal pheromone system.
Materials and Methods
PCR and Sequencing. The intron/exon structure of the mouse TRP2 gene was determined by a comparison of the genomic sequence with various spliced forms of cDNA sequences. We amplified exons 1–13 of TRP2 by PCR from the genomic DNAs of chimpanzee (Pan troglodytes), gorilla (Gorilla gorilla), orangutan (Pongo pygmaeus), gibbon (Hylobates leucogenys), baboon (Papio hamadryas), guereza (Colobus guereza), tamarin (Saguinus oedipus), squirrel monkey (Saimiri sciureus), owl monkey (Aotus trivirgatus), saki (Pithecia irrorata), and spider monkey (Ateles geoffroyi). Most of the DNA samples were purchased from Coriell (Camden, NJ), and the rest were collected over the years from multiple sources. PCR was conducted with Master-Taq under conditions recommended by the manufacturer (Eppendorf, Hamburg, Germany), and the products were purified and sequenced from both directions by using the dideoxy chain termination method with automated sequencer. Because V1R genes do not contain introns, the entire sequences of the orthologous genes of human V1RL1–5 were amplified from the genomic DNAs of the chimpanzee, gorilla, and orangutan, and were cloned into pCR4TOPO vector before sequencing. Multiple (more than five) colonies were sequenced. For human population studies, PCR products were directly sequenced without cloning. PCR primers are available on request.
DNA Sequence Analysis. Phylogenetic trees were reconstructed with mega2 (13) by using the neighbor-joining method (14) with 1,000 bootstrap replications (15) to test the orthology of the gene sequences. The number of synonymous substitutions per synonymous site (dS) and the number of nonsynonymous substitutions per nonsynonymous site (dN) between homologous gene sequences were computed by using the modified Nei–Gojobori method (16, 17).
Half-Life (t1/2) of a V1R Gene Under No Functional Constraint. An intact V1R ORF will eventually be interrupted if it is under neutral evolution without functional constraints. We estimated the rate of this pseudogenization process by using computer simulation. The rate is determined mainly by the sequence of the ORF, the rate of point mutations, and the rate of indel (insertion/deletion) mutations. We used a point mutation rate of 1.25 × 10-9 per site per year because genomic sequence analyses revealed a rate of 1.0–1.5 × 10-9 per site per year in hominoids and OW monkeys (18). We found that the relative mutational frequencies among the four nucleotides have only a negligible effect on the simulation result and assumed that they are equal. We estimated the indel mutation rate from a genomic comparison between the human and chimpanzee [0.78 × 106 nucleotides (19)] and that between the human and baboon [1.4 × 106 nucleotides (20)] to be 1.1 × 10-10 and 0.88 × 10-10 indel per site per year, respectively, and thus used 1.0 × 10-10 per site per year in our simulation. We assumed that all indels with sizes that are multiples of three nucleotides do not disrupt an ORF. This simplifies our simulation but does not affect our results, because the majority of indels generated by mutations have small sizes [≤6 nucleotides, (19, 20)]. In the above two genomic data sets, ≈17% of the total 6,902 indels are of sizes that are multiples of three nucleotides. A simulation was then performed for 20,000 replications with a V1R ORF and the above parameters. Under no functional constraints, the substitution rate is identical to the mutation rate and mutations are assumed to be random. An ORF is interrupted when an indel of a size that is not a multiple of three nucleotides or a nonsense point mutation occurs. We thus determined t1/2 of an ORF, or the time required for an intact ORF to be interrupted in one-half of the simulation replications. The computer program for simulation was written in C language and is available on request. We found that different V1R genes have slightly different t1/2 values. For instance, the t1/2 of human V1RL1–5 is 4.49, 4.01, 5.06, 5.24, and 4.50 million years (MY), respectively, with a median value of 4.5 MY. We also estimated the t1/2 of representative mouse V1R genes [one from each of the 12 subgroups (21)] and found that the median value is 5.3 MY. We thus used (4.5 + 5.3)/2 = 4.9 MY as the t1/2 for V1R genes of ancestral OW monkeys and hominoids. Events other than point mutations and indel mutations, such as insertions of transposable elements and gene duplication, have relatively small effects on t1/2 and are often cancelled out. Given t1/2 and the number (N0) of V1R ORFs at the start of the pseudogenization process, the number of ORFs left (N) after time T is a Poisson random variable with the mean and variance both equal to λ=N0(1/2)T/t1/2. The hypothesis that the five human V1R genes are relics of a pseudogenization process that started 23–35 MY ago cannot be rejected at the 5% significance level when t1/2 is in the range of 3.8–9.3 MY (see Fig. 3, which is published as supporting information on the PNAS web site, www.pnas.org). We also considered the uncertainty of the size of the V1R repertoire in ancestral higher primates (anthropoids) and found that the relic hypothesis is consistent with the observation even when the ancestral V1R repertoire was as small as 70 genes (with t1/2 in the range of 4.5–12.7 MY) or as large as 280 genes (with t1/2 in the range of 3.3–7.3 MY). Note that the mouse genome has ≈140 potentially functional V1R genes (21). Given t1/2, N0, and N, the likelihood of T is given by
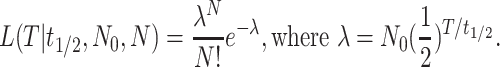 |
It can be shown that the maximum likelihood estimate of T, time since the pseudogenization process started, equals t1/2 ln(N0/N)/ln 2. Likelihood ratio tests can be used to test a null hypothesis on T. Here we found that T < 17.1 or >29.9 MY can be rejected at the 5% significance level, with the assumption of t1/2 = 4.9 MY, N = 5, and N0 = 140.
Results and Discussion
TRP2 Genes of New World (NW) Monkeys (Platyrrhines) Are Functional. The mouse TRP2 gene has several splicing forms. One of them is exclusively and abundantly expressed in VNO and is believed to represent the functional form of TRP2 (22). The 13 exons of this splicing form are named exons 1–13. The human TRP2 gene is an expressed pseudogene with frame-shifting mutations and premature stop codons seen in the cDNA sequence (23). This is further confirmed by our analysis of the draft human genome sequence (Fig. 1). To determine when TRP2 was pseudogenized, we sequenced some of the 13 exons from four non-human hominoids (chimpanzee, gorilla, orangutan, and gibbon) and two OW monkeys (baboon and guereza) and sequenced all 13 exons from five NW monkeys (tamarin, squirrel monkey, owl monkey, saki, and spider monkey). The species were chosen to represent a wide phylogenetic diversity (Fig. 1), including all major lineages of higher primates (anthropoids) (24).
Fig. 1.

Structure of the TRP2 gene in 12 higher primates. The tree represents the current consensus on the phylogeny of these species. The branch lengths are not drawn to scale except for the timing of the two nodes with black dots. For each exon, “+” stands for the presence of an ORF, whereas “S” stands for the interruption of the reading frame by a premature stop codon, which may have resulted from either indels or point mutations. Shared premature stop codons among species within an exon are shown by “S,” whereas unshared ones are shown by “S*.” Exons with no signs are not sequenced. All primate TRP2 gene structures were determined by comparisons of the genomic DNA sequences with the mouse cDNA sequence.
The entire TRP2 retains an intact ORF in each of the five NW monkeys surveyed (Fig. 1), suggesting that it is functional in these species. Further evidence is gained by an examination of the number of nonsynonymous substitutions per nonsynonymous site (dN) and that of synonymous substitutions per synonymous site (dS) between the TRP2 sequences. A functional gene is under functional constraint and purifying selection, which prevents deleterious nonsynonymous substitutions from fixation but generally does not affect synonymous substitutions, leading to a dN/dS ratio <1 (25). By contrast, a pseudogene is not under functional constraint and has a dN/dS ratio of ≈1. We found that the dN/dS ratio for TRP2 (mean = 0.34) is significantly lower than 1 between each pair of the five NW monkeys (P < 0.01), confirming that this gene is indeed under functional constraint. While this manuscript was under preparation, Liman and Innan (26) published a study of primate TRP2 based on incomplete coding sequences from no more than five exons and inferred that NW monkey TRP2 genes are probably not pseudogenes. We here obtained the complete TRP2 coding sequences of NW monkeys and provided direct evidence.
TRP2 Genes of OW Monkeys and Hominoids (Catarrhines) Are Pseudogenes. In contrast, the TRP2 gene contains premature stop codons in all of the hominoids and OW monkeys studied, indicating that it is a pseudogene in these organisms. In particular, none of the three possible reading frames is open in exon 13 for any of these species. Using the reading frame of NW monkey and mouse TRP2, a premature stop codon TGA of exon 13 is found to be shared by all hominoids and OW monkeys, with the exception of the orangutan, which has an arginine codon CGA at the position (Fig. 2). This stop codon is nicknamed “Adam” in our lab, because it is probably the first premature stop codon that appeared in the pseudogenization of TRP2 (see below). It was named “mutation 6” in ref. 26 according to its location in the gene. Given the phylogenetic distribution of Adam, it is most parsimonious to infer that Adam first appeared in the common ancestor of hominoids and OW monkeys but was secondarily lost in orangutan, an event that most likely occurred after the separation of orangutans from African apes (humans, chimpanzees, and gorillas). Another premature stop codon apparently occurred 10 codons upstream of Adam in orangutan (Fig. 2). Adam is located within a predicted coiled-coil domain of TRP2 (5). The disruption of this domain potentially abolishes protein–protein interactions that it mediates. Although Adam is in the final exon of TRP2, there are 106 codons downstream of it. We found that the dN/dS ratio (0.38) for this 106-codon region is significantly smaller than 1 (P < 0.01) between the mouse and rat TRP2 sequences, suggesting that this region is under purifying selection and is probably indispensable in a functional TRP2. Thus, with the appearance of Adam, not only is the coiled-coil domain disrupted, but TRP2 now also lacks the functionally important 106 amino acids in its C terminus. We therefore infer that TRP2 was unlikely to be functional after the appearance of Adam. Interestingly, our TRP2 sequences, although not complete for most of the OW monkey and hominoid species studied, are sufficient for us to deduce that Adam is probably the only premature stop codon that appeared before the separation of hominoids and OW monkeys (Fig. 1). Since then, additional mutations damaging the gene have apparently accumulated in various species, as is evident in Fig. 1. Fossil records and molecular dating suggested a divergence time of 23 MY between hominoids and OW monkeys, and 35 MY between these organisms and NW monkeys (24, 27). The pseudogenization of TRP2 likely took place between 23 and 35 MY ago, and probably shortly before 23 MY ago, because only one premature stop codon was found to predate the divergence of hominoids and OW monkeys. A similar result was independently obtained recently (26).
Fig. 2.

Alignment of the first 120 nucleotides of the TRP2 exon 13 sequences from higher primates, rat, and mouse. Dots show identical nucleotides as in the human sequence. Premature stop codons are boxed. The entire exon has 393 nucleotides in the mouse. The sequences from the human, rat, and mouse are retrieved from GenBank (accession nos. X89067, NM_022638, and NM_011644).
Pseudogenization of Catarrhine Pheromone Receptor Genes. Without a functional TRP2, the vomeronasal pheromone signal transduction pathway was impaired; other protein components of the pathway, if not used in additional physiological processes, would be released from functional constraints and their genes would gradually incorporate indels and nonsense mutations randomly. Indeed, the mouse genome contains ≈140 potentially functional V1R pheromone receptor genes (21), but the human genome has only five V1R genes that retain ORFs (28). Dozens of human V1R pseudogenes have been reported, but it is still unknown exactly how many exist in the human genome (29, 30). Assuming that the genome of the higher primate ancestor had 140 functional V1R genes as in the mouse genome, we ask whether it is possible to have five V1R ORFs left in the present-day human genome simply by chance, without the presence of any functional constraints on them. By using computer simulation, we determined that the average half-life of a V1R gene under no selection is ≈4.9 MY (see Materials and Methods). If functional relaxation started 35 MY ago, one can compute that the probability for an ORF to remain intact today is 0.5(35/4.9) = 0.708%. Therefore, the expected number of intact ORFs at present is 140 × 0.708% = 1.0, with a 95% confidence interval of 0–3 (see Materials and Methods). If the functional relaxation started 23 MY ago, the expected number of intact ORFs at present is 5.4, with a 95% confidence interval of 2–10. The observation of five V1R ORFs in the human genome is within the latter interval, suggesting that the human V1R ORFs may not possess physiological functions; they may simply be relics of an ongoing pseudogenization process. This conclusion is robust to various assumptions made in the computation such as the half-life of V1R genes and the number of V1R genes in the higher primate ancestor (see Materials and Methods). By comparing the observed and expected numbers of intact ORFs left today, one may also estimate when the relaxation of functional constraints on V1R genes started. Our maximum likelihood estimate of this date is 23.6 MY ago (see Materials and Methods), consistent with the observation from the TRP2 gene.
The hypothesis of the absence of functional constraints on the five human V1R genes leads to the predication that they are subject to random inactivation in evolution. We tested this prediction by sequencing their orthologous genes in the chimpanzee, gorilla, and orangutan. We found that none of the five genes maintain intact ORFs in all of the apes (Table 1). Because of the evolutionary proximity of the apes to humans, we are unlikely to have been misled by paralogous genes. In fact, we obtained these gene sequences by sequencing multiple colonies after the PCR products were cloned and verified that the gene sequences show the expected phylogenetic relationships among the species. Furthermore, for the orthologous sequences with intact ORFs, dN/dS is ≈1 (Table 1), consistent with the above result of no functional constraints on them. Giorgi and Rouquier (31) recently conducted a similar study of one of the five genes and obtained the same result. We also examined intraspecific variations of these five V1R genes in 11 humans of different geographic origins (one African, three African Americans, three Europeans, three Asians, and one Amerindian) in an attempt to test the hypothesis that these genes are under neutral evolution in humans. Interestingly, at V1RL5, we found an allele that has a C→T change at position 136, generating a premature stop codon (CAA→TAA; see Fig. 4, which is published as supporting information on the PNAS web site). This allele was found seven times (in Europeans, Africans, and African Americans) from our sample of 22 chromosomes, thus reaching a frequency of ≈32%. More surprisingly, at V1RL3, all 22 chromosomes sequenced contain a deletion of 17 nucleotides (from position 287 to 303), in comparison to GenBank sequence AF336873 (see Fig. 5, which is published as supporting information on the PNAS web site). This deletion generated multiple premature stop codons and rendered V1RL3 nonfunctional. Furthermore, we sequenced V1RL3 from 39 additional human individuals (1 African, 10 African Americans, 10 Europeans, 11 Asians, and 7 Amerindians), and all are found to be homozygous for the deletion-containing alleles. A further scrutiny shows that the draft human genome sequence also contains this deletion, but sequences from two research groups (GenBank accession no. AF336873 and ref. 28) do not contain the deletion. It is unlikely that our sequence and AF336873 represent two gene loci in the genome and that AF336873 was not amplified in our experiment, because our primers match the AF336873 sequence perfectly. One possibility is that the deletion-containing allele is predominant, whereas the allele of the AF336873 sequence has a low frequency in human populations. Because we sequenced 100 chromosomes, the draft human genome sequence contains at least one chromosome, and sequences from AF336873 and ref. 28 each represent one chromosome, the total number of chromosomes sequenced is ≈103. The frequency of the deletion-containing allele is 101/103 = 98.05% at V1RL3. In sum, of the five human V1R genes previously reported to have intact ORFs, two are in the process of pseudogenization, with the pseudogene alleles reaching relatively high frequencies, and no indication of selection was found from the other three genes. Thus, in contrast to the previous belief that the five V1R genes of humans may be functional (28), the molecular evolutionary and population genetic data presented here strongly suggest they are not. There is a second family of pheromone receptor genes named V2R in the mammalian genome (7–9). Because of the presence of introns, it is not easy to identify all V2R genes and pseudogenes in a draft genome sequence (21, 28). Nevertheless, although ≈100 V2R genes have been suggested to exist and many have been identified in mice and rats, no intact ORFs of this family are known in humans, despite multiple computational and experimental searches (7–9, 30). We predict that a similar pseudogenization process has taken place in the V2R family of hominoids and OW monkeys.
Table 1. Inactivations of the five V1R genes in humans and great apes.
| Species | V1RL1 | V1RL2 | V1RL3 | V1RL4 | V1RL5 |
|---|---|---|---|---|---|
| Human | + | + | +/- (0.98) | + | +/- (0.32) |
| Chimpanzee | - | - | + | - | - |
| Gorilla | + | - | - | - | + |
| Orangutan | - | - | - | + | - |
| dN/dS | 0.96 | 1.17 | 1.14 | 1.13 |
These five genes are the only V1R genes of humans known to have intact ORFs. +, ORF intact; -, ORF disrupted; +/-, alleles for the intact and disrupted ORFs are segregating within species, with the frequency of the disrupted ORF allele given in parentheses. One individual per species was examined for the great apes, 11 individuals of humans were examined for V1RL1-4, and 50 individuals were used for V1RL5. The dN/dS ratios were computed for orthologous intact ORFs, and none were significantly different from 1
Why Is the Catarrhine VNO Pheromone Transduction Pathway Dispensible? Earlier anatomical and physiological studies showed that adults of hominoids and OW monkeys do not have VNOs or functional VNOs, whereas NW monkeys and prosimians clearly have them (1–3, 32, 33). Observations of presumably pheromone-based reproductive or social behaviors are largely consistent with this dichotomy (1, 2, 34), although notable exceptions do exist (35, 36). For example, menstrual cycles tend to synchronize among female humans living in close proximity (37, 38). The molecular basis of the synchronization, however, is unclear, and it may not be mediated by VNO. In fact, certain non-VNO-mediated behavioral responses to putative pheromones have been reported in pigs and rabbits (39, 40). Our evolutionary genetic data from two components of the vomeronasal pheromone transduction pathway are consistent with each other and are in general congruent with the aforementioned anatomical, physiological, and behavioral data. It may be concluded that the VNO-mediated pheromone sensitivity was lost in the common ancestor of hominoids and OW monkeys not long before 23 MY ago. From an analysis of the dN/dS ratio of TRP2 among rodents, lemurs, and NW monkeys, Liman and Innan proposed that the purifying selection on TRP2 was reduced but not entirely removed in NW monkeys (26). This interesting suggestion requires further scrutiny, because their incomplete gene sequences are relatively short, and weaker selective constraints in higher primates than in rodents appear a general phenomenon rather than unique to TRP2, possibly due to the difference in population size between primates and rodents (41, 42).
Females of many OW monkeys and hominoids are known to develop, around the time of ovulation, a prominent reddening and swelling of the sexual skin surrounding the perineum, whereas NW monkeys do not have true sexual skin (43). This vision-based signaling mechanism may have in part replaced the chemical-based pheromone signaling in the former group of organisms (43), because sexual swelling can be perceived from a distance, whereas pheromone perception is probably through physical contact (44). It is interesting to relate this observation to the duplication of the X-chromosome-linked red/greensensitive opsin gene that also occurred in the common ancestors of OW monkeys and hominoids after they were separated from NW monkeys, although a more precise date of the duplication has yet to be obtained (45, 46). The gene duplication and subsequent functional divergence led to the emergence of a trichromatic color vision (with the blue, green, and red opsins) in both sexes, in contrast to the situation before the duplication when only females may have had trichromacy due to the existence of functionally polymorphic alleles at the red/green opsin locus (46, 47). Trichromacy allows a clear distinction in perceiving yellow, orange, pink, and red hues and may have been fixed because it helped primates in detecting young leaves and ripened fruits against dappled foliage (46). Once males have trichromatic vision, they can sense the subtle color changes of female sexual skins, which may have provided the selective force for the evolution of sexual swelling; the new visual-based signaling-sensory mechanism probably made pheromone perception dispensable. In this respect, it is interesting to mention the analogy in birds, which have tetrachromatic color vision and develop colorful plumages at sexual maturity, but lack VNOs (1, 2). Changes in pheromone signaling/perception and color vision have also been shown to cause premating isolation and speciation in moths and fishes (48, 49). Although similar impacts of such changes have yet to be proven in hominoids and OW monkeys, the gain of color vision and loss of vomeronasal pheromone perception apparently have had profound consequences in the biology of these organisms, including humans.
Supplementary Material
Acknowledgments
We thank Priscilla Tucker, the member editor, and three anonymous reviewers for valuable comments. This work was supported by a startup fund and a Rackham Fellowship from the University of Michigan, and by National Institutes of Health Grant GM67030 (to J.Z.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: OW, Old World; NW, New World; MY, million years; VNO, vomeronasal organ.
Data deposition: The sequences reported in this paper have been deposited in the GenBank database (accession nos. AY302599–AY302698 and AY312463–AY312491).
References
- 1.Keverne, E. B. (1999) Science 286 716-720. [DOI] [PubMed] [Google Scholar]
- 2.Stoddart, D. M. (1980) The Ecology of Vertebrate Olfaction (Chapman & Hall, London).
- 3.Loo S. K. (1973) Folia. Primatol. 20 410-422. [DOI] [PubMed] [Google Scholar]
- 4.Zufall, F., Kelliher, K. R. & Leinders-Zufall, T. (2002) Microsc. Res. Tech. 58 251-260. [DOI] [PubMed] [Google Scholar]
- 5.Liman, E. R., Corey, D. P. & Dulac, C. (1999) Proc. Natl. Acad. Sci. USA 96 5791-5796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dulac, C. & Axel, R. (1995) Cell 83 195-206. [DOI] [PubMed] [Google Scholar]
- 7.Ryba, N. J. & Tirindelli, R. (1997) Neuron 19 371-379. [DOI] [PubMed] [Google Scholar]
- 8.Herrada, G. & Dulac, C. (1997) Cell 90 763-773. [DOI] [PubMed] [Google Scholar]
- 9.Matsunami, H. & Buck, L. B. (1997) Cell 90 775-784. [DOI] [PubMed] [Google Scholar]
- 10.Stowers, L., Holy, T. E., Meister, M., Dulac, C. & Koentges, G. (2002) Science 295 1493-1500. [DOI] [PubMed] [Google Scholar]
- 11.Leypold, B. G., Yu, C. R., Leinders-Zufall, T., Kim, M. M., Zufall, F. & Axel, R. (2002) Proc. Natl. Acad. Sci. USA 99 6376-6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Del Punta, K., Leinders-Zufall, T., Rodriguez, I., Jukam, D., Wysocki, C. J., Ogawa, S., Zufall, F. & Mombaerts, P. (2002) Nature 419 70-74. [DOI] [PubMed] [Google Scholar]
- 13.Kumar, S., Tamura, K., Jakobsen, I. B. & Nei, M. (2001) Bioinformatics 17 1244-1245. [DOI] [PubMed] [Google Scholar]
- 14.Saitou, N. & Nei, M. (1987) Mol. Biol. Evol. 4 406-425. [DOI] [PubMed] [Google Scholar]
- 15.Felsenstein, J. (1985) Evolution (Lawrence, Kans.) 39 783-791. [DOI] [PubMed] [Google Scholar]
- 16.Nei, M. & Gojobori, T. (1986) Mol. Biol. Evol. 3 418-426. [DOI] [PubMed] [Google Scholar]
- 17.Zhang, J., Rosenberg, H. F. & Nei, M. (1998) Proc. Natl. Acad. Sci. USA 95 3708-3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yi, S., Ellsworth, D. L. & Li, W. H. (2002) Mol. Biol. Evol. 19 2191-2198. [DOI] [PubMed] [Google Scholar]
- 19.Britten, R. J. (2002) Proc. Natl. Acad. Sci. USA 99 13633-13635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Silva, J. C. & Kondrashov, A. S. (2002) Trends Genet. 18 544-547. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez, I., Del Punta, K., Rothman, A., Ishii, T. & Mombaerts, P. (2002) Nat. Neurosci. 5 134-140. [DOI] [PubMed] [Google Scholar]
- 22.Hofmann, T., Schaefer, M., Schultz, G. & Gudermann, T. (2000) Biochem. J. 351 115-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vannier, B., Peyton, M., Boulay, G., Brown, D., Qin, N., Jiang, M., Zhu, X. & Birnbaumer, L. (1999) Proc. Natl. Acad. Sci. USA 96 2060-2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goodman, M., Porter, C. A., Czelusniak, J., Page, S. L., Schneider, H., Shoshani, J., Gunnell, G. & Groves, C. P. (1998) Mol. Phylogenet. Evol. 9 585-598. [DOI] [PubMed] [Google Scholar]
- 25.Nei, M. & Kumar, S. (2000) Molecular Evolution and Phylogenetics (Oxford Univ. Press, New York).
- 26.Liman, E. R. & Innan, I. (2003) Proc. Natl. Acad. Sci. USA 100 3328-3332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Glazko, G. V. & Nei, M. (2003) Mol. Biol. Evol. 20 424-434. [DOI] [PubMed] [Google Scholar]
- 28.Rodriguez, I. & Mombaerts, P. (2002) Curr. Biol. 12 R409-R411. [DOI] [PubMed] [Google Scholar]
- 29.Giorgi, D., Friedman, C., Trask, B. J. & Rouquier S. (2000) Genome Res. 10 1979-1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kouros-Mehr, H., Pintchovski, S., Melnyk, J., Chen, Y. J., Friedman, C., Trask, B. & Shizuya, H. (2001) Chem. Senses 26 1167-1174. [DOI] [PubMed] [Google Scholar]
- 31.Giorgi, D. & Rouquier, S. (2002) Chem. Senses 27 529-537. [DOI] [PubMed] [Google Scholar]
- 32.Hunter, A. J., Fleming, D. & Dixson, A. F. (1984) J. Anat. 138 217-226. [PMC free article] [PubMed] [Google Scholar]
- 33.Taniguchi, K., Matsusaki, Y., Ogawa, K. & Saito, T. R. (1992) Folia. Primatol. 59 169-176. [DOI] [PubMed] [Google Scholar]
- 34.Abbott, D. H. (1984) Am. J. Primatol. 6 169-186. [DOI] [PubMed] [Google Scholar]
- 35.Meredith, M. (2001) Chem. Senses 26 433-445. [DOI] [PubMed] [Google Scholar]
- 36.McClintock, M. K. (1998) Ann. N.Y. Acad. Sci. 855 390-392. [DOI] [PubMed] [Google Scholar]
- 37.McClintock, M. K. (1984) Physiol. Behav. 32 701-705. [DOI] [PubMed] [Google Scholar]
- 38.Stern, K. & McClintock, M. K. (1998) Nature 392 177-179. [DOI] [PubMed] [Google Scholar]
- 39.Dorries, K. M., Adkins-Regan, E. & Halpern, B. P. (1997) Brain Behav. Evol. 49 53-62. [DOI] [PubMed] [Google Scholar]
- 40.Hudson, R. & Distel, H. (1986) Physiol. Behav. 37 123-128. [DOI] [PubMed] [Google Scholar]
- 41.Zhang, J. (2000) J. Mol. Evol. 50 56-68. [DOI] [PubMed] [Google Scholar]
- 42.Eyre-Walker, A., Keightley, P. D., Smith, N. G. & Gaffney, D. (2002) Mol. Biol. Evol. 19 2142-2149. [DOI] [PubMed] [Google Scholar]
- 43.Dixson, A. F. (1983) Adv. Study Behav. 13 63-106. [Google Scholar]
- 44.Luo, M., Fee, M. S. & Katz, L. C. (2003) Science 299 1196-201. [DOI] [PubMed] [Google Scholar]
- 45.Yokoyama, S. & Yokoyama, R. (1989) Mol. Biol. Evol. 6 186-197. [DOI] [PubMed] [Google Scholar]
- 46.Surridge, A. K., Osorio, D. & Mundy, N. I. (2003) Trends Ecol. Evol. 51 198-205. [Google Scholar]
- 47.Boissinot, S., Tan, Y., Shyue, S. K., Schneider, H., Sampaio, I., Neiswanger, K., Hewett-Emmett, D. & Li, W. H. (1998) Proc. Natl. Acad. Sci. USA 95 13749-13754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roelofs, W. L., Liu, W., Hao, G., Jiao, H., Rooney, A. P. & Linn, C. E., Jr. (2002) Proc. Natl. Acad. Sci. USA 99 13621-13626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Terai, Y., Mayer, W. E., Klein, J., Tichy, H. & Okada, N. (2002) Proc. Natl. Acad. Sci. USA 99 15501-15506. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.