

Abstract
Cytochrome c oxidase, the terminal protein in the respiratory chain, converts oxygen into water and helps generate the electrochemical gradient used in the synthesis of ATP. The catalytic action of cytochrome c oxidase involves electron transfer, proton transfer, and O2 reduction. These events trigger specific molecular changes at the active site, which, in turn, influence changes throughout the protein, including alterations of amino acid side chain orientations, hydrogen bond patterns, and protonation states. We have used IR difference spectroscopy to investigate such modulations for the functional intermediate states E, R2,Pm, and F. These spectra reveal deprotonation of its key glutamic acid E286 in the E and in the Pm states. The consecutive deprotonation and reprotonation of E286 twice within one catalytic turnover illustrates the role of this residue as a proton shuttle. In addition, the spectra point toward deprotonation of a redox-active tyrosine, plausibly Y288, in the F intermediate. Structural insights into the molecular mechanism of catalysis based on the subtle molecular changes observed with IR difference spectroscopy are discussed.
Keywords: electron transfer, infrared attenuated total reflection, membrane protein, bacteriorhodopsis, glutamic acid
The respiratory chain generates an electrochemical gradient which drives production of ATP, our immediate, most readily available energy source. This team of enzymes governs the traffic of protons and electrons across the inner mitochondrial membrane of eukaryotes and the cytoplasmic membrane of prokaryotes. Cytochrome c oxidase (CcO) is the terminal respiratory enzyme, catalyzing the reduction of a molecule of dioxygen to two molecules of water, consuming four protons and four electrons. The free energy made available by this reaction is coupled to the translocation of four additional protons (1, 2). The active site of the enzyme is a bimetallic center consisting of a high-spin heme and a copper. Within the heme–copper oxygen reductase family, the electron donor, heme type, and assistant cofactors vary. The heme–copper oxygen reductase from mitochondria and the prokaryotic counterparts such as the oxidase from Rhodobacter sphaeroides employ cytochrome c as the electron donor and hence are better known by the name CcO. Electrons from cytochrome c are funneled into the active site via two other cofactor sites: a two-copper site (CuA) receives single electrons at the outer side of the membrane from cytochrome c, and a low-spin heme (heme a) passes the electrons received from CuA into the active site heme a3 and CuB (Fig. 1).
Fig. 1.

Structural view of CcO from R. sphaeroides (PDB ID 1M56; ref. 9) parallel to the membrane surface, with periplasmic surface on top. (Left) Ribbon diagram highlighting the arrangement of the four subunits (SU I–IV). (Right) A view in which the protein backbone is stripped to show the chemical structure of particularly important catalytically active groups. The gray arrows indicate the two spatially separated proton transfer pathways that contain the critical residues D132 and K362.
Electron transfers are strongly coupled to proton movements within the
enzyme. Two pathways containing hydrophilic amino acids and internal water
molecules facilitate the proton translocation from the bacterial cytoplasm or
mitochondrial matrix into the active site
(Fig. 1). The first of these
two pathways leads to a conserved tyrosine in the active site, Y288 (numbering
for CcO from the bacterium R. sphaeroides). This tyrosine appears to
play a critical role in catalyzing the splitting of the  bond by
providing a proton or a hydrogen atom to O2 after it binds to
reduced heme a3 at the active site
(3). If the tyrosine donates
both a proton and an electron to O2, a tyrosyl radical must form
(4,
5). Its formation correlates
with the finding that in the x-ray structures of several heme–copper
oxygen reductases [Paracoccus denitrificans
(6), Bos taurus
mitochondrial (7), and
Thermus thermophilus ba3
(8)], that the tyrosine is
posttranslationally cross-linked to a histidine that also serves as a ligand
to CuB. The second proton translocation pathway extends to a a
glutamic acid, E286 (Fig. 2),
≈11 Å from the active site
(9). Although the role of E286
is not fully understood, the presence of a protonatable residue at or near
this position seems to form an essential component of the proton-conducting
pathway, critical for rapid proton delivery to the active site (to form
H2O) and for proton pumping
(10).
bond by
providing a proton or a hydrogen atom to O2 after it binds to
reduced heme a3 at the active site
(3). If the tyrosine donates
both a proton and an electron to O2, a tyrosyl radical must form
(4,
5). Its formation correlates
with the finding that in the x-ray structures of several heme–copper
oxygen reductases [Paracoccus denitrificans
(6), Bos taurus
mitochondrial (7), and
Thermus thermophilus ba3
(8)], that the tyrosine is
posttranslationally cross-linked to a histidine that also serves as a ligand
to CuB. The second proton translocation pathway extends to a a
glutamic acid, E286 (Fig. 2),
≈11 Å from the active site
(9). Although the role of E286
is not fully understood, the presence of a protonatable residue at or near
this position seems to form an essential component of the proton-conducting
pathway, critical for rapid proton delivery to the active site (to form
H2O) and for proton pumping
(10).
Fig. 2.

Qualitative overview of the molecular steps taking place in the active center of CcO during catalysis (adapted from refs. 9 and 37 and modified). In the initial state, all four redox centers (CuA, heme a, heme a3, and CuB) are oxidized (O). Successive electron input steps from cytochrome c initiate a series of molecular changes (in red) that finally lead to oxygen cleavage (in blue) and proton transfer across the membrane (green arrows). The first electron to the binuclear center reduces CuB to form the E state. The uptake of a second electron from cytochrome c reduces heme a3 (R2), and dioxygen can bind to the central iron of heme a3. Both initial electron transfer reactions are coupled to net proton uptake by the protein. In the Pm state, bound dioxygen is cleaved and tyrosine 288 (Y288) is proposed to be a radical. The uptake of the third electron with a proton forms the F state. The final step (F to O) is driven by the uptake of the fourth electron. It is concluded from the present work that E286 is protonated in the O, R2, and F state but deprotonated in the E and the Pm intermediate. The side chain of a tyrosine, presumed to be Y288, is a phenolate radical in Pm, a phenolate in F but a phenol in the other states. For simplicity, only those intermediates are included that are investigated in the present study. Although CcO maneuvers a total of eight protons during the cycle (four destined to form H2O and four pumped protons), stoichiometric balance of these protons is not shown in the figure because not all proton donors and acceptors within the protein have been identified.
Proton and electron transfers must trigger specific molecular changes at the active site, influencing changes throughout the protein, including alterations of amino acid side chain orientations, hydrogen bond lengths, and protonation states. Particulars of the coupling between electron transfer and proton pumping remain controversial (11, 12). Solving the molecular mechanism hinges on acquiring more detailed structural information about the catalytic intermediates. IR spectroscopy resolves singular proton transfer events between cofactors, amino acid side chains and internal water molecules. The success of this technique is demonstrated by its elucidation of the sequence of proton transfer steps in bacteriorhodopsin, a prototypical transmembrane proton pump (13).
In this work, time-resolved and steady-state IR spectroscopy were combined
to observe protonation changes in a series of CcO intermediates
(Fig. 2). The first of these,
O, is the state in which all four metal cofactors are fully oxidized. Before
dioxygen binds, two electrons are received externally, passed from
CuA via heme a to the active site. Each step exhibits
rigorous control: electrons are received one at a time and key internal
electron transfer rates are limited by proton transfers. We use the terms E
and R2 to designate forms of the enzyme with one and two electrons,
respectively, in the binuclear center. Net proton uptake from solution is
evident during both O-to-E and E-to-R2 transitions
(14), though the destinations
and eventual fate of these protons (e.g., pumped vs. water formation) have
remained unclear. Proton translocation takes place in the E-to-R2
transition (11). It is noted
that when the enzyme is reduced by two-electrons, the population of the enzyme
with both electrons at the binuclear center (i.e., R2) is small in
the absence of a stabilizing ligand (such as CO). Dioxygen binds to the
two-electron reduced enzyme, and rapid cleavage of the bond is performed
in such way as to prevent release of harmful partially reduced forms of
O2 (5). In the
reaction of R2 with dioxygen, the first spectroscopically
detectable intermediate with partially reduced dioxygen bound to the active
site is Pm. In this state, the bond has been split and one of
the oxygen atoms forms an oxoferryl intermediate with heme
a3, i.e.,
Fe+4a3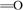 2–
(15). The heterolytic bond splitting on O2 reaction with the two-electron reduced form of
the enzyme requires an additional reducing equivalent, presumed to come from
the active site tyrosine Y288, forming a tyrosyl radical
(4). A second oxoferryl
intermediate, F, is formed when an electron is donated to Pm from
an external donor, accompanied by another proton uptake. The electron is
thought to reduce the radical formed in Pm
(6). The Pm-to-F
transition is almost certainly coupled to pumping one proton
(16,
17). When the F intermediate
receives an externally donated electron, the
Fe+4a32–
species is reduced to Fe3+ and the oxygen atom is lost
as a water molecule, another proton is pumped, O is regenerated, and the cycle
is completed. We recorded IR difference spectra for three sets of
intermediates of CcO from R. sphaeroides. Differences between
intermediates R2 (with bound CO) and E were recorded by using
time-resolved IR difference spectroscopy, whereas differences between
Pm and O as well as F and O were observed with perfusion-induced IR
spectroscopy. Most previous IR experiments have focused on the vibrational
differences between the oxidized (O) and the four-electron reduced
(R4) states of the enzyme
(18–20),
i.e., in the absence of dioxygen, and there is one recent report of the
Pm-O difference spectrum of bovine oxidase
(21). Our study is the first
to demonstrate protonation state changes of functionally relevant amino acid
side chains in states of the enzyme identified as catalytic intermediates.
2–
(15). The heterolytic bond splitting on O2 reaction with the two-electron reduced form of
the enzyme requires an additional reducing equivalent, presumed to come from
the active site tyrosine Y288, forming a tyrosyl radical
(4). A second oxoferryl
intermediate, F, is formed when an electron is donated to Pm from
an external donor, accompanied by another proton uptake. The electron is
thought to reduce the radical formed in Pm
(6). The Pm-to-F
transition is almost certainly coupled to pumping one proton
(16,
17). When the F intermediate
receives an externally donated electron, the
Fe+4a32–
species is reduced to Fe3+ and the oxygen atom is lost
as a water molecule, another proton is pumped, O is regenerated, and the cycle
is completed. We recorded IR difference spectra for three sets of
intermediates of CcO from R. sphaeroides. Differences between
intermediates R2 (with bound CO) and E were recorded by using
time-resolved IR difference spectroscopy, whereas differences between
Pm and O as well as F and O were observed with perfusion-induced IR
spectroscopy. Most previous IR experiments have focused on the vibrational
differences between the oxidized (O) and the four-electron reduced
(R4) states of the enzyme
(18–20),
i.e., in the absence of dioxygen, and there is one recent report of the
Pm-O difference spectrum of bovine oxidase
(21). Our study is the first
to demonstrate protonation state changes of functionally relevant amino acid
side chains in states of the enzyme identified as catalytic intermediates.
Materials and Methods
Wild-type and mutant CcO from R. sphaeroides was grown and purified as described (22). R2CO was created by depositing 10 μl of ≈300 μM oxidase in 100 mM KCl, 50 mM phosphate (pH 8.5), 0.05% dodecylmaltoside on a BaF2 cuvette, and deoxygenating by repeated cycles of evacuation and CO exposure in a custom-built anaerobic chamber (23). After 2 min under 1 bar CO (1 bar = 100 kPa), the cuvette was sealed with a second BaF2 window. The final optical path length was 10 μm. The presence of the R2CO state was verified by UV/visible spectrophotometry (Fig. 3). The shoulder on the band at 448 nm indicates some overreduction after the time-resolved IR experiments. All IR experiments were performed on an IFS 66v spectrometer (Bruker, Rheinstetten, Germany). Photo-induced dissociation of carbon monoxide from R2CO was achieved by a nanosecond laser pulse (frequency doubled output from a Nd:YAG laser at 532 nm, 40 mJ per pulse per cm–2, 5-Hz repetition rate). To slow the kinetics and to ensure long-term stability of the R2CO state, the sample was thermostated at 5°C. The trace shown represents an average of 30,000 difference spectra recorded 30 ms after photolysis in rapid-scan mode with an optical resolution of 4.5 cm–1. IR difference spectroscopy of the Pm and F state were performed with the attenuated total reflection (ATR) technique (20). CcO was reconstituted with dimyristoyl-phosphatidylcholine (DMPC) lipids and adhered to the surface of a diamond internal reflection element at 20°C. A flow-cell enabled exchange of the bathing buffer to induce Pm and F in steady-state. Films of CcO were examined spectrophotometrically under simulated conditions of the ATR IR experiments by drying the protein–lipid solution onto the inner wall of a conventional 1-cm path length quartz cuvette. The film was then immersed in oxygenated buffer containing 50 mM phosphate, 100 mM KCl (pH 8.5), and an O spectrum (Shimadzu UV–2101PC) was recorded. For Pm, the buffer was saturated with CO and O2 at pH 8.5. Likewise, fully reduced spectra were recorded in the presence of a degassed solution of 50 mM phosphate, 90 mM KCl, and 10 mM Na2S2O4 (pH 8.5). F was created via perfusion with 50 mM phosphate, 1 mM H2O2 (pH 8.5) (24). The concentration of H2O2 was calibrated spectrophotometrically by using the extinction coefficient 40 mM–1·cm–1 at 240 nm (24). For each intermediate, the films were cycled back to O with several washes of unsaturated 50 mM phosphate, 100 mM KCl (pH 8.5), and the measurements were repeated. In each case, nine 5,000-scan cycles at an optical resolution of 2 cm–1 were averaged. Further details about the sample preparation and spectroscopic methods have been described elsewhere (20).
Fig. 3.

Difference spectroscopy of the reaction intermediates of CcO. (A)
Absorption difference spectra in the visible wavelength range. The
two-electron reduced CO-bound state (R2CO) was generated by
bubbling CO through a detergent solubilized solution of CcO. The visible
spectrum (red trace) exhibits characteristic maxima at 590 nm of the
α-band and at 432 nm in the Soret region. The shoulder on the latter
band at 448 nm indicates some overreduction after the time-resolved IR
experiments. Pm was created by applying a saturated solution of
equimolar amounts of CO and O2 to a protein–lipid film
adhered to the inner surface of a quartz cuvette. The corresponding difference
spectrum (Pm-O, middle trace) shows positive bands at 437 and 607
nm. F was established by the action of H2O2 on the
protein-lipid film. The F-O difference spectrum (black trace, bottom) shows
characteristic maxima at 434 and 580 nm. (B) Infrared spectra of CcO
from R. sphaeroides, showing the E-R2CO (red trace, top),
Pm-O (blue trace, middle), and F-O (black trace, bottom)
differences. (C) Enlarged view (×10) of the difference bands of
wild-type CcO in the region where carbonyl vibrations of protonated carboxylic
acids are expected. The dashed spectra are the corresponding spectra of the
E286D mutant measured under identical conditions. The dashed vertical line
indicates the frequency of the  stretching vibration of the carboxylic
acid side chain of E286. For comparison, the change in the carbonyl stretch of
E286 in the transition from the oxidized to the fully reduced state
(R4-O) is shown in green [top traces; the fully reduced state has
been prepared as published
(20) but at pH 8.5].
stretching vibration of the carboxylic
acid side chain of E286. For comparison, the change in the carbonyl stretch of
E286 in the transition from the oxidized to the fully reduced state
(R4-O) is shown in green [top traces; the fully reduced state has
been prepared as published
(20) but at pH 8.5].
Results and Discussion
The E-to-R2CO Transition. As with other
hemoproteins, carbon monoxide can be bound in place of dioxygen under
anaerobic conditions. In CcO, the binding site is the central iron of the
reduced heme a3. The visible difference spectrum (red
trace in Fig. 3A)
exhibits characteristic maxima at 590 nm of the α-band and at 432 nm in
the Soret region for the two-electron reduced CO-bound state
(R2CO). A fortuitous feature of the
Fea3–CO bond is that it is photolabile, hence
dissociable with a laser pulse. After photodissociation of CO from heme
a3, the reduction potential of heme a is higher
relative to that of heme a3, and electron transfer between
heme a3 and heme a ensues
(23). When this occurs, a
state of the enzyme is transiently created in which the heme
a3-CuB binuclear center carries only one
electron, the distal heme ligand (CO) is absent, and heme a is
reduced. We refer to this as the E state. Time-resolved IR difference spectra
were acquired across the range from 4,000 to 800
cm–1. The dissociation of CO from the heme
a3 Fe was monitored by the negative band at 1,967
cm–1 of the  stretching vibration (data not
shown). This frequency is indicative for the presence of the two-electron
reduced state. A slight overreduction, which appears at the end of the
time-resolved Fourier transform IR experiment (see shoulder at 448 nm in the
top spectrum of Fig.
3A), would shift the band toward 1,965
cm–1
(25). In the region below
1,800 cm–1, numerous difference bands appear (red
spectrum in Fig. 3B).
The absorbance changes are much bigger than those observed for
photodissociation of CO from fully reduced CcO
(25), particularly in the
amide I (≈1,650 cm–1) and the amide II regions
(≈1,540 cm–1) where the peptide bond has
vibrational modes. This fact indicates that conformational changes of the
protein backbone take place during the E-to-R2 reaction, i.e., when
an electron is transferred from heme a to the binuclear center. Such
conformational changes alter the microenvironment of particular residues. For
acidic residues such as glutamic acid, the modulation can decrease the
apparent pKa and effect proton dissociation. Deprotonation in the
E-to-R2CO transition is indeed observed for E286. A negative band
at 1,745 cm–1 appears in the E-R2CO
difference spectrum of the wild-type enzyme (continuous red trace in
Fig. 3C) but
disappears in the spectrum on mutation of glutamic acid 286 to an aspartatic
acid (E286D; dashed red trace in Fig.
3C). Consequently, this band is assigned to the
stretching vibration of E286. Band fitting reveals that the full width at half
maximum of the 1,745 cm–1 band is 8
cm–1. This value is almost identical to that of
the stretch of D96 of bacteriorhodopsin, i.e., 9
cm–1, observed for the N-BR difference spectrum of
the E194Q mutant (26). This
agreement supports the finding that the negative band at 1,745
cm–1 in Fig.
3C is solely attributable to the deprotonation of E286. A
conformational change of this residue would lead to a sigmoidal shaped band
feature where the apparent band width of the negative part is ≈5
cm–1. Even stronger evidence in favor of a
deprotonation would be the assignment of the vibrational modes of the
corresponding carboxylate. However, the symmetric and asymmetric carboxylate
modes at 1,300–1,420 cm–1 and
1,550–1,610 cm–1, respectively, are
difficult to disentangle because of the strong overlap with other bands in
these frequency domains (27).
These considerations also apply to the Pm-O difference spectrum
discussed below. The possible presence of overreduced protein does not
influence the conclusion of the deprotonation of E286 because the fully
reduced enzyme does not exhibit any changes in the region around 1,745
cm–1
(25).
stretching vibration (data not
shown). This frequency is indicative for the presence of the two-electron
reduced state. A slight overreduction, which appears at the end of the
time-resolved Fourier transform IR experiment (see shoulder at 448 nm in the
top spectrum of Fig.
3A), would shift the band toward 1,965
cm–1
(25). In the region below
1,800 cm–1, numerous difference bands appear (red
spectrum in Fig. 3B).
The absorbance changes are much bigger than those observed for
photodissociation of CO from fully reduced CcO
(25), particularly in the
amide I (≈1,650 cm–1) and the amide II regions
(≈1,540 cm–1) where the peptide bond has
vibrational modes. This fact indicates that conformational changes of the
protein backbone take place during the E-to-R2 reaction, i.e., when
an electron is transferred from heme a to the binuclear center. Such
conformational changes alter the microenvironment of particular residues. For
acidic residues such as glutamic acid, the modulation can decrease the
apparent pKa and effect proton dissociation. Deprotonation in the
E-to-R2CO transition is indeed observed for E286. A negative band
at 1,745 cm–1 appears in the E-R2CO
difference spectrum of the wild-type enzyme (continuous red trace in
Fig. 3C) but
disappears in the spectrum on mutation of glutamic acid 286 to an aspartatic
acid (E286D; dashed red trace in Fig.
3C). Consequently, this band is assigned to the
stretching vibration of E286. Band fitting reveals that the full width at half
maximum of the 1,745 cm–1 band is 8
cm–1. This value is almost identical to that of
the stretch of D96 of bacteriorhodopsin, i.e., 9
cm–1, observed for the N-BR difference spectrum of
the E194Q mutant (26). This
agreement supports the finding that the negative band at 1,745
cm–1 in Fig.
3C is solely attributable to the deprotonation of E286. A
conformational change of this residue would lead to a sigmoidal shaped band
feature where the apparent band width of the negative part is ≈5
cm–1. Even stronger evidence in favor of a
deprotonation would be the assignment of the vibrational modes of the
corresponding carboxylate. However, the symmetric and asymmetric carboxylate
modes at 1,300–1,420 cm–1 and
1,550–1,610 cm–1, respectively, are
difficult to disentangle because of the strong overlap with other bands in
these frequency domains (27).
These considerations also apply to the Pm-O difference spectrum
discussed below. The possible presence of overreduced protein does not
influence the conclusion of the deprotonation of E286 because the fully
reduced enzyme does not exhibit any changes in the region around 1,745
cm–1
(25).
The exchange from glutamic to aspartic acid usually leads to a frequency
shift of the stretch which is indeed observed in the fully
reduced-minus-oxidized (R4-O) difference spectrum of the
R. sphaeroides E286D oxidase (green traces in
Fig. 3C; see
(20) for a more detailed
description). The R4-O difference spectrum of the wild-type CcO
(continuous green trace in Fig.
3C) reveals that this transition involves only a change
in hydrogen bonding (20). Such
a shift is not observed, however, in the E-R2CO difference spectrum
(red trace in Fig.
3C). Because the difference band of D286 is small
in the R4-O difference (dashed green trace in
Fig. 3C), the
corresponding band may be below the detection limit of the E-R2CO
difference experiment. Nevertheless, it seems that the exact location of the
carboxylic group at position 286 is very critical for the kinetics of proton
pumping in CcO. It is important to note that the E286D mutant, although
somewhat slower than the wild-type enzyme, appears fully functional and pumps
protons (28). The
R2CO-to-E transition has been shown to be associated with proton
release from the enzyme (28).
Because proton release is supposed to take place also from the E286Q mutant
(29), E286 cannot be the
origin of the proton but a proton from a different group is released to the
external medium. We attribute the deprotonation observed in the
E-R2CO IR experiments to an internal transfer of the E286 proton to
an unidentified proton acceptor on electron transfer from heme
a3 to heme a.
The E-R2CO difference spectrum of the R. sphaeroides enzyme (red trace in Fig. 3B) agrees well with that reported for the CcO from P. denitrificans (30). However, we assign the negative band specifically to the deprotonation of E286 rather than to an unspecified conformational change. Our experiments demonstrate that E286 is deprotonated in state E and is reprotonated when an electron is transferred from hemea to hemea3. Amide difference bands indicate that significant protein backbone changes accompany this transition.
The Pm-to-O Transition. Vibrational differences for intermediates Pm and F were examined with ATR technique, which employs a protein sample in contact with buffer that can be exchanged (20). Perfusion of CcO with buffers of differing chemical composition enables stable steady-state generation of oxygen intermediates Pm and F. Pm was trapped by applying a saturated solution of equimolar amounts of CO and O2. The establishment of the reaction intermediate was again verified by visible absorbance spectroscopy (Fig. 3A, blue middle trace) with characteristic difference bands at 607 nm (+) and at 414 (–)/439 nm (+) (31). The corresponding IR difference spectrum acquired with the perfusion-induced ATR technique is presented in Fig. 3B (blue middle trace). This spectrum essentially agrees with that previously observed for bovine CcO (21), although no role for glutamic acid E286 could be definitively concluded from that work. We are clearly able to detect a negative band at 1,745 cm–1 of the Pm-O difference spectrum (blue continuous trace in Fig. 3C), which downshifts in D2O by 6 cm–1 (data not shown), as expected for a carboxylic acid. The assignment was again performed with the E286D mutant and, as was the case for the E-R2CO difference spectrum, the signal at 1,745 cm–1 disappears (blue dashed trace in Fig. 3C). The absence of significant positive features in this wavelength range signifies that E286 deprotonates during the transition from O to Pm.
It is expected that the active site tyrosine (Y288) forms a neutral radical
in state Pm. Results from EPR
(32) and radioactive iodide
labeling experiments (4) have
supported this conclusion. It should be stressed that IR spectroscopy is not
the optimal method to detect radicals in a protein. However, three positive
bands in the Pm-O difference spectrum indicate the presence of a
tyrosine radical in Pm. The 1,587-cm–1
band concurs with the  stretching vibration observed at 1,587
cm–1 in the deprotonated neutral radical form of
ortho-imidazole-bound para-cresol, a model compound for the
crosslinked histidine–tyrosine
(33). Either of the two
positive bands at 1,528 cm–1 and 1,517
cm–1 in the Pm-O difference spectrum
can be assigned to the C–C stretch of the radical form of cross-linked
histidine-phenol observed at 1,522 cm–1
(34). The
1,479-cm–1 band could correspond to the
stretching vibration observed at 1,587
cm–1 in the deprotonated neutral radical form of
ortho-imidazole-bound para-cresol, a model compound for the
crosslinked histidine–tyrosine
(33). Either of the two
positive bands at 1,528 cm–1 and 1,517
cm–1 in the Pm-O difference spectrum
can be assigned to the C–C stretch of the radical form of cross-linked
histidine-phenol observed at 1,522 cm–1
(34). The
1,479-cm–1 band could correspond to the  stretch of a tyrosyl radical of Y288
(35). Bands at similar
frequency have also been observed in the radical forms of model compounds
(33,
34). However, it must be noted
that although these bands are consistent with the formation of a tyrosyl
radical, contributions from vibrations of a variety of other groups, in
particular from heme modes, cannot be excluded. Similar features are also
present in the Pm-O difference spectrum of the bovine enzyme
(21), subject to the same
uncertainties.
stretch of a tyrosyl radical of Y288
(35). Bands at similar
frequency have also been observed in the radical forms of model compounds
(33,
34). However, it must be noted
that although these bands are consistent with the formation of a tyrosyl
radical, contributions from vibrations of a variety of other groups, in
particular from heme modes, cannot be excluded. Similar features are also
present in the Pm-O difference spectrum of the bovine enzyme
(21), subject to the same
uncertainties.
It should be noted that time-resolved visible absorption spectroscopy studies (28, 29) have examined the P-to-F and F-to-O transitions in the reaction of the fully reduced oxidase with O2. The data suggest that proton transfer from E286 is rate-limiting in each of these steps. However, the P intermediate in this reaction, termed Pr, differs from Pm in that the heme–copper binuclear center is reduced by one more electron. The active-site tyrosine is thought not to be a neutral radical in the Pr state, but to be reduced to either tyrosine or tyrosinate (36). Hence, because the electron distribution is different for these two versions of the P intermediate, the protonation state of residues (E286 in particular) may also be different in Pr and Pm.
The F-to-O Transition. Although the Pm and F states of
the enzyme both have oxoferryl structure
(Fe+4a32–),
their resonance-Raman and visible absorbance spectra differ considerably
(37). F can be generated by
exposing a solution of fully oxidized protein (state O) to an excess of
H2O2. When formed under alkaline conditions, the
H2O2-generated F intermediate has a visible absorbance
band at 580 nm (24). The black
spectrum in Fig. 3A
demonstrates that F is established under the conditions used for
perfusion-induced IR difference spectroscopy. The corresponding IR difference
spectrum (black trace at the bottom of Fig.
3B) of F shows spectral changes as small as those of the
O to Pm transition (blue trace). The most obvious difference
between these two spectra is the absence of a negative band at 1,745
cm–1 in the F-O difference spectrum
(Fig. 3C). Because
E286 is protonated in state O
(20), the absence of a change
in the E286 vibration in the F-O difference spectrum indicates that E286 is
protonated in F. It is therefore concluded that E286 is reprotonated during
the Pm to F transition.
The strongest difference band in the F-O spectrum is the sharp negative
band at 1,515 cm–1 (black trace in Figs.
3B and
4). This band matches with the
frequency expected for the phenyl ring stretching mode of protonated tyrosine,
which downshifts by 16 cm–1 on formation of the
tyrosinate (38). Indeed, there
is a positive band (i.e., associated with state F) at 1,499
cm–1, which is D2O-insensitive, as
expected if this features is caused by a tyrosinate/tyrosine (F/O) transition
(Fig. 4, red trace). Moreover,
the direct comparison to the pH-induced difference spectrum of the free amino
acid tyrosine reveals that there is also another band, namely at 1,248
cm–1, that coincides with a pH-sensitive tyrosine
vibration (Fig. 4, green
trace). Characteristically, the phenyl stretch at 1,515
cm–1 is down-shifted by only 2
cm–1 in D2O (red trace), whereas the
band at 1,249 cm–1 is up-shifted by 14
cm–1 because the coupling to the CO-H bending is
removed on H/D exchange leaving the pure stretch to be observable at
1,262 cm–1
(39). To account for the
possible presence of a covalent linkage of Y288 to H284, the spectrum of a
model compound is depicted where imidazol is bound via the N
(1) to p-cresol in
ortho position (40).
Indeed, the strong negative band at 1,546 cm–1,
which is not present in the pH-induced difference spectrum of tyrosine,
appears in the IR absorption spectrum of the model compound
(Fig. 4, blue spectrum). This
band has been assigned to a coupled Tyr and His ring mode with large
contributions from the CN of the covalent bond between both ring systems
(40). Although the pH-induced
difference spectrum of the model compound is not yet available, it is
reasonable to assume that the frequency of this mode is changed on
deprotonation of the phenol. Because the bands at 1,515 and 1,455
cm–1 of the F–O difference spectrum of CcO
also coincide with bands of the Tyr–His model compound, the data are in
line with the presence of a covalently linked Y288-H284 as observed for CcO
from other organisms
(6–8).
However, the recently published structure of CcO from R. sphaeroides
(9) lacks support for this
cross-link. Apart from the tyrosine modes, either cross-linked or not, the F-O
difference spectrum exhibits negative bands 1,687, 1,656, and 1,612
cm–1. These are essentially insensitive to
H2O/D2O exchange and may reflect heme modes.
Fig. 4.

Infrared F-O difference spectra of CcO compared with the pH-induced difference of tyrosine. The red trace is the F-O difference in D2O. The black trace corresponds to the sample measurement in H2O (same as in Fig. 3B but displayed across a broader frequency range and enlarged in the absorbance axis). The green spectrum is the vibrational difference spectrum between a 2 mM solution of tyrosine at pH 12 (Tyr-O–) and pH 8 (Tyr-OH). The IR spectrum of the synthetic compound 2-(4-methyl-1H-imidazol-1-yl)-4-methylphenol, which models the covalently linked amino acids Y288-H284, is depicted in blue. Data have kindly been provided by W. Woodruff (see ref. 40 for details).
Conclusions
The IR difference spectra presented here are consistent with the role of E286 as a proton shuttle. Taken together, the IR data indicate that E286 changes its protonation state at least four times during the course of the cycle. The residue(s) playing the role of proton donor and acceptor may be different for different transitions, and they remain as yet unidentified. The pumping steps from E-to-R2 and Pm-to-F occur from a state where E286 is deprotonated whereas the pumping step from F-to-O occurs from a state where E286 is apparently protonated. This demonstrates the fact that proton pumping clearly cannot be explained by the protonation state of E286 alone. The roles of other residues and their sequence of protonation states remain to be elucidated before this vectorial proton pump can be understood. The acidity of the E286 side chain is subject to large changes depending on the redox status of the metal centers. The shift in the pKa of E286 implies the need for a conformational change that will result in a stabilization of the deprotonated form of the glutamate, which is in a hydrophobic environment in both the O and R2 states of the enzyme. This situation is reminiscent of that for D96 in bacteriorhodopsin (13, 41), where conformational changes introduce water to link the residue to enable proton transfer to the Schiff base and also transiently stabilize the deprotonated form of D96.
In addition to the central role of E286, the IR difference spectra provide experimental support for Y288 playing an active role in catalysis. This tyrosine is protonated in the fully oxidized enzyme, is a neutral radical in state Pm, and is reduced to the anionic tyrosinate in state F. The IR data are also consistent with the presence of the cross-linked histidine-tyrosine in the R. sphaeroides oxidase.
Based on the status of E286 and Y288 in the various intermediate states, we suggest the following reaction scheme of catalysis by CcO (Fig. 2). The first electron transfer to reduce CuB in the E state leads to the dissociation of the bound hydroxide. The high proton affinity of the OH anion abstracts a proton from E286 to form water. The proton taken up during this reaction (42), is used in the subsequent reaction step. The second electron to reduce heme a3 in the E-to-R2 step triggers the reprotonation of E286. Because proton translocation occurs during this transition (11), the uptaken proton might be the translocated one. The conformational change of the protein backbone, which is deduced from the observed amide I differences (Fig. 3B red spectrum), initiates either of the two proton transfer reactions. Additionally, it is quite plausible that an electrostatic rearrangement drives the changes in protonation state (43). The R2-to-Pm transition is accompanied by neither proton uptake nor translocation. However, binding and fission of dioxygen drives internal proton rearrangements, such as deprotonation of E286 and Y288, the latter becoming a radical. Of the two protons, one is required for the establishment of the proposed hydroxy ligand of CuB. The other may be transferred to a residue of the proton exit pathway, such as the ring D propionate of heme a3 (9). The third electron transforms the radical of Y288 into the phenolate. The uptake of a proton during the lifetime of the F intermediate instigates the reprotonation of E286. Proton release to the extracellular surface takes place from the aforementioned residue of the exit pathway. Thus, proton translocation is accomplished during the Pm-to-F transition. In the final reaction steps from F to O, the fourth electron reduces heme a3. Concomitant protonation of the oxo-ferryl shifts the hydroxy ligand to CuB. Associated with these steps are reprotonation of Y288 to form the phenolic tyrosine and vectorial proton translocation across the protein.
Finally, it is important to point out that the states examined in the current work, E, Pm, F and O, are really starting and ending points of electron transfer-induced transitions that result in proton pumping. The states that are critically involved in proton pumping are the transient states that form during these transitions, such as from F to O. Virtually nothing is known about these states of CcO. The current work is a significant step along the way toward defining the spectroscopic features needed to exploit time-resolved Fourier transform IR spectroscopy which will ultimately reveal the dynamics of the proton pump in action.
Acknowledgments
We are grateful to W. Woodruff (Los Alamos, NM) for providing the IR spectrum of the Tyr–His model compound and for helpful discussions. This work was supported National Institutes of Health Grant HL16101 (to R.B.G.) and Training Grant 5-T32 GMO8276-13 (to R.M.N.), a grant from the Deutscher Akademischer Austauschdienst (to R.M.N.), Deutsche Forschungsgemeinschaft Grant SFB-189, Project C6 (to J.H.), and a grant from the Volkswagen Foundation (to J.H.).
This paper was submitted directly (Track II) to the PNAS office.
Abbreviations: CcO, cytochrome c oxidase; ATR, attenuated total reflection.
References
- 1.Zaslavsky, D. & Gennis, R. B. (2000) Biochim. Biophys. Acta 1458, 164–179. [DOI] [PubMed] [Google Scholar]
- 2.Babcock, G. T. & Ferguson-Miller, S. (1996) Chem. Rev. 96, 2889–2907. [DOI] [PubMed] [Google Scholar]
- 3.Gennis, R. B. (1998) Biochim. Biophys. Acta 1365, 241–248. [DOI] [PubMed] [Google Scholar]
- 4.Proshlyakov, D. A., Pressler, M. A., DeMaso, C., Leykam, J. F., DeWitt, D. L. & Babcock, G. T. (2000) Science 290, 1588–1591. [DOI] [PubMed] [Google Scholar]
- 5.Fabian, M., Wong, W. W., Gennis, R. B. & Palmer, G. (1999) Proc. Natl. Acad. Sci. USA 96, 13114–13117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ostermeier, C., Harrenga, A., Ermler, U. & Michel, H. (1997) Proc. Natl. Acad. Sci. USA 94, 10547–10553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tsukihara, T., Aoyama, H., Yamashita, E., Tomizaki, T., Yamaguchi, H., Shinzawa-Itoh, K., Nakashima, R., Yaono, R. & Yoshikawa, S. (1996) Science 272, 1136–1144. [DOI] [PubMed] [Google Scholar]
- 8.Soulimane, T., Buse, G., Bourenkov, G. P., Bartunik, H. D., Huber, R. & Than, M. E. (2000) EMBO J. 19, 1766–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Svensson-Ek, M., Abramson, J., Larsson, G., Tornroth, S., Brzezinski, P. & Iwata, S. (2002) J. Mol. Biol. 321, 329–339. [DOI] [PubMed] [Google Scholar]
- 10.Verkhovskaya, M. L., Garcia-Horsman, A., Puustinen, A., Rigaud, J. L., Morgan, J. E., Verkhovsky, M. I. & Wikström, M. (1997) Proc. Natl. Acad. Sci. USA 94, 10128–10131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruitenberg, M., Kannt, A., Bamberg, E., Fendler, K. & Michel, H. (2002) Nature 417, 99–102. [DOI] [PubMed] [Google Scholar]
- 12.Wikström, M. & Verkhovsky, M. (2002) Biochim. Biophys. Acta 1555, 128. [DOI] [PubMed] [Google Scholar]
- 13.Heberle, J. (2000) Biochim. Biophys. Acta 1458, 135–147. [DOI] [PubMed] [Google Scholar]
- 14.Mitchell, R. & Rich, P. R. (1994) Biochim. Biophys. Acta 1186, 19–26. [DOI] [PubMed] [Google Scholar]
- 15.Proshlyakov, D. A., Ogura, T., Shinzawa-Itoh, K., Yoshikawa, S. & Kitagawa, T. (1996) Biochemistry 35, 8580–8586. [DOI] [PubMed] [Google Scholar]
- 16.Wikström, M. (1989) Nature 338, 776–778. [DOI] [PubMed] [Google Scholar]
- 17.Michel, H. (1999) Biochemistry 38, 15129–15140. [DOI] [PubMed] [Google Scholar]
- 18.Hellwig, P., Rost, B., Kaiser, U., Ostermeier, C., Michel, H. & Mäntele, W. (1996) FEBS Lett. 385, 53–57. [DOI] [PubMed] [Google Scholar]
- 19.Lübben, M. & Gerwert, K. (1996) FEBS Lett. 397, 303–307. [DOI] [PubMed] [Google Scholar]
- 20.Nyquist, R. M., Heitbrink, D., Bolwien, C., Wells, T. A., Gennis, R. B. & Heberle, J. (2001) FEBS Lett. 505, 63–67. [DOI] [PubMed] [Google Scholar]
- 21.Iwaki, M., Breton, J. & Rich, P. (2002) Biochim. Biophys. Acta 1555, 116–121. [DOI] [PubMed] [Google Scholar]
- 22.Mitchell, D. M. & Gennis, R. B. (1995) FEBS Lett. 368, 148–150. [DOI] [PubMed] [Google Scholar]
- 23.Brzezinski, P. & Malmström, B. G. (1985) FEBS Lett. 187, 111–114. [DOI] [PubMed] [Google Scholar]
- 24.Jünemann, S., Heathcote, P. & Rich, P. R. (2000) Biochim. Biophys. Acta 1456, 56–66. [DOI] [PubMed] [Google Scholar]
- 25.Heitbrink, D., Sigurdson, H., Bolwien, C., Brzezinski, P. & Heberle, J. (2002) Biophys. J. 82, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zscherp, C., Schlesinger, R. & Heberle, J. (2001) Biochem. Biophys. Res. Commun. 283, 57–63. [DOI] [PubMed] [Google Scholar]
- 27.Dioumaev, A. K. (2001) Biochemistry 66, 1269–1276. [DOI] [PubMed] [Google Scholar]
- 28.Ädelroth, P., Karpefors, M., Gilderson, G., Tomson, F. L., Gennis, R. B. & Brzezinski, P. (2000) Biochim. Biophys. Acta 1459, 533–539. [DOI] [PubMed] [Google Scholar]
- 29.Ädelroth, P., Svensson-Ek, M., Mitchell, D. M., Gennis, R. B. & Brzezinski, P. (1997) Biochemistry 36, 13824–13829. [DOI] [PubMed] [Google Scholar]
- 30.Rost, B., Behr, J., Hellwig, P., Richter, O. M., Ludwig, B., Michel, H. & Mäntele, W. (1999) Biochemistry 38, 7565–7571. [DOI] [PubMed] [Google Scholar]
- 31.Wikström, M. & Morgan, J. E. (1992) J. Biol. Chem. 267, 10266–10273. [PubMed] [Google Scholar]
- 32.MacMillan, F., Kannt, A., Behr, J., Prisner, T. & Michel, H. (1999) Biochemistry 38, 9179–9184. [DOI] [PubMed] [Google Scholar]
- 33.Aki, M., Ogura, T., Naruta, Y., Le, T. H., Sato, T. & Kitagawa, T. (2002) J. Phys. Chem. A 106, 3436–3444. [Google Scholar]
- 34.Cappuccio, J. A., Ayala, I., Elliott, G. I., Szundi, I., Lewis, J., Konopelski, J. P., Barry, B. A. & Einarsdottir, O. (2002) J. Am. Chem. Soc. 124, 1750–1760. [DOI] [PubMed] [Google Scholar]
- 35.Uchida, T., Mogi, T. & Kitagawa, T. (2000) Biochemistry 39, 6669–6678. [DOI] [PubMed] [Google Scholar]
- 36.Karpefors, M., Ädelroth, P., Namslauer, A., Zhen, Y. & Brzezinski, P. (2000) Biochemistry 39, 14664–14669. [DOI] [PubMed] [Google Scholar]
- 37.Proshlyakov, D. A., Pressler, M. A. & Babcock, G. T. (1998) Proc. Natl. Acad. Sci. USA 95, 8020–8025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Venyaminov, S. Y. & Kalnin, N. N. (1990) Biopolymers 30, 1243–1257. [DOI] [PubMed] [Google Scholar]
- 39.Hienerwadel, R., Boussac, A., Breton, J., Diner, B. A. & Berthomieu, C. (1997) Biochemistry 36, 14712–14723. [DOI] [PubMed] [Google Scholar]
- 40.Tomson, F., Bailey, J. A., Gennis, R. B., Unkefer, C. J., Li, Z., Silks, L. A., Martinez, R. A., Donohoe, R. J., Dyer, R. B. & Woodruff, W. H. (2002) Biochemistry 41, 14383–14390. [DOI] [PubMed] [Google Scholar]
- 41.Zscherp, C., Schlesinger, R., Tittor, J., Oesterhelt, D. & Heberle, J. (1999) Proc. Natl. Acad. Sci. USA 96, 5498–5503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ruitenberg, M., Kannt, A., Bamberg, E., Ludwig, B., Michel, H. & Fendler, K. (2000) Proc. Natl. Acad. Sci. USA 97, 4632–4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kannt, A., Lancaster, C. R. & Michel, H. (1998) Biophys. J. 74, 708–721. [DOI] [PMC free article] [PubMed] [Google Scholar]